
Abstract
Endometriosis (EMS) is a disease that shows immune dysfunction and chronic inflammation characteristics, suggesting a role of complement system in its pathophysiology. To find out the hub genes and pathways involved in the pathogenesis of EMs, three raw microarray datasets were recruited from the Gene Expression Omnibus database (GEO). Then, a series of bioinformatics technologies including gene ontology (GO), Hallmark pathway enrichment, protein–protein interaction (PPI) network and gene co-expression correlation analysis were performed to identify hub genes. The hub genes were further verified by the Real-time quantitative polymerase chain reaction (RT-PCR) and Western Blot (WB). We identified 129 differentially expressed genes (DEGs) in EMs, of which 78 were up-regulated and 51 were down-regulated. Through GO functional enrichment analysis, we found that the DEGs are mainly enriched in cell adhesion, extracellular matrix remodeling, chemokine regulation, angiogenesis regulation, epithelial cell proliferation, et al. In Hallmark pathway enrichment analysis, coagulation pathway showed great significance and the terms in which included the central complement factors. Moreover, the genes were dominating in PPI network. Combined co-expression analysis with experimental verification, we found that the up-regulated expression of complement (C1S, C1QA, C1R, and C3) was positively related to tissue factor (TF) in EMs. In this study, we discovered the over expression complement and the positive correlation between complement and TF in EMs, which suggested that interaction of complement and coagulation system may play a role within the pathophysiology of EMS.
Similar content being viewed by others


Introduction
Endometriosis (EMS), characterized by the presence of functional endometrial tissue outside the uterine cavity, is a common disease affecting up to 10–15% of reproductive-age women and is closely related to dysmenorrhea, chronic pelvic pain and infertility1. Among the hypotheses that have been proposed to explain the pathology of EMS, Sampson’s theory of retrograde menstruation is the most widely accepted etiology2. “Immunosurveillance” dysfunction was a critical facilitator of ectopic endometrial tissue implantation and growth3. EMS was considered as a chronic inflammatory disease4. One of the important immune mechanisms involved in the peritoneal clearance, inflammation and autoimmune in EMS was the complement system5,6,7. The close association of coagulation and inflammatory conditions has been known for a long time8. Current studies have showed that women with EMS appeared to be in a state of hypercoagulability and this coagulation dysfunction potentially attributed to the inflammatory nature of the ectopic lesions9,10,11. In recent years, the massive data of gene expression profile generated from high-throughput sequencing technology make it possible for us to explore the intrinsic mechanism of diseases at the molecular level12. Some meaningful researches have been conducted to identify genes involved in the development of EMS13,14. In this study, we integrated data sets from multiple studies and performed deep bioinformatic analyses to identify differentially expressed genes (DEGs) and important pathways participating in the development of EMS. Through pathway enrichment analysis, the coagulation process containing the central complement factors was indicated as the significant component of EMS. Real-time quantitative polymerase chain reaction (RT-PCR) and Western Blot (WB) were adopted to validate these results in women with or without EMS. After evaluating and comparing the expression of hub genes in EMS and control women, we found that the overexpressed state of complement was positively related to tissue factor (TF) in EMS. These suggested the existence of crosstalk between complement and coagulation in EMS.
Method
Data acquistion and differentially expressed genes (DEGs) identification
Three datasets (GSE23339, GSE7305, and GSE25628) were selected for our analysis. In detail, the gene set GSE23339 was annotated by GPL6102 platform, which included 10 endometriosis tissue samples and 9 normal endometrial tissue samples (as of control samples); GSE7305 was annotated by GPL570 platform, the data in which consist of 10 endometriosis tissue samples and 10 normal endometrial tissue samples; GSE25628 was annotated by GPL571 platform, which included 16 endometriosis and 6 normal endometrial samples. The platform annotation and series matrix files were downloaded for follow-up analyses.
Next, the orignial file obtained from GEO databases were preprocessed in R software. Samples of each dataset were normalized by the Robust Multi-array Average (RMA) function in the Affy package15. And then we used SVA package to eliminate the inter-batch difference between groups16. Multiple probes relating to the same gene were summarized as the median value for further analysis17. In our analysis, DEGs were defined with a threshold of p value < 0.05 and |log (FC)|> 1. DEGs in each database were displayed with the volcano map. Overlapping DEGs within three databases were shown with Venn diagrams.
Protein–protein interaction (PPI) network analysis
The PPI of DEGs-encoded proteins was confirmed by STRING18 (version 11.0), with search limitation to Homo sapiens and a score > 0.400 correspondingsto high confidence and statistical significance. After that, Cytoscape software (version 3.7.1) was used to construct the network19. Moreover, the cytohubba plug-in unit of Cytoscape software was used for the identification of the top 20 hub genes according to the count of interactions between genes.
Gene ontology and pathway enrichment analysis
Gene ontology (GO) Biological Processes of DEGs were analyzed using "clusterProfiler" package in R software20. P-value < 0.05 was defined as the cutoff value. GO considers annotates gene function from three aspects: molecular function(MF), cellular component(CC) and biological process(BP)21. Among these, Molecular functions define molecular processes, Cellular components define locations where molecular processes occur and Biological processes define biological programs comprised of regulated factors. To remove repetition and ensure the authenticity of the results, we merged the terms using simplify function in the GOplot package. The Hallmark pathway enrichment analysis was performed in Metascape16 (https://metascape.org/gp/index.html#/main/step3)(22). P-value < 0.05 was defined as the cutoff value.
Gene co-expression correlation analysis
To identify the correlation between hub genes, GSE141549 was downloaded as a validation set. GSE141549 was annotated by GPL10558 platform, the data in which consist of 198 endometriosis samples and 147 normal samples. We used R to build a co-expression network and visualize it.
Clinical sample collection
Eutopic endometrial tissues and control endometrium were obtained respectively from three patients with ovarian endometriosis (OE) and three women with benign ovarian teratoma, which were diagnosed by pathology after surgery. According to the American Fertility Society revised (AFS-r), these 3 EMs cases were all stage IV. All participants have written informed consent under the study protocol approved by the Ethics Committee of Changzhou Second People's Hospital (Changzhou, China). All of the endometrial tissues were collected at the proliferative phase during the menstrual cycle. None of the recruited women smoked or had taken any anticoagulant drugs or steroid hormones 3 months prior to the operation. There was no significant difference in age between the two groups (P > 0.05).
Quantitative-Polymerase chain reaction (qPCR)
Firstly, we isolated total RNA by using Trizol (Invitrogen) according to the manufacturer's protocol. The PrimeScript RT Master Mix (Takara, JPN) was used for the synthesis of cDNA. Quantitative-Polymerase chain reaction was performed using the SYBR Green assay to evaluate the expression of hub genes on the basis of manufacturer's instructions (Applied Biosystems, USA). The typical qPCR cycle were as followed: Pre‑denaturation at 95˚C for 30 s, followed by 40 cycles of denaturation at 95˚C for 5 s, annealing at 60˚C for 10 s and extension at 70–72˚C for 30 s23. We used GAPDH to normalize the expression of hub genes. And then, The relative expression level of the target amplicon was calculated between the EMs group and the control group. Hub genes and GAPDH primers were designed and synthesized by Sangon Biotech Co., Ltd. (Shanghai, China). The primers’ sequences were showed in Table 1.
Proteins extraction and Western Blot (WB)
Total proteins were extracted from human EMs tissue with Western and IP lysis buffer (Beyotime, P0013; Beijing). Protein concentration was measured using the BCA reagent kit (Pierce, 23,227). The proteins were resolved by 8%-12% SDS-PAGE and then blotted onto polyvinylidene fluoride (PVDF) membranes. Membranes were blocked in TBS/0.1% Tween-20 (TBST) containing 5% skimmed milk powder for 1 h at room temperature. Hub proteins and GAPDH antibodies were diluted with 1:300 (AtaGenix, Wuhan) and 1:2000 (AtaGenix, Wuhan) before incubation with 2 h at room temperature. The secondary antibody [anti-rabbit IgG (H + L) biotinylated antibody (CST, USA)] was incubated with 2 h at room temperature24.
Statistical analysis
All data statistics are analyzed using R software. P value < 0.05 is considered statistically significant. Independent t-test was used to compare the continuous variables of normal distribution and Mann Whitney U-test was used to compare the continuous variables of skew distribution.
Ethics approval, informed consent and experimental approval
This study was approved by the Ethics Committee of the Affiliated Changzhou No.2 People’s Hospital of Nanjing Medical University, China. All patients have signed the informed consent for the study protocol and reserve the right to withdraw at any time. The experimental scheme was approved by the academic committee of Ethics Committee of the Affiliated Changzhou No.2 People’s Hospital of Nanjing Medical University, and the experimental methods were carried out in accordance with the guidelines of the academic committee. The study was conducted according to common standard guidelines for clinical trials (Declaration of Helsinki).
Results
Identification of differentially expressed genes (DEGs) in EMS using integrated bioinformatics
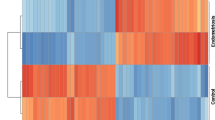
Samples in each dataset (GSE7305, GSE23339, and GSE25628) were normalized using Robust Multi-array Average (RMA) function in the Affy package (Fig. 1). All datasets were normalized using SVA package to eliminate potential inter-batch difference (Fig. 2). Then we used the Limma package to perform differential expression analysis on the three data sets with a definition criterion based on p < 0.05 and |log (FC)|> 1 to select statically statistically DEGs. In GSE7305, 1262 DEGs were identified, of which 694DEGs were up-regulated and 568 down-regulated (Fig. 3A); In GSE23339,1241 DEGs were identified, of which 676DEGs were up-regulated and 565 down-regulated (Fig. 3B); In GSE25628, 1644 DEGs were identified, of which 549DEGs were up-regulated and 1095 down-regulated (Fig. 3C). We identified 129 DEGs, of which 78 were up-regulated and 51 down-regulated (Fig. 3D,E and Table 2). Then, we converted the chart into the heat map to show the expression of DEGs intuitively (Fig. 4).

Box plot graph of value distribution of GSE7305 (A), GSE2339 (B) and GSE25628 (C) The left side is before normalization, the right side is after normalization.

Principal Component Analysis for combined expression profile of each data set. (A) raw PCA for combined expression profile before eliminating the inter-batch difference. (B) combat PCA for combined expression profile after eliminating the inter-batch difference.

Volcano plots and Venn diagrams of DEGs in endometriosis microarray datasets. Volcano plots showing DEGs in GSE7305 (A), GSE23339 (B), and GSE25628 (C). DEGs are those genes with P-value < 0.05 and |log (FC)|> 1. Red indicates relative up-regulated genes and green indicates down-regulated genes. Venn diagrams of up-regulated (D) or down-regulated (E) DEGs from these three datasets, as indicated.

Heat maps of the DEGs in endometriosis microarray datasets. The vertical column is the name of the data set, the horizontal column is the gene name. DEGs are those genes with P value < 0.05 and |log (FC)|> 1. Red indicates up-regulation and blue indicates down-regulation.
Protein–protein interaction (PPI) network analysis in DEGs
PPI network was performed using the online STRING database and then Cytoscape software was used for visualization. After removing partly connected and isolated nodes, we constructed a grid network using the Cytoscape-software (Fig. 5A). Then, we used the Cytohubba module of Cytoscape to calculate the top 20 hub genes with the highest scores, including SERPING1, P2RY14, C1S, C3, C1QA, C1R, GNG4, CCL21, CXCL12, COLEC11, AGTR1, LRP1, THBS1, BGN, FMOD, THBS2, DSP, CD24, CLDN5, CLDN7 (Fig. 5B).

(A) PPI network analysis of DEGs. (B) PPI network analysis of the top 20 hub genes with the highest scores. The size of the circle is related to the score. (C) GO analysis of DEGs in endometriosis. The result of GO analysis was divided into three parts: biological process (BP), cellular component (CC), and molecular functions (MF). The color of the circle was related to the P-value and the size of the circle was related to the count. (D) Hallmark pathway enrichment of DEGs in endometriosis. Pathway enrichment of DEGs in endometriosis was visualized on a bar chart, showing -Log10 (P-value).
Gene Ontology (GO) functional enrichment in DEGs
Thereafter, we performed gene ontology (GO) enrichment analysis of DEGs in endometriosis by using clusterprofiler package. The results were divided into three parts: biological process (BP), cellular component (CC), and molecular functions (MF). Then, we use R to import the top ten results of every sector into the figure (Fig. 5C). In the BP, DEGs were mainly involved in cell adhesion, epithelial cell proliferation, granulocyte chemotaxis regulation, and extracellular matrix remodeling. In the CC, cell connection, cell adhesion, and extracellular matrix collagen-containing were included. In the MF, terms were mainly involved in binding and extracellular matrix structural constituent conferring compression resistance.
Hallmark signaling pathway enrichment in DEGs
We use Metascape16 to perform signaling pathway enrichment of DEGs in endometriosis, and then we submit the most significantly enriched pathways to Hallmark genes hit analysis. Through Hallmark pathway enrichment analysis, epithelial-mesenchymal transition (EMT), coagulation, estrogen response late, KRAS signaling up, and apical junction were identified (Fig. 5D). Among them, EMT and coagulation pathways showed great significance according to the P Value (Table 3).
Bioinformatics analysis shows that SERPING1, C1S, C1QA, C1R, C3, and TF are up-regulated in EMs while P2RY14 is down-regulated in EMs group
Combined Top20 hub genes with pathway enrichment analysis, coagulation pathway showed great significance. Meanwhile, SERPING1, P2RY14, C1S, C1QA, C1R, and C3 showed better coagulation pathway correlation and EMs correlation. Among them, SERPING1, C1S, C1QA, C1R, and C3 were up-regulated in EMs while P2RY14 was down-regulated (Figure S2a). To verify whether the expression of genes involved in the coagulation cascade was synchronized with complement factors, we selected tissue factor (TF) for verification, which was the important factor in the exogenous coagulation pathway and EMs25,26. After analyzing the data set, TF was found to be up-regulated in EMs (P = 0.044) (Figure S2b).
The expression of SERPING1, C1S, C3, C1QA, C1R, and TF are up-regulated while P2RY14 is down-regulated at mRNA level and protein level in EMs group
We used qPCR to analyze the mRNA expression level of the hub genes in the coagulation pathway. The expression of SERPING1, C1S, C1QA, C1R, and C3 were up-regulated in EMs group (Fig. 6A–E) while P2RY14 was down-regulated (Fig. 6G). This result was also validated by western blotting at the protein level (Fig. 7). The verification of the hub genes both at mRNA level and protein level suggested the abnormal status of complement in EMs. We also found that the expressions of TF were up-regulated both at the mRNA and protein levels of EMs group (Fig. 6F, 8).

The mRNA expression of hub genes between groups by qPCR. (A–E) the expression of SERPING1, C1S, C1QA, C1R, and C3 were frequently up-regulated in the EMs group compared with the normal group. (F) the expression of TF was up-regulated in EMs group compared with the normal group. (G) the expression of P2RY14 was down-regulated in EMs group compared with the normal group.

The protein expression of hub genes between groups by WB. The expression of SERPING1, C1S, C1QA, C1R, C3, and TF were up-regulated in EMs group compared with the normal group. The expression of P2RY14 was down-regulated in EMs group compared with the normal group.

Gene co-expression correlation analysis. The expression of central complement factors was positively related to TF while SERPING1 and P2RY14 were negatively related to TF. Red represents a positive correlation and blue indicates a negative correlation.
Bioinformatics analysis shows that Central complement factors (C1S, C1QA, C1R, C3) are positively related to TF
Further, we analyzed the correlation between the expression of hub genes and TF in GSE141549 data set. In co-expression correlation analysis, the complement showed a positive correlation with TF while SERPING1 and P2RY14 showed a negative correlation with TF. Interestingly, SERPING1 was up-regulated in EMs synchronized with complement components (Fig. 8). Also, these genes were involved in the coagulation, proposing the existence of crosstalk between complement and coagulation cascades in EMs.
Discussion
Here, through integration and analysis of the data from 3 microarray datasets, we identified 129 DEGs, of which 78 up-regulated and 51 down-regulated in EMs. GO functional enrichment analysis showed that DEGs are mainly enriched in cell adhesion, extracellular matrix remodeling, chemokine regulation, angiogenesis regulation, epithelial cell proliferation, which have been proved the essential steps in the development of EMs26,27,28,29. In this study, Hallmark pathway enrichment analysis disclosed coagulation and epithelial-mesenchymal transition (EMT) were the most significant pathways in EMs30,31,32. To EMT, many studies have revealed its main role in the development of EMS and Yihua Wang et al. have also confirmed this by bioinformatic analysis33.
So, in this study, we focused on the coagulation pathway of EMS and found that complement factors were enriched in this way according Hallmark pathway enrichment analysis. It is widely known that the relationship between the immune system and EMS is intimate34,35. Swati et al. revealed that chronic inflammation in endometriosis is dominated by complement7. Sikora et al. had proved that abnormal activation of the complement pathway occurred in EMS and resulted in the overexpression of C1Q, MBL, C1INH36. In our study, we found that the up-regulated expression of the central complement factor (C1S, C1QA, C1R, and C3) in EMS, indicating that the abnormal status of complement might be involved in the pathogenesis of EMS. Meanwhile, Vigano et al. confirmed the existence of hypercoagulable state in EMS patients37. Researches conducted by Guo et al. also revealed that stromal cells in EMS lesions could secrete thrombin and Thromboxane A2 (TXA2) to induce platelet activation and participate in coagulation38,39. And then, Thrombin in the endometrial matrix could be activated in response to repeated bleeding from highly vascularized lesions in EMs and combined with PAR-1 to mediate thrombin-induced angiogenesis40,41. By now, few researches focused on EMs indicated the existence of the crosstalk between complement and coagulation.
Though, the crosstalk between the complement and coagulation cascades has been proved in septicemia. Rittirsch revealed that complement activation could enhance coagulation through up-regulating the expression of tissue factor42. Hence, we proposed that TF might be the interaction between complement and coagulation and the crosstalk between complement and coagulation might also play an essential role in the development of EMS. Previously, Ding et al. found that the concentration of TF in the peritoneal fluid of EMS patients is higher than that of normal women43. TF had also been proved to be overexpressed in ectopic lesions44. Lin et al. found that TF was overexpressed in epithelial cells and stromal cells of EMs45. Graciela et al. found that markedly increased expression of TF in glandular epithelial cells of ectopic and eutopic endometrium derived from patients with EMS25. It is generally known that TF could combine with circulating factor VII-a to participate in the exogenous coagulation pathway. Therefore, we considered that TF might be a bridge link between complement and coagulation in EMS. Two mechanisms might account for such an association. Firstly, when membrane attack complex (MAC) was anchored on the cell surface, MAC could stimulate oxidation of protein disulfide isomerase and induce exposure of functionally active procoagulant TF46. Secondly, MAC could enhance the platelet procoagulant reaction by generating procoagulant particles or inverting the membrane to expose phosphatidylserine47. Importantly, in addition to participating in blood clotting, activated platelets could be recruited to lesions and derive TGF- β to promote the development of diseases32,48.
Moreover, TF could induce the formation of blood vessels through AKT-ETS1 and promotes the stability of blood vessels through PAR2-SMAD3, which might be beneficial to the survival of lesions49,50. Nowadays, TF targeted treatments have shown a good therapeutic effect, which could significantly inhibit the EMS lesion51,52,53. In our experimental verification, TF was also found to be overexpressed in EMS. Combined with co-expression analysis, we demonstrated that the up-regulated expression of complement factors was positively related to TF. The result indicated the existence of crosstalk between the complement and coagulation cascade. So far, however, there is still little evidence focused on the interaction of complement and coagulation cascade in EMS. Subsequently, the encouraging result discovered by our study may provide a novel direction for future researches.
Besides, SERPING1 and P2RY14 were also genes of great significance in our analysis. SERPING1 is the coding gene of C1NH, which is the inhibitor of the complement pathway54,55. And, SERPING1 was found to be overexpressed in EMS synchronized with C1S, C1QA, C1R, and C3, which showed a dual tendency to pro-inflammatory and anti-inflammatory. It is widely considered that EMS is a chronic inflammatory disease while Zhou et al. proposed that anti-inflammatory was equally important in EMS and that Anti-inflammatory factors involved in EMS could promote the growth, invasion, angiogenesis and immune escape of lesions56. This phenomenon has also been reported at the cell level. In general, the M2 macrophage phenotype accounts for anti-inflammation, whereas the M1 macrophage phenotype is proinflammatory. Interestingly, the ratio of M2 increased as EMS progressed57. Up-regulated expression of SERPING1 suggested that there may be protective mechanisms for ectopic lesions in vivo. P2RY14 is a membrane receptor for UDP-glucose, which is exclusively expressed in the epithelium. It could induce innate mucosal immunity in the female reproductive tract independent of the pathogen recognition pattern58. In our research, P2RY14 was down-regulated, which might be related to immune surveillance escape.
Through bioinformatics analysis, our research proposed that TF might be a significant bridge link between complement and coagulation in EMS. Meanwhile, we proved the abnormal state of complement in EMS again. To the best of our knowledge, this is an important study concentrating on the interaction of TF and the central complement factor in EMS. However, our study inevitably had certain limitations. Firstly, the amount of data published in GEO was limited, which might lead to potential errors or bias. In subsequent studies, the samples should be further expanded to reduce bias. Secondly, there were regional differences in data sources, so the research results might not apply to all patients. Hence, an in-depth research is needed to be verified.
Conclusion
Taken as a whole, through serial bioinformatics analysis and verification experiments, we confirmed the abnormal status of complement in EMs. And then, SERPING1, P2RY14, C1S, C1QA, C1R, and C3 might become potential immunotherapy targets for EMs. Besides, TF connected the central complement factors and coagulation cascades and was confirmed to be positively related to complement. Hence, the crosstalk between the complement and coagulation might play an essential role in the development of EMs.
References
Zondervan, K. T., Becker, C. M. & Missmer, S. A. Endometriosis. N. Engl. J. Med. 382, 1244–1256 (2020).
Vercellini, P. et al. Asymmetry in distribution of diaphragmatic endometriotic lesions: evidence in favour of the menstrual reflux theory. Hum. Reprod. (Oxford, England) 22, 2359–2367 (2007).
PaulDmowski, W. & Braun, D. P. Immunology of endometriosis. Best Pract. Res. Clin. Obstetr. Gynaecol. 18, 245–263 (2004).
Jiang, L., Yan, Y., Liu, Z. & Wang, Y. Inflammation and endometriosis. . Front. Biosci. (Landmark edition) 21, 941–948 (2016).
Blom, A. M. The role of complement inhibitors beyond controlling inflammation. J. Intern. Med. 282, 116–128 (2017).
Lubbers, R., van Essen, M. F., van Kooten, C. & Trouw, L. A. Production of complement components by cells of the immune system. Clin. Exp. Immunol. 188, 183–194 (2017).
Suryawanshi, S. et al. Complement pathway is frequently altered in endometriosis and endometriosis-associated ovarian cancer. Clin. Cancer Res. 20, 6163–6174 (2014).
Esmon, C. T. The interactions between inflammation and coagulation. Br. J. Haematol. 131, 417–430 (2005).
Wu, Q., Ding, D., Liu, X. & Guo, S. W. Evidence for a hypercoagulable state in women with ovarian endometriomas. Reprod. Sci. (Thousand Oaks, Calif.) 22, 1107–1114 (2015).
Ding, D., Liu, X. & Guo, S. W. Further Evidence for Hypercoagulability in Women With Ovarian Endometriomas. Reprod. Sci. (Thousand Oaks, Calif.) 25, 1540–1548 (2018).
Ottolina, J. et al. Assessment of coagulation parameters in women affected by endometriosis: validation study and systematic review of the literature. Diagnostics (Basel, Switzerland) 10, 1 (2020).
Huang, X., Liu, S., Wu, L., Jiang, M. & Hou, Y. High throughput single cell RNA sequencing, bioinformatics analysis and applications. Adv. Exp. Med. Biol. 1068, 33–43 (2018).
Zhang, Z., Ruan, L., Lu, M. & Yao, X. Analysis of key candidate genes and pathways of endometriosis pathophysiology by a genomics-bioinformatics approach. Gynecol. Endocrinol. 35, 576–581 (2019).
Tan, A., Luo, R., Liang, H., Li, M. & Ruan, P. Bioinformatics approach reveals the key role of C-X-C motif chemokine receptor 2 in endometriosis development. Mol. Med. Rep. 18, 2841–2849 (2018).
Gautier, L., Cope, L., Bolstad, B. M. & Irizarry, R. A. affy–analysis of Affymetrix GeneChip data at the probe level. Bioinformatics (Oxford, England) 20, 307–315 (2004).
Leek, J. T., Johnson, W. E., Parker, H. S., Jaffe, A. E. & Storey, J. D. The sva package for removing batch effects and other unwanted variation in high-throughput experiments. Bioinformatics (Oxford, England) 28, 882–883 (2012).
Tarafdar, A. et al. CML cells actively evade host immune surveillance through cytokine-mediated downregulation of MHC-II expression. Blood 129, 199–208 (2017).
Szklarczyk, D. et al. STRING v11: protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucl. Acids Res. 47, D607-d613 (2019).
Doncheva, N. T., Morris, J. H., Gorodkin, J. & Jensen, L. J. Cytoscape stringapp: network analysis and visualization of proteomics data. J. Proteome Res. 18, 623–632 (2019).
Yu, G., Wang, L. G., Han, Y. & He, Q. Y. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS 16, 284–287 (2012).
Thomas, P. D. The gene ontology and the meaning of biological function. Methods Mol. Biol. (Clifton, N.J.) 1446, 15–24 (2017).
Zhou, Y. et al. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun. 10, 1523 (2019).
Miller, J. R. & Andre, R. Quantitative polymerase chain reaction. Br. J. Hosp. Med. (Lond. Engl.) 75, 188–192 (2014).
B. Kim, Western Blot Techniques. Methods in molecular biology (Clifton, N.J.) 1606, 133–139 (2017).
Krikun, G., Schatz, F., Taylor, H. & Lockwood, C. J. Endometriosis and tissue factor. Ann. N. Y. Acad. Sci. 1127, 101–105 (2008).
G. Krikun, Endometriosis, angiogenesis and tissue factor. Scientifica 2012, 306830 (2012).
Tsai, H. W. et al. Decoy receptor 3 promotes cell adhesion and enhances endometriosis development. J. Pathol. 244, 189–202 (2018).
Yang, W. V. et al. Matrix remodeling and endometriosis. Reprod. Med. Biol. 4, 93–99 (2005).
Agic, A., Djalali, S., Diedrich, K. & Hornung, D. Apoptosis in endometriosis. Gynecol. Obstet. Invest. 68, 217–223 (2009).
D. M. Gonzalez, D. Medici, Signaling mechanisms of the epithelial-mesenchymal transition. Sci. Signal. 7, re8 (2014).
A. Misra, C. Pandey, S. K. Sze, T. Thanabalu, Hypoxia activated EGFR signaling induces epithelial to mesenchymal transition (EMT). PloS One 7, e49766 (2012).
Zhang, Q., Duan, J., Liu, X. & Guo, S.-W. Platelets drive smooth muscle metaplasia and fibrogenesis in endometriosis through epithelial–mesenchymal transition and fibroblast-to-myofibroblast transdifferentiation. Mol. Cell. Endocrinol. 428, 1–16 (2016).
Chen, M. et al. Bioinformatic analysis reveals the importance of epithelial-mesenchymal transition in the development of endometriosis. Sci. Rep. 10, 8442 (2020).
Symons, L. K. et al. The Immunopathophysiology of Endometriosis. Trends Mol. Med. 24, 748–762 (2018).
Poli-Neto, O. B., Meola, J., Rosa, E. S. J. C. & Tiezzi, D. Transcriptome meta-analysis reveals differences of immune profile between eutopic endometrium from stage I-II and III-IV endometriosis independently of hormonal milieu. Sci. Rep. 10, 313 (2020).
Sikora, J. et al. The role of complement components C1q, MBL and C1 inhibitor in pathogenesis of endometriosis. Arch. Gynecol. Obstet. 297, 1495–1501 (2018).
P. Viganò et al., Coagulation Status in Women With Endometriosis. Reprod. Sci. (Thousand Oaks, Calif.) 25, 559–565 (2018).
S. W. Guo, Y. Du, X. Liu, Endometriosis-Derived Stromal Cells Secrete Thrombin and Thromboxane A2, Inducing Platelet Activation. Reprod. Sci. (Thousand Oaks, Calif.) 23, 1044–1052 (2016).
Ding, S. et al. Is there a correlation between inflammatory markers and coagulation parameters in women with advanced ovarian endometriosis?. BMC Womens Health 19, 169 (2019).
Tsopanoglou, N. E. & Maragoudakis, M. E. Inhibition of angiogenesis by small-molecule antagonists of protease-activated receptor-1. Semin. Thromb. Hemost. 33, 680–687 (2007).
Hirota, Y. et al. Possible involvement of thrombin/protease-activated receptor 1 system in the pathogenesis of endometriosis. J. Clin. Endocrinol. Metab. 90, 3673–3679 (2005).
Rittirsch, D., Flierl, M. A. & Ward, P. A. Harmful molecular mechanisms in sepsis. Nat. Rev. Immunol. 8, 776–787 (2008).
Ding, D., Liu, X., Duan, J. & Guo, S. W. Platelets are an unindicted culprit in the development of endometriosis: clinical and experimental evidence. Hum. Reprod. (Oxford, England) 30, 812–832 (2015).
Key, N. S. Platelet tissue factor: how did it get there and is it important?. Semin. Hematol. 45, S16-20 (2008).
Lin, X. et al. Hypoxia Promotes Ectopic Adhesion Ability of Endometrial Stromal Cells via TGF-β1/Smad Signaling in Endometriosis. Endocrinology 159, 1630–1641 (2018).
Subramaniam, S. et al. Distinct contributions of complement factors to platelet activation and fibrin formation in venous thrombus development. Blood 129, 2291–2302 (2017).
Conway, E. M. Complement-coagulation connections. Blood Coagulat. Fibrinol. Int. J. Haemostas. Thrombosis 29, 243–251 (2018).
Zhang, Q., Liu, X. & Guo, S. W. Progressive development of endometriosis and its hindrance by anti-platelet treatment in mice with induced endometriosis. Reprod. Biomed. Online 34, 124–136 (2017).
Arderiu, G., Espinosa, S., Peña, E., Aledo, R. & Badimon, L. PAR2-SMAD3 in microvascular endothelial cells is indispensable for vascular stability via tissue factor signaling. J. Mol. Cell Biol. 8, 255–270 (2016).
Arderiu, G., Peña, E., Aledo, R. & Badimon, L. Tissue factor-Akt signaling triggers microvessel formation. J. Thrombos. Haemostas. JTH 10, 1895–1905 (2012).
Wang, Y. et al. ENMD-1068, a protease-activated receptor 2 antagonist, inhibits the development of endometriosis in a mouse model. Am. J. Obstet. Gynecol. 210(531), e531-538 (2014).
Krikun, G. et al. The immunoconjugate “icon” targets aberrantly expressed endothelial tissue factor causing regression of endometriosis. Am. J. Pathol. 176, 1050–1056 (2010).
Z. Hu, Therapeutic Antibody-Like Immunoconjugates against Tissue Factor with the Potential to Treat Angiogenesis-Dependent as Well as Macrophage-Associated Human Diseases. Antibodies (Basel, Switzerland) 7, 1 (2018).
Ponard, D. et al. SERPING1 mutation update: Mutation spectrum and C1 Inhibitor phenotypes. Hum. Mutat. 41, 38–57 (2020).
Rekker, K. et al. High-throughput mRNA sequencing of stromal cells from endometriomas and endometrium. Reproduction (Cambridge, England) 154, 93–100 (2017).
Zhou, W. J. et al. Anti-inflammatory cytokines in endometriosis. Cellular and molecular life sciences : CMLS 76, 2111–2132 (2019).
Hogg, C., Horne, A. W. & Greaves, E. Endometriosis-associated macrophages: origin, phenotype, and function. Front. Endocrinol. 11, 7 (2020).
T. Arase et al., The UDP-glucose receptor P2RY14 triggers innate mucosal immunity in the female reproductive tract by inducing IL-8. J. Immunol. (Baltimore, Md.: 1950) 182, 7074–7084 (2009).
Acknowledgements
This work was supported by Jiangsu Provincial Commission of Health and Family Planning scientific research project (H2018017), Jiangsu Provincial Women's and Children's Health key talent project (RC201709).
Author information
Authors and Affiliations
Contributions
R.X. collected the data. Y.L. performed all analysis and wrote the manuscript. S.H. did the experiment. Z.Y. & W.X. provided guidance on research directions. All the authors participated in the data analysis and approved the final version of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Yu, L., Shen, H., Ren, X. et al. Multi‐omics analysis reveals the interaction between the complement system and the coagulation cascade in the development of endometriosis. Sci Rep 11, 11926 (2021). https://doi.org/10.1038/s41598-021-90112-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-021-90112-x
This article is cited by
-
Identifying hub genes of sepsis-associated and hepatic encephalopathies based on bioinformatic analysis—focus on the two common encephalopathies of septic cirrhotic patients in ICU
BMC Medical Genomics (2024)
-
Characterizing the Extracellular Matrix Transcriptome of Endometriosis
Reproductive Sciences (2024)
-
CEP55: an immune-related predictive and prognostic molecular biomarker for multiple cancers
BMC Pulmonary Medicine (2023)
-
Identification of STEAP3-based molecular subtype and risk model in ovarian cancer
Journal of Ovarian Research (2023)
-
Exploring the prognostic function of TMB-related prognostic signature in patients with colon cancer
BMC Medical Genomics (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
