
Abstract
Mitochondrial-derived peptides are encoded by mitochondrial DNA but have biological activity outside mitochondria. Eight of these are encoded by sequences within the mitochondrial 12S and 16S ribosomal genes: humanin, MOTS-c, and the six SHLP peptides, SHLP1-SHLP6. These peptides have various effects in cell culture and animal models, affecting neuroprotection, insulin sensitivity, and apoptosis, and some are secreted, potentially having extracellular signaling roles. However, except for humanin, their importance in normal cell function is unknown. To gauge their importance, their coding sequences in vertebrates have been analyzed for synonymous codon bias. Because they lie in RNA genes, such bias should only occur if their amino acids have been conserved to maintain biological function. Humanin and SHLP6 show strong synonymous codon bias and sequence conservation. In contrast, SHLP1, SHLP2, SHLP3, and SHLP5 show no significant bias and are poorly conserved. MOTS-c and SHLP4 also lack significant bias, but contain highly conserved N-terminal regions, and their biological importance cannot be ruled out. An additional potential mitochondrial-derived peptide sequence was discovered preceding SHLP2, named SHLP2b, which also contains a highly conserved N-terminal region with synonymous codon bias.
Similar content being viewed by others


Introduction
In this paper we evaluate the notion that peptides encoded by short open reading frames (ORFs) necessarily have important biological functions1. In particular, we focus on the mitochondrial-derived peptides (MDPs), of which humanin is the most well-known. When humanin was discovered over twenty years ago, it was the first of its kind, a peptide encoded by mitochondrial DNA but with biological activity outside the mitochondrion. It was found in a screen for neuroprotective peptides using a cDNA library created from the surviving neurons of an Alzheimer’s disease patient2. Prior to this, the mitochondrial genome was thought to only encode 13 proteins, which all remain inside the mitochondria. However, humanin was found to inhibit apoptosis, interacting with the pro-apoptosis proteins BAX, BIM, BID and IGFBP-3 in the cell cytosol5,6,7,8. Furthermore, secreted humanin was detected in cell culture medium and extracellular humanin was found to interact with cell-surface receptor proteins FPRL-1 and CNTFR/WSX-1/gp130, which are involved in metabolism and inflammation signaling2,9,10. Interestingly, its anti-apoptotic property can also potentially be detrimental, as beyond a certain threshold, inhibiting cell death can promote tumor growth in cancer11,12,13.
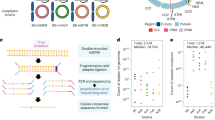
Humanin is unique in another way; it is encoded by a nested, overlapping gene within an RNA gene. The sequence encoding humanin lies within MT-RNR2, the gene for the 16S subunit of the mitochondrial ribosome. Recent searches of the mitochondrial 16S and 12S RNA genes have identified seven additional MDPs with potential biological activity. These are the six SHLPs (Small Humanin-Like Peptides), SHLP1-SHLP6, found within MT-RNR2, and MOTS-c (Mitochondrial Open-reading-frame of the Twelve S rRNA type-c), within the mitochondrial 12S RNA gene MT-RNR114,15. A diagram showing the locations of their ORFs within the 16S and 12S RNA genes is shown in Fig. 1. Several potential biological activities for the SHLP peptides have been observed, including enhancement of cell survival by SHLP2 and SHLP3, cell proliferation by SHLP4, and induction of apoptosis by SHLP614. MOTS-c production is increased by exercise, and treatment with exogenous peptide confers many of the same benefits as exercise in cell and animal models, including enhancement of insulin sensitivity15,16.

Location of MDPs in mtDNA. The locations of the MDP coding sequences in the 16S and 12S RNA genes (green) is indicated, with numbers indicating the SHLP peptides, SHLP1-SHLP6, and humanin (HN) and MOTS-c indicated. The arrows indicate coding by the mtDNA H-strand (pointing left) or L-strand (pointing right). Below, a diagram of the full circular mtDNA is shown for comparison. The tRNA sequences are in light green, the D-loop in red, and the protein genes in shades of blue and purple.
To demonstrate a peptide is biologically important, one can show that lowering its amount leads to dysfunction in the cell. While there is currently no way to knock out mitochondrial ORFs, they can be knocked down with siRNA (small interfering RNA). This has been done for humanin, where knockdown reduced its anti-apoptotic and neuroprotective effects5,17. However, such results for the other MDPs have not been forthcoming. One can also show the importance of a peptide using a more straightforward test which relies on evolution. Natural selection will conserve the amino acids important for the function of the peptide, so one can look for sequence conservation across species. Unfortunately, because the coding sequences of the MDPs lie within ribosomal genes, sequence conservation alone is insufficient as it might instead reflect the importance of the corresponding RNA bases for ribosome function. A complementary approach is to look for synonymous codons in the peptide-coding sequences. Like amino acid sequence conservation, synonymous codon bias is evidence of purifying, or negative, natural selection that has been “weeding out” non-synonymous amino acid mutations over the course of evolution. On the other hand, for non-peptide-coding regions of the RNA genes, there should be no bias. Thus, in this study, in addition to looking for sequence conservation, we examine synonymous codon bias in the MDP coding sequences. In doing so, not only do we determine which MDPs have been conserved by evolution but also identify shared features that offer clues to their biological roles.
Results
Humanin is conserved across vertebrates with many residues showing synonymous codon bias
Figure 2 shows the consensus base and amino acid sequence logos for the primate, mammal, and vertebrate humanin alignments, with the corresponding fsyn values below. Amino acid positions with fsyn ≥ 0.5, that is, with equal or more synonymous mutations than non-synonymous mutations, are highlighted. Additional details on the number of mutations and sequence conservation are given in supplemental Fig. S1. Figure 2 and the figures following show the results using the standard DNA code. Results using the vertebrate mitochondrial DNA code are also included in figure S1, with the only major difference being that the codon in position 22 becomes a stop codon, yielding a 21 amino acid peptide instead of 24. A curious feature of the humanin sequences is that start codon methionine is not particularly well-conserved. Mutations of the start codon occur across all vertebrate branches with an overall conservation rate of 79%, ranging from 96% in birds to 46% in reptiles. The stop codon in position 25 is even less well conserved, with an overall conservation of 58%.

Synonymous codon bias in humanin. (A) Humanin sequence in humans. Sequence logos of the base and amino acid sequence alignments with the synonymous codon bias fsyn below in (B) primates (number of species = 252), (C) mammals (number of species = 148), and (D) vertebrates (number of species = 359). The base and amino acid letter heights indicate how conserved they are across species. Invariant bases and amino acids with invariant codons are indicated with asterisks. The “X” symbol stands for stop codon. Values of fsyn ≥ 0.5 are highlighted in red. Because methionine has only one codon in the standard DNA code, it has no fsyn value. The color code for the amino acids is hydrophobic (black), acidic (red), basic (blue), and neutral hydrophilic (green for glycine and hydroxyl-containing residues and magenta for amide-containing).
The results for vertebrates show two regions of highly conserved or invariant bases, corresponding to the codons for C8, L9 and E15, I16. This conservation might be due to these bases being critical for ribosome function, or it could be due to the corresponding amino acids being critical for humanin function, or a combination of both. On the other hand, there are highly conserved amino acids with high fsyn values in regions with less well conserved base sequences, A2, L10, D17, L18 and V20, suggesting that whereas these amino acids are important for humanin function, the corresponding bases are less important for ribosome function. The C-terminal region has highly conserved residues in mammals but shows more variation in non-mammals. Especially interesting is position 22, which in mammals is nearly always arginine, but in non-mammals is usually lysine, suggesting that a positive charge in this position is important for humanin function or processing. Some amino acids have high fsyn values in primates, F6 and R23, but not in other vertebrates, and vice versa, G5 and D17. This could reflect some specialization of function involving those residues present in one branch of vertebrates but absent in others, or the high fsyn values could be due to chance. The probability of a fsyn > 0.5 happening by chance is estimated empirically to be in the range 0.035–0.065, as described in a later section that examines the statistical significance of the MDP analyses (see Statistical analysis).
MOTS-c contains a highly conserved pentapeptide sequence, MGYIF, but has almost no synonymous codon bias
Figure 3 shows the consensus base and amino acid sequence logos for the primate, mammal, and vertebrate MOTS-c alignments, with the corresponding fsyn values below. Additional details on the number of mutations, sequence conservation, and differences using the vertebrate mitochondrial DNA code are given in supplemental Fig. S2. The most striking feature of the MOTS-c alignments is the highly conserved pentapeptide MGYIF in the middle of the sequence. The N-terminal region shows only moderate sequence conservation, and the C-terminal region shows even less. In contrast to humanin, nearly all the fsyn values are small, apart from G7 in the alignment for mammals. Thus, full-length MOTS-c does not show compelling evidence for natural selection in vertebrates. However, given the very high conservation of the MGYIF pentapeptide with its high fsyn value for G7 in mammals, it is possible that the pentapeptide is biologically important, at least in mammals.

Synonymous codon bias in MOTS-c. (A) MOTS-c sequence in humans. Sequence logos of the base and amino acid sequence alignments with the synonymous codon bias fsyn below in (B) primates (number of species = 254), (C) mammals (number of species = 178), and (D) vertebrates (number of species = 348). The base and amino acid letter heights indicate how conserved they are across species. Invariant bases and amino acids with invariant codons are indicated with asterisks. The “X” symbol stands for stop codon. Values of fsyn ≥ 0.5 are highlighted in red. Because methionine and tryptophan have only one codon in the standard DNA code, they have no fsyn values. The color code for the amino acids is hydrophobic (black), acidic (red), basic (blue), and neutral hydrophilic (green for glycine and hydroxyl-containing residues and magenta for amide-containing).
SHLP1, SHLP3, & SHLP5 show poor start codon conservation and almost no synonymous codon bias
Figure 4 shows the consensus base and amino acid sequence logos for primates for the SHLP1, SHLP3 and SHLP5 peptides, with the corresponding fsyn values below. Additional details on the number of mutations, sequence conservation, and differences using the vertebrate mitochondrial DNA code are given in supplemental Fig. S3. None of these SHLP peptides show compelling evidence of natural selection in primates. The highlighted positions with high fsyn values, one per peptide, could easily be due to chance. In addition, their start codons are poorly conserved, 33% for SHLP1, 18% for SHLP3, and 30% for SHLP5, further arguing against important biological roles for these peptides. To confirm that SHLP1, SHLP3, and SHLP5 are not conserved in vertebrates, the human sequences were aligned with 19 non-primate vertebrate species (supplemental Table S37), again showing poor sequence conservation, especially with regard to the start codons. Because of this, full analyses with mammal and vertebrate multiple sequence alignments were not performed. Also, it should be noted that the poor conservation of the SHLP3 base sequence is somewhat misleading; it contains two regions with insertions/deletions of varying length, and when aligned allowing gaps, the base sequence conservation improves dramatically.

Synonymous codon bias in SHLP1, SHLP3, and SHLP5. (A) The human SHLP1 sequence above the corresponding sequence logos of the base and amino acid sequence alignments and synonymous codon bias fsyn values below in primates (number of species = 217). (B) The human SHLP3 sequence above the corresponding sequence logos of the base and amino acid sequence alignments and synonymous codon bias fsyn values below in primates (number of species = 221). (C) The human SHLP5 sequence above the corresponding sequence logos of the base and amino acid sequence alignments and synonymous codon bias fsyn values below in primates (number of species = 216). The base and amino acid letter heights indicate how conserved they are across species. Invariant bases and amino acids with invariant codons are indicated with asterisks. The “X” symbol stands for stop codon. Values of fsyn ≥ 0.5 are highlighted in red. Because methionine and tryptophan have only one codon in the standard DNA code, they have no fsyn values. The color code for the amino acids is hydrophobic (black), acidic (red), basic (blue), and neutral hydrophilic (green for glycine and hydroxyl-containing residues and magenta for amide-containing).
SHLP2 shows poor sequence conservation and almost no synonymous codon bias but is preceded by a sequence with high synonymous codon bias, SHLP2b
Initial analysis of the SHLP2 sequence alignment revealed a cluster of codons with high fsyn values preceding SHLP2 in the same open reading frame. This cluster starts with a standard methionine ATG start codon in most primate species, and this new, putative peptide is named SHLP2b. Because they overlap, the consensus base and amino acid sequence logos for SHLP2 and SHLP2b are shown together in Fig. 5, with the corresponding fsyn values below. Additional details on the number of mutations, sequence conservation, and differences using the vertebrate mitochondrial DNA code are given in supplemental Fig. S4. Humans have the alternate start codon GTG at the start of the putative SHLP2b peptide, as do most non-primate vertebrates18. In humans, the stop codon for SHLP2b would be the same as for SHLP2, but for primates in general, the stop codon would more often be in position 24 in Fig. 5, yielding a peptide 23 residues long.

Synonymous codon bias in SHLP2 and SHLP2b. (A) SHLP2b (blue line) and SHLP2 (green line) sequences in humans. Sequence logos of the base and amino acid sequence alignments with the synonymous codon bias fsyn below in (B) primates (number of species = 219), (C) mammals (number of species = 174), and (D) vertebrates (number of species = 369). The base and amino acid letter heights indicate how conserved they are across species. Invariant bases are indicated with asterisks. The “X” symbol stands for stop codon. Values of fsyn ≥ 0.5 are highlighted in red. Because methionine has only one codon in the standard DNA code, it has no fsyn value. The color code for the amino acids is hydrophobic (black), acidic (red), basic (blue), and neutral hydrophilic (green for glycine and hydroxyl-containing residues and magenta for amide-containing).
In primates SHLP2 has only one amino acid, in position 29 in Fig. 5, with an fsyn value greater than 0.5, but two others, in positions 15 and 16, have values close to 0.5. Because the choice of 0.5 as the fsyn threshold was somewhat arbitrary, SHLP2 was also analyzed at the mammal and vertebrate levels; however, the fsyn values were even lower. Thus, SHLP2 does not show compelling evidence of purifying selection. In contrast, the cluster of conserved, high fsyn value codons at the beginning of SHLP2b persists even at the vertebrate level, which suggests the putative SHLP2b peptide might be biologically important. Note that, like SHLP3, SHLP2 contains a region with insertion/deletions soon after the start codon, making the base sequence conservation appear misleadingly low in the figure.
SHLP4 contains a well-conserved N-terminal region with residues showing synonymous codon bias
Figure 6 shows the consensus base and amino acid sequence logos for the primate, mammal, and vertebrate SHLP4 alignments, with the corresponding fsyn values below. Additional details on the number of mutations, sequence conservation, and differences using the vertebrate mitochondrial DNA code are given in supplemental Fig. S5. In mammals, including primates, the N-terminal region is highly conserved, with many invariant bases, and is flanked by amino acids, L2 and R11, for example, with high fsyn values. The results suggest that the N-terminal region of SHLP4, residues 1–12, might have undergone purifying selection in mammals. In contrast, the rest of the peptide is more poorly conserved. The alignments of SHLP4 have a region of insertions/deletions in the middle of the sequence, which accounts for the poorer sequence conservation. In non-mammal vertebrates, the start codon is not well-conserved, but M5 is still highly conserved, and nearly all mutations that do occur for it are to the alternate start codon GTG. The results including all vertebrates are not as compelling as for mammals alone, but it cannot be ruled out that residues 5–12 have also undergone natural selection in non-mammals, with M5 serving as the start codon.

Synonymous codon bias in SHLP4. (A) SHLP4 sequence in humans. Sequence logos of the base and amino acid sequence alignments with the synonymous codon bias fsyn below in (B) primates (number of species = 215), (C) mammals (number of species = 144), and (D) vertebrates (number of species = 339). The base and amino acid letter heights indicate how conserved they are across species. Invariant bases and amino acids with invariant codons are indicated with asterisks. The “X” symbol stands for stop codon. Values of fsyn ≥ 0.5 are highlighted in red. Because methionine has only one codon in the standard DNA code, it has no fsyn value. The color code for the amino acids is hydrophobic (black), acidic (red), basic (blue), and neutral hydrophilic (green for glycine and hydroxyl-containing residues and magenta for amide-containing).
SHLP6 is the most conserved MDP with several residues showing synonymous codon bias
Figure 7 shows the consensus base and amino acid sequence logos for the primate, mammal, and vertebrate SHLP6 alignments, with the corresponding fsyn values below. Additional details on the number of mutations, sequence conservation, and differences using the vertebrate mitochondrial DNA code are given in supplemental Fig. S6. SHLP6 has the most highly conserved sequence of the MDPs presented here, and unlike the other MDPs, its stop codon is also highly conserved. In addition, it has several high fsyn value residues, thus the results suggest that the SHLP6 peptide has undergone purifying selection.

Synonymous codon bias in SHLP6. (A) SHLP6 sequence in humans. Sequence logos of the base and amino acid sequence alignments with the synonymous codon bias fsyn below in (B) primates (number of species = 242), (C) mammals (number of species = 147), and (D) vertebrates (number of species = 348). The base and amino acid letter heights indicate how conserved they are across species. Invariant bases and amino acids with invariant codons are indicated with asterisks. The “X” symbol stands for stop codon. Values of fsyn ≥ 0.5 are highlighted in red. Because methionine has only one codon in the standard DNA code, it has no fsyn value. The color code for the amino acids is hydrophobic (black), acidic (red), basic (blue), and neutral hydrophilic (green for glycine and hydroxyl-containing residues and magenta for amide-containing).
Statistical analysis
As already mentioned, high fsyn values can happen by chance. The base sequences before and after the MDPs can be used to get an empirical estimate of the baseline probability that fsyn ≥ 0.5 occurs by chance because these regions lie outside the ORFs. In the sequence alignments, 12–15 additional bases were included before the start codon and after the stop codon, that is, 4–5 additional “codons” before and after each peptide. Using all the MDPs (for SHLP2/SHLP2b, only the bases before SHLP2b and after SHLP2 are used) this yields 77 additional “codons,” five of which have values fsyn ≥ 0.5, giving a probability estimate of 0.065. This empirical approach makes no assumptions about the neutrality of synonymous mutations except that they tend to occur more frequently in conserved peptide coding regions than non-coding regions. Because SHLP1, SHLP3 and SHLP5 show no evidence of natural selection, they can be used as a consistency check. Their sequence alignments each have just one amino acid position with fsyn ≥ 0.5. SHLP1 and SHLP5 are 24 amino acids long, and SHLP3 is 38. Assuming these high fsyn values are due to chance, this gives an average probability of 0.035, which suggests the probability of 0.065 calculated with the sequences before and after the MDPs could be an overestimate. The regions used for calculating the two baseline probability estimates consist of 231 bases before and after the MDPs ORFs, and 258 bases of SHLP1, 3 and 5, totaling 489 bases. The 16S gene length is 1558 bases; thus, a considerable amount of the RNA genes has been sampled to obtain the baseline probability estimates. Varying base conservation, presence of insertions/deletions, and proximity to ORFs could influence the baseline probability and might account for some of the difference in the two baseline values.
Table 1 shows the p-values for each sequence alignment using the binomial distribution (Eq. 2), using the more stringent baseline probability of 0.065. In other words, by using the higher estimate of the baseline probability, more residues of the MDP must have fsyn ≥ 0.5 to achieve statistical significance. Only humanin and SHLP6 have p-values less than 0.05, the usual criterion for considering results statistically robust, though SHLP4 is close. Keep in mind that because the value 0.065 could be an overestimate of the baseline probability, the p-values could also be overestimates.
Discussion
Our evaluation of synonymous codon bias has revealed that at least two MDPs, humanin and SHLP6, show evidence of natural selection, implying these peptides have biological roles that have been important during the course of vertebrate evolution. For humanin the synonymous codon bias analysis in vertebrates shows robust evidence of purifying selection (p < 0.00063). Although the importance of humanin has already been demonstrated by knockdown experiments5,17, the result here serves as more than just a positive control. In humans there are 13 humanin homologs in the nuclear genome, 10 of which have been confirmed to be transcribed in various human tissues19. Nuclear DNA sequences originating from mitochondria are called numts (NUclear sequence of MiTochondrial origin), and they are common20. Thus, hypothetically, it could be that RNA interference with the nuclear versions, and not mitochondrial humanin, led to dysfunction in the knockdown experiments. Nevertheless, the result here suggests the mitochondrial version of humanin must be important because its peptide sequence has been conserved throughout vertebrate evolution. The nuclear versions might also be important, and they warrant further study, such as using CRISPR techniques to alter or knock out the numts, and methods of introducing base changes into mitochondrial genes are on the horizon21.
SHLP6 also shows synonymous codon bias (p < 0.037 in vertebrates) satisfying the significance threshold of p < 0.05. Perhaps the results for SHLP4 (p < 0.085 in mammals) and SHLP2b (p < 0.19 in vertebrates) also reflect purifying selection in their highly conserved N-terminal regions, but there are too few residues with high fsyn values to conclude this statistically. Similarly, this analysis cannot rule out that the highly conserved MGYIF pentapeptide in MOTS-c has undergone purifying selection to conserve its sequence, but the region is too short, with only G7 having a high fsyn value in mammals, to conclude significance. While this study did not include invertebrate species, searches of their mitochondrial DNA reveal sequences homologous to humanin, SHLP6 and the highly conserved regions of SHLP2b, SHLP4 and MOTS-c; so MDPs do not appear to be unique to vertebrates (supplemental Fig. S7).
For the other MDPs: SHLP1, SHLP2, SHLP3 and SHLP5, the analyses show poor sequence conservation and no evidence of synonymous codon bias. SHLP peptides were first identified by analyzing the human mitochondrial 16S gene MT-RNR2 for all ORFs greater or equal to 20 amino acids long, and it is certainly possible that some of them arose by chance and do not correspond to biologically important peptides. The authors know of no reports showing biological activities for SHLP1 and SHLP5. In the original paper describing them, SHLP2 and SHLP3 were shown to be cytoprotective in two cell models, one of human and one of murine origin. Curiously, mice (Mus musculus) do not seem to have homologs of SHLP2 and SHLP3 (supplemental Fig. S8), thus, at least for the mouse cell model, the cytoprotective effect would seem serendipitous. It is also possible that SHLP1, SHLP2, SHLP3 and SHLP5 are biologically important, but they arose too recently in human evolution for synonymous codon analyses to be useful.
An earlier analysis of MOTS-c sequences in 14 species (13 mammals and 1 fish) claimed to show evidence of “positive” selection15. Positive selection typically refers to a recently introduced beneficial mutation that sweeps through a population, and it is unclear how this should be interpreted across species. Four residues were claimed to show positive selection, Q4, E5, G7 and I9, and the base sequences and the codon counts for these residues for the 14 species are reanalyzed here (supplemental Fig. S9). The bases for G7 are invariant, whereas those of Q4 are quite variable. The previously reported data showed ratios of non-synonymous to synonymous mutation rates, with larger ratios indicating positive selection, but because G7 is invariant, both rates are zero and it is unclear how the reported ratio was obtained. It is possible the results are artifacts due to the small sample size. In any case, none of the four residues has synonymous codons in the 14 species. Interestingly, G7 does have synonymous mutations in sloths (supplemental Table S10), but no sloths were included in the earlier analysis.
One feature that distinguishes humanin and SHLP6 from the other MDPs of the 16S gene MT-RNR2 is the direction of their translation, with humanin and SHLP6 being coded by the H-strand of the mtDNA and the others being coded by the L-strand (Fig. 1). MOTS-c is also coded by the H-strand. Both strands of mtDNA are transcribed into RNA polycistronically over nearly their whole lengths. However, there are no genes coded by the L-strand region complementary to the 16S and 12S ribosomal genes, and transcription of the L-strand often terminates before reaching there22. This means that the L-strand coded MDPs would be less transcribed than the H-strand coded ones. MDP RNA sequences, including sequences with 3’ polyadenylated tails, have been observed in cell and plasma samples using humanin, MOTS-c and SHLP6 gene sequence probes in Northern blot and RT-PCR experiments14,19,23,24, but no such experiments have yet been reported for the other SHLP peptides.
SHLP6 is unique among the MDPs in that both its start and stop codons are nearly invariant. The relatively poor conservation of the start codon of humanin has been reported before25, and is confirmed here, with only 79% conservation over all vertebrate species. Even less conserved are the other MDP stop codons, with a 58% conservation rate for humanin, and only 17% in SHLP4, though SHLP4 has an earlier stop codon at position 23 in 71% of mammals. The arginine codon in position 22 of humanin is a stop codon in the vertebrate mitochondrial DNA code, and in mammals, it is nearly invariant with an fsyn value of 0.86 for the stop codon (supplemental Fig. S1). This suggests that purifying selection might be conserving the stop codon, but only if humanin is translated inside mitochondria. In fact, one study saw colocalization of the humanin peptide inside mitochondria in synovial cells from rheumatoid arthritis patients26, consistent with mitochondria being the site of humanin expression; however, more study is needed to determine whether this holds true under non-pathological conditions and in other cell types.
On the other hand, humanin has also been localized outside mitochondria, as well as extra-cellularly, and is known to act on both cytosolic and cell surface proteins5,6,7,9,10,27, thus, cytosolic translation is also a possibility. If humanin is translated in the cytosol, the resulting peptide can vary greatly in length; for instance, human humanin has 24 residues whereas rat humanin, also known as rattin, has 38. Intriguingly, humanin residues K21, R22, and R23, which are highly conserved in mammals, resemble the dibasic cleavage sites important in the processing of many secreted hormones28. SHLP4 residues R10 and R11, at the end of its highly conserved N-terminal region, also resemble a dibasic cleavage site, as do MOTS-c residues R13 and K14. Post-translational proteolytic processing could help explain why MDP stop codons are so variable; as long as the cleavage site is conserved, the sequence past it may be of little importance. However, it is not currently known whether proteolytic processing is important for MDP biological function. Also not known is whether MDPs are predominantly translated in mitochondria or in the cytosol. One argument for cytosolic translation is the fact that the second codon of MOTS-c is a stop codon in the mitochondrial DNA code, thus full-length MOTS-c must be translated in the cytosol15. However, the conserved pentapeptide MGYIF starting at position 6 of MOTS-c has its own start codon, so it could be translated inside mitochondria. The questions of proteolytic processing and mitochondrial versus cytosolic translation of MDPs clearly warrant further study.
One concern regarding the synonymous codon bias analysis is that out of the 55 residues with fsyn ≥ 0.5 in the MDP sequences, 15 are leucine residues. Leucine is one of three amino acids with six codons in the standard DNA code, and it is possible leucine is over-represented because it has a greater chance that a random mutation is synonymous. On the other hand, serine also has six codons, but only one of the residues with fsyn ≥ 0.5 is serine, so perhaps the effect is small. In fact, the reason for the higher number of leucine residues could be biological; the L9R and L10D mutants of humanin cannot be secreted from cells29, and perhaps leucine residues could be similarly important for other MDPs.
These shared features, variable peptide length, possible dibasic cleavage sites in humanin, SHLP4, and MOTS-c, and high leucine frequency in humanin and SHLP4 are consistent with these MDPs being processed and secreted, thereby implying roles in intercellular signaling. Indeed, both humanin and MOTS-c have already been detected outside cells2,15,29, though there are no reports yet on SHLP4. Conversely, the well-conserved stop codon of SHLP6 with a lack of any apparent dibasic cleavage site suggests its role might be intracellular. Intriguingly, if translated in mitochondria, humanin also has a highly conserved stop codon, replacing the dibasic cleavage site, suggesting humanin might play dual roles, intracellular for the peptide expressed in mitochondria and extracellular when expressed cytosolically.
The search for MDPs has focused on longer peptides, at least 20 residues in the case of the mitochondrial 16S gene, but the results presented here imply shorter peptides might also be important, especially if proteolytic processing occurs. In particular, the N-terminal regions of SHLP2b and SHLP4, and the MGYIF pentapeptide of MOTS-c are highly conserved and include residues with high fsyn values, suggesting they might have important biological roles. There are many biologically important shorter peptides, gonadotropin-releasing hormone with ten residues, oxytocin with nine, and the enkephalins with five, to name a few. MDPs need not occur only in RNA genes, and the synonymous codon bias analysis could be extended to identify candidate MDPs throughout the mitochondrial genome. One analysis suggested there could be over 80 ORFs leading to MDPs in the mitochondrial genome30, and an MDP outside the ribosomal genes, SHMOOSE, has already been identified31, as well as two microproteins, gau and Mtaltnd43,4. In addition, by analyzing larger regions of the mitochondrial genome, including more non-peptide-coding base sequences, the statistical analysis can be improved.
The peptides belonging to the family of MDPs are fascinating, and they offer a new paradigm to regulate cell signaling, metabolism, inflammation, and cell death from within the mitochondrial genome. Some of them, such as humanin and MOTS-c have been studied more extensively than the others for their impact on the above cellular functions. Because of their reported beneficial effects, humanin and MOTS-c are already being marketed as anti-aging therapeutics and performance enhancers in humans32, in spite of the possible cancer promoting properties of humanin. This highlights the need to evaluate the potential validity of these peptides at the genome level. One outcome from such an evaluation is to ensure that the form of the peptide targeted for therapeutics is the relevant one. For instance, the results presented here suggest that the MOTS-c peptide as it is currently being studied (and marketed) might not be in its most biologically relevant form. Plus, identifying the important and conserved residues will aid optimization of the therapeutic performance of these peptides. At the fast rate of which these peptides are being developed for potential treatments of diseases against the backdrop of their potentially negative effects, it is only prudent to urge that they be subjected to the rigorous scrutiny of the broader scientific community.
Methods
Multiple sequence alignments
Sequence alignments of the base sequences coding the MDPs were assembled using nucleotide BLAST. Because the sequences correspond to open reading frames, the alignments contain no insertions or deletions. For the primate alignments, comprehensive, though not exhaustive, searches of all primate species were performed. For the vertebrate alignments, the sequences were limited to species whose nuclear genomes have also been sequenced, which yielded numbers of species comparable to the primate alignments. The resulting vertebrate alignments consisted of roughly 10% primates, 30% other mammals, 20% other land vertebrates, and 40% fish. For the mammal alignments, non-mammal sequences were deleted from the vertebrate list. For species with more than one version of the sequence, presumably due to polymorphisms, the most common version was used. For species that had a mutation of an otherwise invariant base, or a frame shifting insertion/deletion that resulted in mutation of otherwise invariant amino acids, the search was expanded to include any additional species with mutations at that site. If no other species with a mutation at that site were found, then the sequence was removed. As a consequence, the number of species in the alignments is variable, and the specific numbers are given in the figure legends. All the sequence alignments also included 12–15 bases before the start codon and 12–15 bases after the stop codon to provide a comparison with adjacent, presumably non-peptide-coding regions. All sequence alignments are included in the supplemental information (supplemental Tables S1–S36).
Synonymous codon bias
To calculate the fraction of synonymous mutations, fsyn, for each amino acid position, the most frequent codon of the consensus amino acid was taken as the “ancestral” codon, and the total number of synonymous mutations of that codon, nsyn, is divided by the total number of mutations, synonymous plus non-synonymous, nnon, at that position,
Thus, a result of 0.0 indicates all mutations are non-synonymous, 0.5 indicates an equal number of synonymous and non-synonymous mutations, and 1.0 indicates all the mutations are synonymous for that position. For comparison, the ratio of synonymous to non-synonymous rates, which is another common way to express synonymous codon bias1, ranges from 0 for all non-synonymous mutations, 1 for an equal number of synonymous and non-synonymous mutations, to +\(\infty \) for all synonymous mutations.
The one-tailed p-values for the number of occurrences of fsyn ≥ 0.5 in each sequence alignment were calculated using the binomial distribution,
where X is the number of residues with fsyn ≥ 0.5, n is the length of the peptide and p is the random probability of fsyn ≥ 0.5. The probability p was calculated using non-peptide-coding regions of the vertebrate sequence alignments, except for SHLP1, SHLP3, and SHLP5, where the primate sequence alignments were used. The consensus sequence logo figures were made with Weblogo33.
Data availability
All data generated or analyzed during this study are included in this published article and its supplementary information files.
References
Andrews, S. & Rothnagel, J. Emerging evidence for functional peptides encoded by short open reading frames. Nat. Rev. Genet. 15, 193–204 (2014).
Hashimoto, Y. et al. "A rescue factor abolishing neuronal cell death by a wide spectrum of familial Alzheimer’s disease genes and Aβ. Proc. Natl. Acad. Sci. U S A 98, 6336–6341 (2001).
Faure, E. et al. Probable presence of an ubiquitous cryptic mitochondrial gene on the antisense strand of the cytochrome oxidase I gene. Biol. Direct. 6, 56 (2011).
Kienzle, L. et al. A small protein coded within the mitochondrial canonical gene nd4 regulates mitochondrial bioenergetics. BMC Biol. 21(1), 111 (2023).
Guo, B. et al. Humanin peptide suppresses apoptosis by interfering with Bax activation. Nature 423(6938), 456–461 (2003).
Ikonen, M. et al. Interaction between the Alzheimer’s survival peptide humanin and insulin-like growth factor-binding protein 3 regulates cell survival and apoptosis. Proc. Natl. Acad. Sci. U S A 100(22), 13042–13047 (2003).
Luciano, F. et al. Cytoprotective peptide humanin binds and inhibits proapoptotic Bcl-2/Bax family protein BimEL. J. Biol. Chem. 280(16), 15825–15835 (2005).
Morris, D., Johnson, S., Bleck, C., Lee, D. & Tjandra, N. Humanin selectively prevents the activation of pro-apoptotic protein BID by sequestering it into fibers. J. Biol. Chem. 295(52), 18226–18238 (2020).
Ying, G. et al. Humanin, a newly identified neuroprotective factor, uses the G protein-coupled formylpeptide receptor-like-1 as a functional receptor. J. Immunol. 172(11), 7078–7085 (2004).
Hashimoto, Y., Kurita, M., Aiso, S., Nishimoto, I. & Matsuoka, M. Humanin inhibits neuronal cell death by interacting with a cytokine receptor complex or complexes involving CNTF receptor alpha/WSX-1/gp130. Mol. Biol. Cell 20(12), 2864–2873 (2009).
Maximov, V., Martynenko, A., Hunsmann, G. & Tarantul, V. Mitochondrial 16S rRNA gene encodes a functional peptide, a potential drug for Alzheimer’s disease and target for cancer therapy. Med. Hypotheses 59(6), 670–673 (2002).
Gottardo, M. et al. Baculovirus-based gene silencing of Humanin for the treatment of pituitary tumors. Apopotosis 23(2), 14–151 (2018).
Moreno Ayala, M. et al. Humanin promotes tumor progression in experimental triple negative breast cancer. Sci. Rep. 10(1), 8542 (2020).
Cobb, L. et al. Naturally occurring mitochondrial-derived peptides are age-dependent regulators of apoptosis, insulin sensitivity, and inflammatory markers. Aging 8(4), 796–809 (2016).
Lee, C. et al. The mitochondrial-derived peptide MOTS-c promotes metabolic homeostasis and reduces obesity and insulin resistance. Cell Metab. 21(3), 443–454 (2015).
Reynolds, J. et al. MOTS-c is an exercise-induced mitochondrial-encoded regulator of age-dependent physical decline and muscle homeostasis. Nat. Commun. 12, 470 (2021).
Jung, J. et al. The mitochondria-derived peptide humanin improves recovery from intracerebral hemorrhage: Implication of mitochondria transfer and microglia phenotype change. J. Neurosci. 40(10), 2154–2165 (2020).
Ivanov, I., Firth, A., Michel, A., Atkins, J. & Baranov, P. Identification of evolutionarily conserved non-AUG-initiated N-terminal extensions in human coding sequences. Nucleic Acids Res. 39(10), 4220–4234 (2011).
Bodzioch, M. et al. Evidence for potential functionality of nuclearly-encoded humanin isoforms. Genomics 94(4), 247–256 (2009).
Hazkani-Covo, E., Zeller, R. & Martin, W. Molecular poltergeists: Mitochondrial DNA copies (numts) in sequenced nuclear genomes. PLoS Genet. 6(2), e1000834 (2010).
Kar, B., Castillo, S., Sabharwal, A., Clark, K. & Ekker, S. Mitochondrial base editing: Recent advances towards therapeutic opportunities. Int. J. Mol. Sci. 24(6), 5798 (2023).
D’Souza, A. & Minczuk, M. Mitochondrial transcription and translation: Overview. Essays Biochem. 62(3), 309–320 (2018).
Tajima, H. et al. Evidence for in vivo production of Humanin peptide, a neuroprotective factor against Alzheimer’s disease-related insults. Neurosci. Lett. 324(3), 227–231 (2002).
D’Souza, R. et al. Increased expression of the mitochondrial derived peptide, MOTS-c, in skeletal muscle of healthy aging men is associated with myofiber composition. Aging 12(6), 5244–5258 (2020).
Logan, I. Pseudogenization of the Humanin gene is common in the mitochondrial DNA of many vertebrates. Zool. Res. 38(4), 198–202 (2017).
Ijiri, K. et al. Increased expression of humanin peptide in diffuse-type pigmented villonodular synovitis: Implication of its mitochondrial abnormality. Ann. Rheum Dis. 64(6), 816–823 (2005).
Gong, Z. et al. Humanin is an endogenous activator of chaperone-mediated autophagy. J. Cell Biol. 217(2), 635–647 (2018).
Rholam, M. & Fahy, C. Processing of peptide and hormone precursors at the dibasic cleavage sites. Cell. Mol. Life Sci. 666, 2075–2091 (2009).
Yamagishi, Y., Hashimoto, Y., Niikura, T. & Nishimoto, I. Identification of essential amino acids in Humanin, a neuroprotective factor against Alzheimer’s disease-relevant insults. Peptides 24(4), 585–595 (2003).
Robson, B. Extension of the Quantum Universal Exchange Language to precision medicine and drug lead discovery: Preliminary example studies using the mitochondrial genome. Comput. Biol. Med. 117(3), 103621 (2020).
Miller, B. et al. Mitochondrial DNA variation in Alzheimer’s disease reveals a unique microprotein called SHMOOSE. Mol. Psychiatry 1, 1 (2022).
Thevis, M. & Schänzer, W. Emerging drugs affecting skeletal muscle function and mitochondrial biogenesis—Potential implications for sports drug testing programs. Rapid Commun. Mass Spectrom 30(5), 635–651 (2016).
Crooks, G., Hon, G., Chandonia, J. & Brenner, S. WebLogo: A sequence logo generator. Genome Res. 14, 1188–1190 (2004).
Acknowledgements
This work was funded intramurally by the National Heart, Lung and Blood Institute of the National Institutes of Health.
Funding
Open Access funding provided by the National Institutes of Health (NIH).
Author information
Authors and Affiliations
Contributions
J.M.G., D.L.M. and N.T. contributed to the design of the study. J.M.G. carried out the analysis. J.M.G., D.L.M. and N.T. contributed to the manuscript preparation.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Gruschus, J.M., Morris, D.L. & Tjandra, N. Evidence of natural selection in the mitochondrial-derived peptides humanin and SHLP6. Sci Rep 13, 14110 (2023). https://doi.org/10.1038/s41598-023-41053-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-023-41053-0
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
