
Abstract
In a designed study to screen for antimicrobial exhibiting bacteria using molecular aspects, Bacillus species were considered to investigate antibiotic biosynthesis genes. 28 bacterial strains and 3 induced mutants were screened for the presence of subtilosin gene (sbo) and subtilosin through PCR and Mass spectrometry respectively. Sbo gene was detected in 16 out of 28 Bacillus strains. The results from gene sequences deliberated by multiple sequence alignments revealed high-level homology to the sequences of the sbo-alb gene locus of B. subtilis 168 and the other limited reported strains. Hence, this report provided additional strains to support the idea of subtilosin gene predominance amongst Bacillus strains isolated from environment and to find different species containing homologous genes, furthermore the utilization of its conserved region as a means of identifying Bacillus spp. that produce subtilosin. This is the first report to confirm the detection of subtilosin production from B. amyloliquefaciens.
Similar content being viewed by others


Introduction
The soil bacterium Bacillus subtilis represents bacteria that produce a series of peptide antibiotics1. They are members of both classes: the ribosomally synthesized e.g. subtilin2, Ericin3 and sublancin4 and the nonribosomally synthesized such as the lipopeptides surfactin5,6, mycosubtilin7, and fengycin8 Bacilysocin9 and 3,3'-Neotrehalosadiamine 10. Subtilosin A is one of many antibiotics produced by Bacillus strains11,12 its importance and role in Bacillus group is little understood. Subtilosin is a macrocyclic structure (Fig. 1C) with three inter-residual linkages13 that have been elucidated as thioether bonds between cysteine sulphurs and amino acid alpha-carbons14. An acidic isoelectric point differentiates subtilosin from the basic lantibiotics15. In subtilosin, posttranslational linkage of a thiol to the R-carbon of an amino acid residue is unprecedented in ribosomally synthesized peptides or proteins, and very rare in secondary metabolites14,16. The mature product is formed by loss of an unusually short seven amino acid leader peptide, cyclization of the N and C termini, and further modification of Cys, Thr, and Phe residues17. The mature subtilosin peptide is highly resistant to enzymatic proteolysis and is stable to moderate heat and acid treatment. It acts against a variety of Gram-positive bacteria, including Listeria and other pathogens18,19,20,21. The production of mature subtilosin is based on the expression of the sbo-alb gene cluster encompassing the subtilosin structural gene sbo and genes involved in posttranslational modification and processing of presubtilosin and in immunity22,23,24. Expression of the sbo-alb genes occurs under stress conditions25. 16S rRNA gene used for rapid identification of the Bacillus genus was undertaken by Celandroni et al.26. The validity of using a hypervariable region (nucleotides 70–344) of the gene was proven adequate to discriminate between all the species except between B. cereus and B. anthracis and between B. mojavensis and B. atrophaeus. The high 16S rRNA gene sequence similarities between some stains within this genus can even share phenotypic properties27. However, they have been classified as different species based on DNA association values hence, demonstrated the need for a polyphasic approach to the systematics of this genus28,29. This was observed between B. subtilis subsp. subtilis and B. subtilis subsp. spizizenii, which share phenotypic profiles but have segregated based on DNA reassociation values of 58–69%, in addition to minor polymorphisms in the 16S rRNA gene between the type strains26,30. Further, B. mojavensis and B. subtilis subsp. spizizenii have only a 1-bp difference in the 16S rRNA gene and can only be distinguished from each other by sexual isolation, divergence in DNA sequences of the rpoB and gyrA genes, and fatty acid composition31. Stein et al.32 pre-postulate the coding gene subtilosin gene (sbo) to develop evolutionary divergence in B. subtilis subspecies too. This report is to describe subtilosin production by 16 wild-type B. subtilis strains and B. amyloliquefaciens. The sbo genes of these organisms were sequenced in order to analyze the genetic variation between B. subtilis wild-type strains. Finally, we confirm the association between production of subtilosin A and the detection of sbo gene using PCR screening.

(A) RP-HPLC, (B) MALDI-TOF-MS, intensive signals correspond to the molecular mass of protonated subtilosin [M + H]+ (C) 3D structure of subtilosin.
Materials and methods
Strains and media
The following 32 different bacterial strains were tested for their sensitivity to the antibiotic using the agar-well diffusion assay: Bacillus subtilis 168, Bacillus subtilis ATCC, from Bacillus Genetic Stock Center (BGSC) and Bacillus cereus (Lab. collection) and 28 environmental isolates (Table 1). Bacillus fusiformis was routinely used for sensitivity test. All the strains were regularly maintained on nutrient agar , however, for antibiotic production Landy medium (glucose, 20 g L−1; glutamic acid, 2 g L−1, (NH4)2SO4, 2.3 g L−1; yeast extract, 1 g L−1; K2HPO4, 1 g L−1; MgSO4 0.5 g L−1; KCl, 0.5 g L−1; CuSO4, 1.6 mg L−1; Fe2(SO4)3, 1.2 mg L−1; and MnSO4, 0.4 mg L−1) was used.
Bacterial identification
Identification of the isolated strains was carried on by sequence homology of 16S rDNA accompanied by morphological and biochemical characterization. Identification to the species level was defined as a 16S rDNA sequence similarity of ≥ 99% with that of the prototype strain sequence in GenBank; identification at the genus level was defined as a 16S rDNA sequence similarity of ≥ 97% with that of the prototype strain sequence in GenBank. The biochemical profile of test isolates was determined with the API 50 CHB strips following the manufacturer’s instructions (bioMerieux, France). This test allows bacterial strains to be classified according to their ability to ferment 49 different carbohydrates. The results were analyzed with the APILAB Plus software (bioMerieux, France).
Antibiotic assay
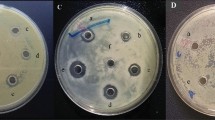
Samples of culture supernatant containing the antibiotic checked for activity using an agar- well diffusion assay33. Fifty µL of Bacillus fusiformis liquid culture of 0.3 OD600 was spread onto the surface of Petri dish containing L-agar. 50 µL antibiotic sample was transferred into the well made in media plates using a sterile cork borer. The sample was allowed to diffuse into the agar and the plate was inverted and incubated at 37 °C until a lawn of the indicator bacteria appeared on the plate (approximately 10–16 h).
DNA isolation, extraction and PCR
Genomic DNA extracted from overnight-inoculated bacterial culture in N-broth at 37 °C with 120 rpm. The extraction carried out using gene extraction kit (Biorad). PCR amplification of ~ 1375-bp consisted of sbo and flanking region was performed successfully with TS13C (GAATTGACACTATCTAGAGAAATGCCG) and TS14 (ATCCGGTGGTGCGGAATTCGATGA)32. While primers 27f. (GAATTGACACTATCTAGAGAAATGCCG) and 1522r (ATCCGGTGGTGCGGAATTCGATGA)34 were used to amplify the 16S rRNA gene. Both sets of primers purchased from Gene Link Inc., USA. 0.5–0.1 ng of chromosomal template DNA and 0.25 µM each primer were added to Master mix (Fermantas). The PCR conditions started with heating at 94 °C for 5 min and passed through 30 cycles as follows: denaturation for 30 s at 94 °C primer annealation for 30 s at 59 °C and extension at 72 °C for 1.2 min. The final extension was at 72 °C for 3 min. PCR fragments were excised from agarose gel electrophoresis followed by extraction with a QIAgen gel extraction kit (Qiagen). The isolated PCR products were sequenced by using an ABI Prism dye terminator cycle sequencing ready reaction kit and an ABI PRISM 377 DNA sequencer (Applied Biosystems). Analyses of DNA sequences were performed by using Prochromas version software (Oxford Molecular, UK).
Production of (-A) No activity mutants
Mutants from strain-7 a confirmed producer of subtilosin were generated from exposed culture to UV light for different intervals. The surviving bacteria were screened for the disruptions in sbo gene through PCR and subtilosin production by MALDI-TOF–MS.
TLC, SPE and RP-HPLC
Bacterial supernatant, recovered by 15,000×g centrifugation for 20 min of 36 h old shaken culture were screened for the presence of subtilosin A. Supernatants were extracted with same amount ethyl acetate (Fishers, USA) and vacuum dried. Residues were dissolved in minimal amount of acidified 20% acetonitrile v/v. Further isolation was performed through SPE columns (CHROMABOND C18ec) purchased from Macherey–Nagel, Germany. First columns were preconditioned with methanol then water and samples were applied under low pressure. The columns were washed with water and four fractions were eluted by consecutive four solutions 40, 60, 80 and 100% acetonitrile. Fractions eluted with 80% were accommodated over short lines in TLC sheets (Merck, Germany) and developed with 1:1:1 v/v/v of n-hexane, chloroform and methanol as mobile phase. The plates were analyzed under UV-Light, however, active fractions were detected using narrow strips of developed sheets by a bioassay method with a sensitive test organism33. These active fractions were scrapped from the TLC plate and extracted with eluent A (0.1% (vol/vol) trifluoroacetic acid and 20% (vol/vol) acetonitrile). Finally, analysis was by reversed-phase HPLC using Thermo Hypersil-Keystone ODS (particle size, 5 µm; column dimensions, 250 by 4.6 mm, Thermo Hypersil, USA). A sample was applied with eluent A and eluted with segmented gradients of eluent B (0.1% (vol/vol) trifluoroacetic acid and 80% (vol/vol) acetonitrile) as follow 40% eluent B for 30 min and 40–100% eluent B for 10 min. Eluents A and B were composed in Milli-Q HPLC grad water.
MALDI-TOF-mass spectrometry
Fractions correlated with Subtilosin A from TLC and Reverse phase HPLC were analyzed using MALDI-TOF–MS. 2 µL of sample mixed with 2 µL matrix solution (2 mg of alpha- hydroxycinnaminic acid per ml in acetonitrile-methanol–water (1:1:1) on the target plate. MALDI-TOF–MS spectra were recorded by using a 337-nm nitrogen laser for desorption and ionization. The mass spectrometer operated in the linear mode at an accelerating voltage of 18 kV with an ion flight path that was 0.7 m long. The delay time was 375 ns. Matrix suppression was also used, and the mass spectra were averaged over 50–100 individual laser shots. The laser intensity was set just above the threshold for ion production. External calibration was performed by using the [M + H]+ signals of renin, adenocorticotropic hormone, insulin oxidized B, and bovine insulin (Sigma-Aldrich Co.) the results were anticipitated as subtilosin A with m/z of 3400.7 and 3406.6. The variance of the m/z of ±0.8 Da was considered acceptable.
Results
Identification of bacterial strains
Strains were identified according to their morphological and biochemical characteristics added by homology to 16S rRNA with the type strains available in NCBI and RIDOM35 the results revealed the distinction between two subsp. subtilis and spizizenii as well as their association with their sources.
Detection and sequencing of sbo gene
Sbo and its flanking region were detected from the environmental strain and Type strains. B. subtilis 168 and B. subtilis ATCC 6633 were used as positive control representing the two classes/subsp. subtilis (class I) and spizizenii (class II) respectively. Results are shown in (Table 1) in which majority of strain designated as B. subtilis were secured sbo class I. All the obtained DNA fragments were sequenced, and the phylogenetic relation was established using Clustal W, EBI (Fig. 2).

Phylogenetic diversity of isolated strains using multiple sequence alignments of sequences sbo and flanking region.
Detection and isolation and of Subtilosin
Subtilosin presence was regularly checked on TLC in reference to match the subtilosin produced by B. subtilis 168 further confirmation was carried on using reverse-phase HPLC (Fig. 1A) and MALDI-TOF-MS (Fig. 1B; Table 1).
Mutation analysis
Mutants produced further selected based on inhibitory activity. Out of 200 mutant 1 strain was isolated with no detectable zone of inhibition. This (-A) strain was not able to produce subtilosin more over the sbo was not amplified using PCR. Hence, it was determined to be functional disruption of encoding gene (Fig. 3B).

(A) Organization of the sboAX locus. the sboA, sboX, and albA region of the sbo-alb gene cluster and flanking region. Arrowheads indicate direction of transcription; (o) is a terminator. (B) PCR amplified fragment of 1375 bp related to sbo gene where M indicates marker (5000, 2000, 850, 400, 100 bp). Positive amplification for lanes 1–3, 5–7 corresponding to different B. subtilis isolates while lane 10 and 11 coorsponds to B. amyloliquefaciens and B. subtilis ATCC6633 respectively. Lane 8 is a positive control (B. subtilis 168) and Lane 9 is a negative control (B. licheniformis).
Nucleotide sequence accession number
The nucleotide sequences reported deposited in NCBI GenBank under accession numbers; FJ151503, FJ151504, FJ151505, FJ151506, FJ151507.
Discussion
It is well established that, the ribosomal peptide antibiotics are synthesized during active growth, while nonribosomal ones are synthesized at later stages. Theories on the role of antibiotic production are yet to be investigated. The best-accepted theory is that nonribosomal antibiotics may play a role in competition with other microorganisms during the starvation phase or spore germination36,37,38. While the role of ribosomal peptides remained undefined. Not so obvious the role of such products in the active life cycle. For example the sublancin gene cluster is not essential for B. subtilis, however, it contains yet unidentified genes mediating resistance against sublancin action. Suggestions of an intrinsic mechanism of gene improvement i.e. utilizing antibiotics as first line of defense for survival rather than a second, question antibiotics as secondary metabolites. Another probability is displayed social behaviors as co-ordinate gene expression and group behavior through different quorum-sensing pathways39. It was determined that interaction of subtilosin with the lipid head group region of bilayer membranes in a concentration dependent manner induced a conformational change in the lipid headgroup and disordering in the hydrophobic region of bilayers that ultimately resulted in membrane permeabilization at high peptide concentrations40. Such adoption may lead to assume a growth control during prosperous stage. Furthermore, under anaerobic conditions an increased by 4- to 90-fold, anticipated that the cell accumulates inactive precursors of subtilosin, which then undergo oxygen-dependent modifications to yield an active peptide when an aerobic environment is encountered41. The widespread occurrence of subtilosin might reflect an important physiological role. A specific function of subtilosin as an antibiotic, killing factor42 or as a pheromone during anaerobiosis43 or biofilm growth of B. subtilis39 could be well thought-out. Never the less the gene encoding subtilosin production has demonstrated a strong biomarker for Bacillus subtilis. The B. subtilis strains have segregated into two subclades, one encompassing strain 168 and the other W23, classified strain 168 as B. subtilis subsp. subtilis and W23-related strains as B. subtilis subsp. spizizenii based on DNA reassociation studies31 and sbo gene analysis32. The W23 and 168 group strains are identical for most phenotypic characteristics. However, cell wall chemistry of the W23 strains and 168 strains were different; the cell wall of the former contained ribitol and glycerol teichoic acids and that of the latter only glycerol teichoic acid44. The sbo-gene of B. subtilis encodes the 43-aminoacid residue comprising the prepropeptide of subtilosin23. The nucleotide sequences of the sbo genes and flanking regions are identical in strains belonging to the same subspecies, and the sequences differ by three nucleotides in the two subspeciesm45. However, the encoded Sbo prepeptides are identical in all cases32. On the other hand sboX, encoded a bacteriocin-like product, a new gene with an unknown function, in strain 16823, which resides in an open reading frame overlapping the coding region of sbo (Fig. 3A). Notably, the expression of sboX would result in a 22-amino-acid curtailed peptide in W23- like strains compared to the peptide produced by 168-like strains, which makes it unlikely that sboX is produced by W23-like strains. These observations were further elaborated to support and to evaluate possible evolutionary relationships among the subtilosin producers, however, A correlation between sbo gene and subtilosin production was not established probably due influence of sboX,. Previous attempts for sboX insertions were not successful as such insertions might render sboA mRNA unstable and explain the reduced subtilosin production in sboX mutant. Alignments has revealed that the sbo genes is highly conserved with those of B. subtilis subsp. subtilis (96–100% amino acid identity), while the remaining were less conserved (83–88% identity). This high and low level of conservation is unprecedented too; for example, thymidylate synthases A (thyA) in B. subtilis subsp. spizizenii ATCC 6633 and W23 and B. subtilis subsp. subtilis (168) exhibit more than 95% amino acid identity46. Even the average level of amino acid identity for the DNA gyrases (gyrA) in seven Bacillus type strains was 95.1%28. With these results, we can confirm subtilosin gene predominance amongst Bacillus strains isolated from environment and the correlation amongst different sub-species containing homologous genes. Furthermore, this article demonstrated the possibility to utilize Sbo conserved region as a mean of identifying Bacillus spp. that produce subtilosin.
References
Tran, C., Cock, I. E., Chen, X. & Feng, Y. Antimicrobial Bacillus: Metabolites and their mode of action. Antibiotics (Basel). 11(1), 88 (2022).
Zhang, Q., Kobras, C. M., Gebhard, S., Mascher, T. & Wolf, D. Regulation of heterologous subtilin production in Bacillus subtilis W168. Microb Cell Fact. 21(1), 57 (2022).
Karagiota, A., Tsitsopoulou, H., Tasakis, R. N., Zoumpourtikoudi, V. & Touraki, M. Characterization and quantitative determination of a diverse group of Bacillus subtilis subsp. subtilis NCIB 3610 antibacterial peptides. Probiotics Antimicrob. Proteins. 13(2), 555–570 (2021).
Li, J., Chen, J., Yang, G. & Tao, L. Sublancin protects against methicillin-resistant Staphylococcus aureus infection by the combined modulation of innate immune response and microbiota. Peptides 141, 170533 (2021).
Danevčič, T. et al. Surfactin facilitates horizontal gene transfer in Bacillus subtilis. Front Microbiol. 12, 657407 (2021).
Al-Ajlani, M. M., Sheikh, M. A., Ahmad, Z. & Hasnain, S. Production of surfactin from Bacillus subtilis MZ-7 grown on pharmamedia commercial medium. Microb. Cell Fact. 5(6), 17 (2007).
Yu, C. et al. Mycosubtilin Produced by Bacillus subtilis ATCC6633 Inhibits Growth and Mycotoxin Biosynthesis of Fusarium graminearum and Fusarium verticillioides. Toxins (Basel) 13(11), 791 (2021).
Gao, W. et al. Production of fengycin from D-xylose through the expression and metabolic regulation of the Dahms pathway. Appl. Microbiol. Biotechnol. 106(7), 2557–2567 (2022).
Tojo, S., Tanaka, Y. & Ochi, K. Activation of antibiotic production in Bacillus spp. by cumulative drug resistance mutations. Antimicrob. Agents Chemother. 59(12), 7799–804 (2015).
Saito, N., Nguyen, H. M. & Inaoka, T. Impact of activation of neotrehalosadiamine/kanosamine biosynthetic pathway on the metabolism of Bacillus subtilis. J Bacteriol. 203(9), e00603-e620 (2021).
Théatre, A., Hoste, A. C. R., Rigolet, A., Benneceur, I., Bechet, M., Ongena, M., Deleu, M. & Jacques, P., Bacillus sp.: A remarkable source of bioactive lipopeptides, in Advances in Biochemical Engineering/Biotechnology. Springer, Berlin, Heidelberg. https://doi.org/10.1007/10_2021_182 (2022).
Kaspar, F., Neubauer, P. & Gimpel, M. Bioactive secondary metabolites from Bacillus subtilis: A comprehensive review. J. Nat. Prod. 82(7), 2038–2053 (2019).
Marx, R., Stein, T., Entian, K.-D. & Glaser, S. J. Structure of the Bacillus subtilis peptide antibiotic subtilosin A determined by 1H-NMR and matrix assisted laser desorption/ ionization time-of-flight mass spectrometry. J. Protein Chem. 20, 501–506 (2001).
Kawulka, K. E. et al. Structure of Subtilosin A, a cyclic antimicrobial peptide from Bacillus subtilis with unusual sulfur to R-carbon cross-links: Formation and reduction of R-Thio-R-Amino acid derivatives. Biochemistry 43, 3385–3395 (2004).
Barbosa, J. C. et al. Insights into the mode of action of the two-peptide lantibiotic lichenicidin. Colloids Surf. B Biointerfaces. 211, 112308 (2022).
Himes, P. M., Allen, S. E., Hwang, S. & Bowers, A. A. Production of Sactipeptides in Escherichia coli: Probing the substrate promiscuity of Subtilosin A biosynthesis. ACS Chem. Biol. 11(6), 1737–1744 (2016).
Zheng, G., Yan, L. Z., Vederas, J. C. & Zuber, P. Genes of the sbo-alb locus of Bacillus subtilis are required for production of the antilisterial bacteriocin subtilosin. J. Bacteriol. 181, 7346–7355 (1999).
Yu, W. Q. et al. Draft genome sequence, disease-resistance genes, and phenotype of a Paenibacillus terrae strain (NK3-4) with the potential to control plant diseases. Genome 61(10), 725–734 (2018).
Cavera, V. L., Volski, A. & Chikindas, M. L. The Natural Antimicrobial Subtilosin A Synergizes with Lauramide Arginine Ethyl Ester (LAE), ε-Poly-L-lysine (Polylysine), Clindamycin Phosphate and Metronidazole, Against the Vaginal Pathogen Gardnerella vaginalis. Probiotics Antimicrob. Proteins 7(2), 164–171 (2015).
Sundara Rajan, S. et al. Polyethylene glycol-based hydrogels for controlled release of the antimicrobial subtilosin for prophylaxis of bacterial vaginosis. Antimicrob. Agents Chemother. 58(5), 2747–2753 (2014).
Quintana, V. M. et al. Antiherpes simplex virus type 2 activity of the antimicrobial peptide subtilosin. J. Appl. Microbiol. 117(5), 1253–1259 (2014).
Velho, R. V., Basso, A. P., Segalin, J., Costa-Medina, L. F. & Brandelli, A. The presence of sboA and spaS genes and antimicrobial peptides subtilosin A and subtilin among Bacillus strains of the Amazon basin. Genet. Mol. Biol. 36(1), 101–104 (2013).
Zheng, G., Hehn, R. & Zuber, P. Mutational analysis of the sbo-albsbo-alb locus of Bacillus subtilis: Identification of genes required for subtilosin production and immunity. J. Bacteriol. 182, 3266–3273 (2000).
Zheng, G., Yan, L. Z., Vederas, J. C. & Zuber, P. Genes of the sbo-alb locus of Bacillus subtilis are required for production of the antilisterial bacteriocin subtilosin. J. Bacteriol. 181, 7346–7355 (1999).
Nakano, M. M., Zheng, G. & Zuber, P. Dual control of sbo-alb operon expression by the Spo0 and ResDE systems of signal transduction under anaerobic conditions in Bacillus subtilis. J. Bacteriol. 182(11), 3274–3277 (2000).
Celandroni, F. et al. Identification of Bacillus species: Implication on the quality of probiotic formulations. PLOS ONE 14(5), e0217021 (2019).
Orthuber, W. Information is selection: A review of basics shows substantial potential for improvement of digital information representation. Int. J. Environ. Res. Public Health 17(8), 2975 (2020).
Patel, S. & Gupta, R. S. A phylogenomic and comparative genomic framework for resolving the polyphyly of the genus Bacillus: Proposal for six new genera of Bacillus species, Peribacillus gen. nov., Cytobacillus gen. nov., Mesobacillus gen. nov., Neobacillus gen. nov., Metabacillus gen. nov. and Alkalihalobacillus gen. nov. Int. J. Syst. Evol. Microbiol. 70(1), 406–438 (2020).
Savi, D. C., Aluizio, R., Galli-Terasawa, L., Kava, V. & Glienke, C. 16S-gyrB-rpoB multilocus sequence analysis for species identification in the genus Microbispora. Antonie Van Leeuwenhoek 109(6), 801–815 (2016).
Locher, K. et al. Automated 16S sequencing using an R-based analysis module for bacterial identification. Microbiol Spectr. 11, e0040822 (2022).
Nakamura, L. K., Roberts, M. S. & Cohan, F. M. Relationship of Bacillus subtilis clades associated with strains 168 and W23: A proposal for Bacillus subtilis subsp. subtilis subsp. nov and Bacillus subtilis subsp. spizizenii subsp. nov. Int. J. Syst. Bacteriol. 49, 1211–1215 (1999).
Stein, T., Düsterhus, S., Stroh, A. & Entian, K.-D. Subtilosin production by two Bacillus subtilis subspecies and variance of the sbo-alb cluster. Appl. Environ. Microbiol. 70, 2349–2353 (2004).
Alajlani, M., Shiekh, A., Hasnain, S., Brantner, A. Purification of bioactive lipopeptides produced by Bacillus subtilis strain BIA. Chromatographia 79, 1527–1532 (2016).
Al-Ajlani, M. M. & Hasnain, S. Antagonistic activity against nosocomial clinical isolates with Bacillus subtilis MZ-7. Ann. Microbiol. 57, 419–424 (2007).
Church, D. L. et al. Performance and application of 16S rRNA gene cycle sequencing for routine identification of bacteria in the clinical microbiology laboratory. Clin. Microbiol. Rev. 33(4), e00053-e00119 (2020).
Chevrette, M. G., Himes, B. W. & Carlos-Shanley, C. Nutrient availability shifts the biosynthetic potential of soil-derived microbial communities. Curr. Microbiol. 79(2), 64 (2022).
Popp, P. F. et al. The epipeptide biosynthesis locus epeXEPAB is widely distributed in Firmicutes and triggers intrinsic cell envelope stress. Microb. Physiol. 31(3), 306–318 (2021).
Nechitaylo, T. Y. et al. Incipient genome erosion and metabolic streamlining for antibiotic production in a defensive symbiont. Proc. Natl. Acad. Sci. USA 118(17), e2023047118 (2021).
Algburi, A. et al. Subtilosin prevents biofilm formation by inhibiting bacterial quorum sensing. Probiotics Antimicrob. Proteins 9(1), 81–90 (2017).
Thennarasu, S. et al. Membrane permeabilization, orientation, and antimicrobial mechanism of subtilosin A. Chem. Phys. Lipids 137(1–2), 38–51 (2005).
Stein, T. Oxygen-limiting growth conditions and deletion of the transition state regulator protein Abrb in Bacillus subtilis 6633 result in an increase in Subtilosin production and a decrease in Subtilin production. Probiotics Antimicrob. Proteins 12(2), 725–731 (2020).
Kalamara, M. & Stanley-Wall, N. R. The intertwined roles of specialized metabolites within the Bacillus subtilis biofilm. J. Bacteriol. 203(22), e0043121 (2021).
Dworkin, J. Anaerobiosis: A surfactant helps bacteria breathe a sigh of relief. Curr Biol. 30(6), R278–R280 (2020).
Reuß, D. R., Schuldes, J., Daniel, R. & Altenbuchner, J. Complete genome sequence of Bacillus subtilis subsp. subtilis strain 3NA. Genome Announc. 3(2), e00084-15 (2015).
Pedreira, T., Elfmann, C. & Stülke, J. The current state of SubtiWiki, the database for the model organism Bacillus subtilis. Nucleic Acids Res. 50(D1), D875–D882 (2022).
Sonbarse, P. P., Kiran, K., Sharma, P. & Parvatam, G. Biochemical and molecular insights of PGPR application for the augmentation of carotenoids, tocopherols, and folate in the foliage of Moringa oleifera. Phytochemistry 179, 112506 (2020).
Acknowledgements
Prof. D. Garozzo, Italy, is acknowledge for MALDI-TOF-MS of prepared samples.
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The author declares no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Alajlani, M.M. Characterization of subtilosin gene in wild type Bacillus spp. and possible physiological role. Sci Rep 12, 10521 (2022). https://doi.org/10.1038/s41598-022-13804-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-022-13804-y
This article is cited by
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
