
Abstract
This study aimed to focus on the simultaneous use of antimicrobial photodynamic therapy (aPDT) and sonodynamic antimicrobial chemotherapy (SACT), which is called photo-sonodynamic antimicrobial chemotherapy (PSACT) to attenuate the virulence of Aggregatibacter actinomycetemcomitans. Following the synthesis of Curcumin-decorated nanophytosomes (Cur-NPhs) as a novel photo-sonosensitizer, its particle size, polydispersity, ζ-potential surface morphology, physical stability, drug release, and entrapment efficiency were determined. In the Cur-NPhs-PSACT, the antimicrobial activities of Cur-NPhs against A. actinomycetemcomitans were investigated using cell viability, biofilm killing/degradation, metabolic activity, expression of quorum-sensing-associated qseB and qseC genes, and biofilm-associated rcpA gene under blue laser irradiation plus ultrasonic waves. Characterization tests showed the presence of a sphere-shaped vesicle and the self-closed structure of Cur-NPhs, resulting in a high drug-loading content and encapsulation efficiency. However, the antimicrobial effect of Cur-NPhs-PSACT was dose-dependent, PSACT using the high concentrations of Cur-NPhs (50 × 10–4 g/L) showed significant reductions (P < 0.05) in cell viability (13.6 log10 CFU/mL), biofilm killing/degradation (65%), metabolic activity (89.6%,), and mRNA levels of virulence determinant genes (qseB; 9.8-fold, qseC; 10.2-fold, and recA; 10.2-fold). This study concludes that the Cur-NPhs-PSACT had antimicrobial activities against A. actinomycetemcomitans by downregulating the expression of virulence genes, and may attenuate this bacterium that decreases periodontal disease severity in patients.
Similar content being viewed by others

Introduction
Periodontal disease is a collective form of the inflammatory disorder that is induced by oral microorganisms, dental plaque, or dental biofilm. According to the latest Portuguese Oral National Health Survey (PONHS), the prevalence of periodontitis was 10.8% and 15.3% in adults and the elderly, respectively1. Several factors that increase the risk of periodontal diseases are divided into two groups; 1. Modifiable risk factors such as smoking, poor oral hygiene, hormonal changes in females, diabetes mellitus, medications, and stress, 2. Non-modifiable risk factors including age and hereditary2. In addition, microbial pathogens are one of the most common causative agents of periodontal disease3.
Aggregatibacter actinomycetemcomitans is one of the main causes of periodontal disease in juveniles and adolescents4. The pathology and etiology of periodontal disease have demonstrated several virulence factors such as lipopolysaccharide (LPS), fimbriae, and enzymes can trigger inflammation in periodontal tissues caused by A. actinomycetemcomitans5.
The success of periodontitis treatment depends on eliminating the bacterial load of periodontal pockets to restore biological compatibility of periodontally diseased root surfaces6. The treatment of periodontitis is challenging and routine mechanical or non-surgical periodontal treatments do not diminish effectively the load of the microbial strains. On the other hand, the over-use of systemic and local antibiotics has been the main reason for the emergence of drug-resistant bacteria7. Nevertheless, the application of the alternative approaches for the removal of periopathogens from periodontal pockets is highly essential. Therefore, the development of a new antimicrobial approach with fewer complications is necessary.
Antimicrobial photodynamic therapy (aPDT) and sonodynamic antimicrobial chemotherapy (SACT) as the non-invasive therapeutic modalities are the new antimicrobial strategies to improve periodontal treatment8. SACT is analogous to aPDT, except that drug activation is achieved through ultrasound instead of visible light. As well, photosensitizer as a photosensitizing agent in aPDT is replaced by sonosensitizer as a sonosensitizing agent in SACT9. The antimicrobial effects of aPDT and SACT depend on the production of cytotoxic reactive oxygen species (ROS) including singlet oxygen (1O2), hydroxyl radicals (·OH), peroxyl radical (ROO·), and superoxide (O2−) ions causing damage to the cellular and molecular structures of microorganisms10.
In the present study, for the first time, we aim to examine the antimicrobial effects of the simultaneous use of aPDT and SACT, which called photo-sonodynamic antimicrobial chemotherapy (PSACT) using Curcumin-decorated nanophytosomes (Cur-NPhs) as the photo-sonosensitizer on the regulation of qseB and qseC genes expression in A. actinomycetemcomitans. After confirmation of Cur-NPhs synthesis, we examined the effects of Cur-NPhs-mediated PSACT on cell viability, biofilm degradation, metabolic activity, and qseB and qseC genes expression.
By revealing the interaction of suppression of metabolic activities during treatments with suppression of rcpA expression among A. actinomycetemcomitans, we attempt to explain, at least in part, the suppression of rcpA expression is likely a byproduct of the general suppression of metabolic activities, and the Cur-NPhs-mediated PSACT targeted specifically the bacterial biofilm structure. Our findings might provide additional options for the management of periodontitis and/or pri-implantitis, either as direct treatment strategies or adjuncts to routine methods.
Results
Confirmation of synthesized Cur-NPhs
As shown in Fig. 1a, the size of the Cur-NPhs was 156.42 ± 2.06 nm and the polydispersity index was 0.34 ± 0.02. The presence of a sphere-shaped vesicle and the self-closed structure was indicated in the TEM of the Cur-NPhs complex (Fig. 1b). It was observed that the average particle size and the ζ-potential value were 86.3 ± 2.08 nm and − 34.7 ± 1.3 mV, respectively, which indicate good stability (Fig. 1c,d).

Characterization of synthesized Cur-NPhs. (a) Representative scanning electron micrograph (scale bar represents 10 μm). (b) Representative transmission electron micrograph (scale bar represents 30 nm). (c) Average particle size. (d) ζ-potential value.
High EE is the main factor for the choice of a specific carrier and the encapsulation approach. According to the results, the EE percentage of the Cur in Cur-NPhs was 83.56 ± 0.5%.
In vitro drug release
The release profile showed the release of Cur occurred in a biphasic process in which a rapid initial burst of about 12% within the first hour followed by sustained drug release of about 74% in 4 h (Fig. 2). The drug entrapped on the surface of the phytosomes might be the reason for the observed initial burst release.

Profile of Cur release from Cur-NPhs.
Physical stability of Cur-NPhs
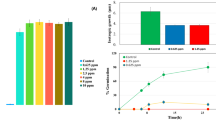
Cur-NPhs stability was measured by particle size over 2 weeks at 4 °C and 37 °C, which showed no significant variation between day 0 and day 14 (P > 0.05; Fig. 3). The results showed no significant changes in polydispersity index and ζ-potential of Cur-NPs during 2 weeks.

Particle size, polydispersity index, and ζ-potential of Cur-NPhs in different conditions; (a) at 4 °C, (b) at 37 °C.
Also, the effects of different pH treatments on the RR of Cur-NPs were shown in Fig. 4. During the 24 and 48 h storage period, Cur-NPhs were more stable under acidic conditions with RR more than 90%, whiles RR of Cur-NPhs decreased to 55% at neutral and alkaline conditions. Meanwhile, no significant changes were achieved in RR of Cur-NPhs after 48 h of storage (P > 0.05). It is therefore concluded that Cur-NPs did not undergo degradation on storage.

Retention rate percentage of Cur-NPhs at different pH; (a) during 24 h and (b) 48 h storage period.
Cytotoxicity effects of treatment groups on A. actinomycetemcomitans viability
A dose-dependent decrease was observed in log10 CFU/mL of A. actinomycetemcomitans (Fig. 5). The log10 CFU/mL reduction was increased to 3.3 ± 0.06, 5.1 ± 0.08, and 6.8 ± 0.05, after treatment with 10, 25, and 50 × 10–4 g/L Cur-NPhs, respectively (P < 0.05). As shown in Fig. 5, cell viability significantly decreased in treated A. actinomycetemcomitans with aPDT and SACT using different concentrations of Cur-NPhs compared with the control group (P < 0.05). The results indicated that 50 × 10–4 g/L Cur-NPhs plus 2 min blue laser irradiation in aPDT group and 50 × 10–4 g/L Cur-NPhs plus ultrasonic waves with the intensity of 1.56 W/cm2 in SACT group could effectively decrease 10.7 ± 0.05 log10 CFU/mL and 11.2 ± 0.03 log10 CFU/mL of microbial cells, respectively (P < 0.05). Interestingly, PSACT was able to reduce the survival rate of bacteria more than the other groups compared to the control group (P < 0.05), so that a 13.6 ± 0.08 log10 CFU/mL reduction was observed in 50 × 10–4 g/L Cur-NPhs-PSACT-treated cells (Fig. 5).

Cell viability of A. actinomycetemcomitans following different treatments. Significant differences according to the control, *P < 0.05.
According to the data obtained in the present study, there was no significant difference in the rate of A. actinomycetemcomitans viability following NPhs, NPhs-mediated aPDT-, SACT-, and PSACT-treatment, blue laser irradiation at a wavelength of 450 ± 5 nm for 2 min, as well as, ultrasonic waves with the intensity of 2 W/cm2 for 2 min at a frequency of 1 MHz (P > 0.05).
Anti-biofilm potency of treatment groups against A. actinomycetemcomitans
The results of the CV assay showed that aPDT, SACT, and PSACT with different concentrations of Cur-NPhs decreased the biofilm forms of A. actinomycetemcomitans compared to that of untreated biofilm (P < 0.05; Fig. 6). According to the results, degradation of A. actinomycetemcomitans biofilms was dose-dependent. PSACT using 50 × 10–4 g/L Cur-NPhs plus ultrasound waves with the intensity of 1.56 W/cm2 was the most effective biofilm degradation against A. actinomycetemcomitans (65% biofilm killing; P < 0.05). In contrast, there was no considerable degradation in biofilms of A. actinomycetemcomitans exposed to the NPhs, Cur-NPhs, blue laser irradiation, ultrasonic waves alone, NPhs-mediated aPDT-, SACT-, and PSACT-treatment (P > 0.05).

Biofilm degradation ability of A. actinomycetemcomitans following different treatments. Significant differences according to the control, *P < 0.05.
Determination of A. actinomycetemcomitans metabolic activity following treatment groups
Although considerable reduction of A. actinomycetemcomitans metabolic activity was observed upon treatment with Cur-NPs-aPDT and Cur-NPs-SACT, therapy employing Cur-NPs-PSACT generated a significant reduction of metabolic activity more than other groups (Fig. 7; P < 0.05). There was a more significant reduction of metabolic activity following Cur-NPhs-PSACT at the concentrations of 10, 25, and 50 × 10–4 g/L to 49.3%, 69.2%, and 89.6%, respectively (all P < 0.05; Fig. 7). In contrast, no significant changes were observed in metabolic activity of A. actinomycetemcomitans after exposure with blue laser irradiation, ultrasonic waves, and different concentrations of NPs and Cur-NPhs alone (P > 0.05), except 50 × 10–4 g/L Cur-NPhs (P < 0.05). Also, there was no significant effect in metabolic activity of A. actinomycetemcomitans following NPhs-mediated aPDT-, SACT-, and PSACT-treatment (P > 0.0.5).

Metabolic activity of A. actinomycetemcomitans following different treatments. Significant differences according to the control, *P < 0.05.
Monitoring of quorum-sensing (QS)-associated qseB and qseC genes expression
The agarose gels of the amplified product showed the single bands corresponding to the predicted amplicon length. The amplification of qseB, qseC, and 16S rRNA yielded no false-positive and unspecific bands in test samples (Fig. 8). As shown in Fig. 8, dissociation melting curves of qseB, qseC, and 16S rRNA amplicons with a single peak demonstrates the specificity of the designed primer pairs.

Specificity of the designed primers; a. A 2% agarose gel electrophoresis for detection of qseB, qseC, and 16S rRNA. L ladder 100 bp, i: qseB, ii: qseC, iii: 16S rRNA. b. Melting curve, i: qseB, ii: qseC, iii: 16S rRNA.
The transcript levels of the biofilm-associated genes, qseB and qseC, were measured by performing qRT-PCR experiments. This study indicated that there was a dose-dependent manner effect of the treatment on the expression levels of qseB and qseC. The results showed that the expression levels of qseB were downregulated 4.2-, 6.5-, and 9.8-fold in treated A. actinomycetemcomitans by PSACT using 10, 25, and 50 × 10–4 g/L Cur-NPhs, respectively (Fig. 9; P < 0.05). Corresponding values in qseC were 4.7-, 7.2-, and 10.2-fold, respectively (P < 0.05).

Gene expression levels of qseB and qseC following different treatments. Significant differences according to the control, *P < 0.05.
As shown in Fig. 9, there were the significant 2.5-, 4.9-, and 6.3-fold downregulation of qseB and 3.1-, 5.4-, and 7.8-fold downregulation of qseC, in response to 10, 25, and 50 × 10–4 g/L increased concentrations of Cur-NPhs during aPDT, respectively (P < 0.05). Also, the transcript levels of qseB were downregulated 3.1-, 5.2-, and 7.3-fold, respectively, in treated A. actinomycetemcomitans by SACT using10, 25, and 50 × 10–4 g/L Cur-NPhs compared with those in the transcript levels of qseC that were 3.6-, 6.0-, and 8.3-fold, respectively (P < 0.05). According to the data obtained in the present study, there was no significant difference in the rate of transcript levels between the two genes in treated A. actinomycetemcomitans with aPDT and SACT (P > 0.05). As shown in Fig. 9, there was no significant changes in gene expression of qseB and qseC following NPhs-mediated aPDT-, SACT-, and PSACT-treatment (P > 0.0.5).
Monitoring of biofilm-associated rcpA gene expression
Significant reductions of rcpA were observed at 10, 25, and 50 × 10–4 g/L of Cur-NPhs-aPDT-, SACT-, and PSACT-treated A. actinomycetemcomitans (4.4-, 5.2-, and 9.7-fold, respectively). No significant change of rcpA mRNA expression was observed in treated groups using NPhs-aPDT, NPhs-SACT, and NPhs-PSACT at the concentrations of 10, 25, and 50 × 10–4 g/L (all P > 0.05; Fig. 10).

Gene expression levels of rcpA following different treatments. Significant differences according to the control, *P < 0.05.
Discussion
PSACT can be defined as the promising approach for eradication of microbial cells through the interaction of photo-sonosensitizer as a non-toxic photo-sonosensitizing agent with visible light at specific wavelength and ultrasonic waves with specific intensity. PSACT minimizes the side effects, maximizes the responses on-target, and can be focused deeply within the target site to a single discrete point in three dimensions11. After the activation of photo-sonosensitizer with the appropriate wavelength of light, electrons are transferred from a low to a higher level of energy which is called the triplet state that can undergo two pathways: transfer directly to the neighboring molecule or cell membrane (type-1 reaction) to form a radical anion which reacts with oxygen to produce ROS; or transfer its energy directly to molecular oxygen (type-2 reaction) to produce excited-state singlet oxygen12,13,14. In addition, the relatively low-intensity ultrasound excites the photo-sonosensitizer at the target site, which leads to microbubble formation through the acoustic cavitation process resulting in the generation of ROS.
The characteristics of the ideal photo-sonosensitizer have been discussed previously15. Briefly, they should be chemical purity non-toxic, absorb the light in the red or far-red wavelengths, eliminate rapidly from normal tissue, be cost-effective and easily available, and generate a large number of cytotoxic products. Based on the literature, the photosensitizers used in the treatment of periodontitis included methylene blue (MB), indocyanine green (ICG), phenothiazine chloride, toluidine blue (TBO), and Cur16,17,18,19,20,21,22.
Curcumin longa, derived from the turmeric rhizomes (Curcuma longa L.), is well known for its wide range of pharmacological properties23. The bioavailability limitations of Cur such as its hydrophobicity, low solubility, instability, poor absorption, rapid metabolism, and fast systemic removal have led to ongoing attempts for developing a new formulation to enhance its solubility, bioavailability, and therapeutic activities24. Evidence from several studies has indicated that phytosomes markedly improve the bioavailability and pharmacokinetics of Cur24,25. As recently reported by Purpura et al.26, phytosomes significantly improve the relative human bioavailability of Cur by 19.2-fold for Cur alone. It has been observed that Cur is directly bound to the polar head of phosphatidylcholine in phytosomes and the covalent bonds between phosphatidylcholine and the phytochemical cargo (i.e., Cur) are the main factor in stability profile of phytosomes compared with many other delivery systems27. Although there are several studies that investigate the efficacy of phytosomal Cur in the treatment of cancer, diabetes, and inflammatory diseases28,29,30,31,32, no study has hitherto assessed the antimicrobial properties of Cur-decorated NPhs especially during PSACT against periopathogenic bacteria.
In the current study, Cur-NPhs alone were able to reduce the planktonic A. actinomycetemcomitans counts significantly but had no inhibitory effect on biofilm growth. The observation of resistance to the Cur-NPhs when A. actinomycetemcomitans was grown in biofilms, suggests that biofilm formation is one of the critical virulence factors for this species, which permits A. actinomycetemcomitans to establish persistent periodontitis and peri-implantitis. Biofilm production by A. actinomycetemcomitans and its role in oral infections remain poorly studied. It has been revealed that the resistance of biofilms to antimicrobial agents is increased 10–1000 times compared with what is normally seen with planktonic bacteria. In fact, when microbial cells exist in a biofilm, may be more resistant to antimicrobial agents because the microorganisms are protected against the action of the agents, with the biofilm impairing the entry of agents by acting as a barrier for diffusion. The biofilm matrix also can react with or adsorb the antimicrobial agents, thereby reducing the number of available agents to interact with microbial cells in the biofilm structure. In addition, microbial cells in biofilm structure have reduced growth rates and metabolic activity. Another possibility that microbial cells in the biofilm are tolerant to antimicrobial agents is conjugation among biofilm cells. This exchange of genes is important for the acquisition of resistance genes and genetic diversity of bacterial communities and favors the exchange of genes that can involve in more antimicrobial resistance33,34.
The results of characterization of synthesized Cur-NPhs as the photo-sonosensitizer in the current study are consistent with a recent report in which strong interaction between Cur and phosphatidylcholine leads to the stability of phytosomes26. The significant reduction of log10 CFU/mL in A. actinomycetemcomitans (13.6 log10 CFU/mL reduction) following PSACT using the high concentrations of Cur-NPhs (50 × 10–4 g/L) combined with the blue laser irradiation at a wavelength of 450 ± 5 nm plus ultrasonic waves with the intensity of 1.56 W/cm2 at a frequency of 1 MHz for 2 min showed that Cur-NPhs-PSACT had an antibacterial effect. The results of this study revealed the activation of Cur-NPhs using both blue laser irradiation and ultrasonic waves enhanced the antimicrobial activity of Cur-NPhs which could be due to increased ROS and singlet oxygen. So, Cur-NPhs-PSACT had higher antibacterial activity than Cur-NPhs-aPDT and Cur-NPhs-SACT against A. actinomycetemcomitans. According to our results, activated NPhs by blue laser irradiation, ultrasonic waves, and both of them in NPhs-aPDT, NPs-SACT, and NPhs-PSACT treatment had no contribution to the observed antimicrobial effects. Notably, our finding that Cur-NPhs-aPDT had an antibacterial effect against A. actinomycetemcomitans is similar to previous reports, which show the antibacterial effect of Cur-aPDT35.
As previously mentioned, biofilm structures are the protected niches for A. actinomycetemcomitans against host defense, hostile environments conditions, and antimicrobial treatment36,37. Nevertheless, some antimicrobial molecules can penetrate throughout the biofilm matrix and lead to biofilm degradation. Our findings are consistent with recent studies that show aPDT using Cur as a photosensitizer is efficient in the reduction of microbial biofilms38. As the results of this study showed, PSACT using 50 × 10–4 g/L Cur-NPhs had the most effective anti-biofilm activity against A. actinomycetemcomitans and could degrade the biofilm cells up to 65%. In addition, the results showed that the reduction of A. actinomycetemcomitans metabolic activity was dose-dependent. There was a more significant reduction of metabolic activity following Cur-NPhs-mediated PSACT at the concentrations of 50 × 10–4 g/L about 90.0%. Additionally, aPDT and SACT with 50 × 10–4 g/L Cur-NPhs could decrease the metabolic activity to 70% and 78%, respectively. The finding indicated the antimetabolite activity of PSAT was more than aPDT and SACT.
The results regarding the reduction of metabolic activity of A. actinomycetemcomitans during aPDT treatment, corroborate with the results found in the literature for this and another pathogen35,39,40. Ari et al.40 studied the effects of bystander responses following aPDT using Cur as a photosensitizer on A. actinomycetemcomitans biofilm growth. Their results showed that bacterial metabolic activity significantly decreased by 42.6% after exposure to bystander effects induced by Cur-aPDT treated whole bacterial cell suspension. In the study reported by Pourhajibagher et al.35, the metabolic activity of treated A. actinomycetemcomitans by nano-chitosan encapsulated ICG-mediated aPDT was assessed. Based on their results, a statistically significant reduction (48%) was observed in the metabolic activity of A. actinomycetemcomitans.
The QseBC two-component systems are characterized as the global regulators of virulence. QseBC is associated with QS and essential for A. actinomycetemcomitans to adapt and respond to various environmental stimuli41. As Novak et al.42 reported, QseBC contributes to the regulation of colonization, biofilm formation, and expression of virulence genes in A. actinomycetemcomitans. Based on the QseBC signaling cascade of A. actinomycetemcomitans, the expression of qseBC in A. actinomycetemcomitans is induced by exogenous autoinducer-2 (AI-2), which may occur via the motility QS regulator43. Also, the expression of rcpA has been linked to biofilm production. RcpA protein has been demonstrated to be involved in outer membrane channel formation that allows for the secretion of the assembled fimbriae to the microbial cell surface. Fimbriae in this species are responsible for biofilm formation which ultimately lead to increased antimicrobial resistance44.
Weigel et al.43 reported the functional outcomes of A. actinomycetemcomitans QseBC have been defined as influencing biofilm growth and virulence. Also, the findings of Novak et al.42 study indicated that qseBC is require for stimulation of biofilm formation of A. actinomycetemcomitans in an animal model of periodontitis. In this study, both qseB and qseC genes were significantly downregulated after treatment with Cur-NPhs-aPDT-, SACT-, and PSACT-treated A. actinomycetemcomitans Cur-NPhs-mediated PSACT compared to the other control groups. Interestingly, the downregulation of qseB and qseC on treated A. actinomycetemcomitans were dose-dependent and may be involved in the attenuation of A. actinomycetemcomitans QS.
Moreover, in the current study, after exposing the biofilm of A. actinomycetemcomitans strains to Cur-NPhs-PSACT, rcpA gene expression was potentially downregulated in a dose-dependent manner which may be the result of the accumulation of Cur-NPhs in the biofilm environment. Since the rcpA gene plays an important role in biofilm formation in A. actinomycetemcomitans, the expression inhibition of this gene following Cur-NPhs-PSACT treatment may cause to decrease bacterial adhesion, biofilm formation, and its pathogenicity. Various recent reports have shown that aPDT can overcome bacterial biofilm formation through affecting rcpA gene expression in A. actinomycetemcomitans6,35. Pourhajibagher et al.35 investigated the efficacy of Cur-aPDT as an alternative antimicrobial approach against A. actinomycetemcomitans. Their results indicated aPDT with Cur leads to significantly decreased A. actinomycetemcomitans cell survival and reduced the expression of the rcpA gene by approximately 8.5-fold. In another study, the outcome of the expression level of A. actinomycetemcomitans rcpA gene following treatment by ICG-aPDT was investigated. The results showed that ICG-mediated aPDT using 125 μg/mL ICG at the fluency of 15.6 J/cm2 of diode laser irradiation could significantly reduce the expression of A. actinomycetemcomitans rcpA gene approximately sixfold6. According to the results of the current study, no change in rcpA, qseB, and qseC mRNA expressions in either of the NPhs and NPhs-treated groups during aPDT-, SACT-, and PSACT-treatment suggests these treatments have no effect on expressions of test genes and therefore on biofilm production in A. actinomycetemcomitans.
Conclusion
This study concludes that the Cur-NPhs-PSACT could reduce the cell viability, metabolic activity, and biofilm growth in A. actinomycetemcomitans by downregulating the expression of rcpA, qseB, and qseC genes, and may attenuate pathogens that decrease disease severity in patients with periodontitis and peri-implantitis. Our findings warrant detailed examination of the interactions between the Cur-NPhs-PSACT as an adjunctive therapy and pathogenicity of the main periodontal pathogens toward efforts to successful treatment of periodontal infections.
Materials and methods
Materials
Cur and phosphatidylcholine were purchased from Sigma-Aldrich, Steinheim, Germany. Dichloromethane was obtained from Merck, Darmstadt, Germany. Brain heart infusion (BHI) agar and BHI broth were purchased from Conda, Torrejon de Ardoz, Spain. Yeast extract, hemin, and menadione were obtained from Merck, Darmstadt, Germany. Phosphate buffered saline (PBS), crystal violet (CV), and acetic acid were purchased from Sigma-Aldrich, Steinheim, Germany. GeneAll Hybrid-R RNA purification kit was purchased from GeneAll Biotechnology Co., Korea. RNase-free DNase I and Revert Aid First Strand cDNA Synthesis Kits were purchased from Thermo Fisher Scientific, US. SYBR Green master mix was obtained from Takara, Kyoto, Japan. Cell Proliferation Kit II (XTT) was obtained from Sigma-Aldrich, Steinheim, Germany. Ethanol and other reagents were all of the analytical grade.
Preparation of Cur-decorated NPhs
The NPhs preparation was done by de-hydration and re-hydration technique of Abdul Azeez et al.45 with minor modifications. Briefly, 0.2 g of phosphatidylcholine was dissolved in 20 mL of dichloromethane as an organic solvent and stored for 2 h. The dichloromethane was then removed under a reduced pressure and temperature using a rotary vacuum evaporator. A thin layer containing phytosome would be formed at the bottom of the flask. After that, the thin layer of phytosome is re-hydrated with distilled water to form micelles that are then probe sonicated to achieve NPhs.
Cur-NPhs were prepared according to the method reported recently with slight modification46. In brief, Cur and prepared NPhs in the molar ratio of 1:1 was dissolved in 20 mL of dichloromethane. The mixtures were thoroughly mixed at room temperature on a magnetic stirrer and the solvent was evaporated under reduced pressure in a rotary evaporator. The obtained thin film was then washed with n-hexane, dried under vacuum, hydrated for 2 h, and ultrasonicated to obtain Cur-NPhs. The resultant Cur-NPhs were placed in amber-colored glass bottle and stored at room temperature.
Characterization of Cur-NPhs
Morphology
The surface morphology, size, and shape of prepared Cur-NPhs were confirmed using a field emission scanning electron microscope (FESEM; Zeiss, Sigma VP, Germany, 15 kV accelerating voltage). Moreover, the core–shell structure of Cur-NPhs was assessed by transmission electron microscope (TEM; Zeiss EM10C) with an accelerating voltage of 100 kV.
Measurement of particle size, polydispersity, and ζ-potential
The Cur-NPhs dispersion was diluted with PBS solution prior analysis. Average particle size, polydispersity, and ζ-potential of Cur-NPhs were determined using a dynamic light scattering particle size analyzer (Malvern Zetasizer Nano ZS system) at 25 °C with a scattering angle of 90°.
Entrapment efficiency (EE)
The percentage of Cur incorporated was determined by centrifuging the Cur-NPhs at 8000 rpm for 30 min. The supernatant was then obtained to measure the absorbance at 425 nm using a UV–visible spectrophotometer. The encapsulation efficiency (EE) was determined using the following equation:
In vitro drug release
The release properties of Cur from Cur-NPhs were obtained as described earlier47. The absorbance of Cur was measured using the UV–visible spectrophotometer at 426 nm. The percentage of released Cur was calculated as:
Physical stability of Cur-NPhs during storage
To evaluate shelf life, Cur-NPhs was stored in the containers at 4 °C and 37 °C in the dark. Changes in the polydispersity index, particle size, and ζ-potential were investigated at different time intervals during storage for 2 weeks. Effects of different pH (pH = 5, 6, 7, 8, 9) on the retention rate (RR) of Cur-NPhs was determined after 24 and 48 h of storage. RR of Cur was measured at 425 nm and calculated as follows:
Bacterial strain and growth conditions
A. actinomycetemcomitans IR-TUMS/BPG4 (Accession number in Genbank: KX108928) was grown at 37 °C in a capnophilic condition (3–5% CO2 and 8–10% O2) in BHI broth supplemented with 0.6% yeast extract and 1 mg/L menadione48.
Light source
A continuous blue laser (ASHA, Iran) at the wavelength of 450 ± 5 nm with an output intensity of 150 mW/cm2, 4.2 V, and 0.34 A was used as a light source. The blue laser was placed approximately 1 mm away from the surface of the bacterial suspension.
Ultrasound system
SACT testing was done as described previously with slight modification49. In brief, a flat microtiterplate containing the bacterial suspension was held in a plastic holder tight to the bottom of a closed dark chamber of an ultrasonic bath containing cooled distilled water. This was done to prevent any external illumination. Sonication was conducted at a frequency of 1 MHz and pulsed repetition frequency of 100 Hz with a spatial average ultrasonic intensity of 2 W/cm2 for 5 min. An ultrasound transducer with a surface area of 7.0 cm2 was applied at the bottom of the chamber of the ultrasonic bath to provide exposure to the bacterial suspension.
Experimental design
To determine the antimicrobial response to Cur-NPhs-mediated PSACT, test groups consisted of A. actinomycetemcomitans subjected to:
-
1.
NPhs: at the concentrations of 10, 25, and 50 × 10–4 g/L.
-
2.
Cur-NPhs: at the concentrations of 10, 25, and 50 × 10–4 g/L.
-
3.
Blue laser: irradiation at a wavelength of 450 ± 5 nm for 2 min.
-
4.
Ultrasonic waves: the intensity of 2 W/cm2 for 2 min at a frequency of 1 MHz.
-
5.
NPhs-aPDT: NPhs (10, 25, and 50 × 10–4 g/L) + blue laser irradiation at a wavelength of 450 ± 5 nm for 2 min.
-
6.
Cur-NPhs-aPDT: Cur-NPhs (10, 25, and 50 × 10–4 g/L) + blue laser irradiation at a wavelength of 450 ± 5 nm for 2 min.
-
7.
NPhs-SACT: NPhs (10, 25, and 50 × 10–4 g/L) + ultrasonic waves with the intensity of 1.56 W/cm2 for 2 min at a frequency of 1 MHz.
-
8.
Cur-NPhs-SACT: Cur-NPhs (10, 25, and 50 × 10–4 g/L) + ultrasonic waves with intensity of 1.56 W/cm2 for 2 min at a frequency of 1 MHz.
-
9.
NPhs-PSACT: NPhs (10, 25, and 50 × 10–4 g/L) + blue laser irradiation at a wavelength of 450 ± 5 nm + ultrasonic waves with the intensity of 1.56 W/cm2 at a frequency of 1 MHz for 2 min.
-
10.
Cur-NPhs-PSACT: Cur-NPhs (10, 25, and 50 × 10–4 g/L) + blue laser irradiation at a wavelength of 450 ± 5 nm + ultrasonic waves with the intensity of 1.56 W/cm2 at a frequency of 1 MHz for 2 min.
-
11.
Control group (bacterial suspension without treatment).
aPDT protocol
To find out the effect of aPDT based on NPhs and Cur-NPhs at different concentrations (10, 25, and 50 × 10–4 g/L; final concentration), 100 μL of each concentration of test materials was added to the well of a 96-well microtiterplate. The wells were then inoculated with a 100 μL/well of fresh BHI broth bacterial cultures (adjusted to the final concentration of 5.0 × 105 CFU/mL). The microtiterplate was incubated for 5 min in the dark at 25 °C. The NPhs and Cur-NPhs treated bacterial suspensions in microtiterplate wells were immediately exposed with the blue laser irradiation at a wavelength of 450 ± 5 nm for 2 min.
SACT protocol
SACT was performed as described previously50. Briefly, 100 μL of NPhs and Cur-NPhs at different concentrations (10, 25, and 50 × 10–4 g/L; final concentration) separately was added to the well of a 96-well microtiterplate. The wells were then inoculated with a fresh BHI broth bacterial suspension (100 μL/well; final concentration of 5.0 × 105 CFU/mL). The microtiterplate was incubated in the dark room (5 min; 25 °C). After that sonication was done with an ultrasound transducer containing a surface area of 7.0 cm2 at a pulsed repetition frequency of 100 Hz.
PSACT protocol
100 μL of bacterial suspension (1.5 × 106 CFU/mL) was transferred to the wells of the 96-well microtiterplate. 100 μL of NPhs and Cur-NPhs at different concentrations (10, 25, and 50 × 10–4 g/L) were separately added to the bacterial suspension. After incubation of the microtiter plate in a dark capnophilic atmosphere for 5 min at room temperature, the bacterial suspension was simultaneously exposed to blue laser at the wavelength of 450 ± 5 nm with an output intensity of 150 mW/cm2 and ultrasonic waves at a frequency of 1 MHz with an ultrasonic intensity of 2 W/cm2 for two min. The schematic diagram of PSACT is shown in Fig. 11.

Schematic apparatus for illumination treatment of sample.
Determination of viability of A. actinomycetemcomitans in planktonic culture following treatment groups
100 μL of NPhs and Cur-NPhs (both at the concentrations of 200 × 10–4 g/L) were added to the well in column one of a 96-well microtiterplate, separately, and was diluted two-fold stepwise to column three after adding 100 μL of 2 × BHI broth to each well. Then, an A. actinomycetemcomitans suspension (100 μL of 1.5 × 106 CFU/mL) was poured into each well. In this experiment, the concentration of test materials was in the range of 10, 25, and 50 × 10–4 g/L. Then microtiterplate was incubated for 24 h under capnophilic condition, in the dark, at 37 °C. Thereafter, 50 μL of each well were cultured in a BHI agar plate supplemented with 0.6% (wt/vol) yeast extract, 5–7% defibrinated sheep blood, 5 mg/L hemin, and 1 mg/L menadione. The BHI agar plates were incubated in a capnophilic condition at 37 °C for 48 h, and the number of CFU/mL was determined.
The effect of NPhs- and Cur-NPhs-mediated aPDT-, SACT-, and PSACT-treatment on A. actinomycetemcomitans in planktonic culture were done according to the method mentioned as previously42. Briefly, NPhs and Cur-NPhs (both 100 μL; 200 × 10–4 g/L) were added to the well in column one of a 96-well microtiterplate, separately, and was diluted two-fold stepwise to column three after adding of 2 × BHI broth (100 μL) to each well. Then, an A. actinomycetemcomitans suspension (100 μL of 1.5 × 106 CFU/mL) was added into each well. The microtiterplate was incubated for 5 min in the dark, at 25 °C. The contents of microtiterplate wells were immediately treated in aPDT-, SACT-, and PSACT-protocols as mentioned in the experimental design section. As mentioned in the above paragraph, the number of CFU/mL of treated A. actinomycetemcomitans in planktonic culture was determined.
Determination of treatment groups efficiency on biofilm killing/degradation
The biofilm killing/ degradation effect of NPhs- and Cur-NPhs-mediated aPDT-, SACT-, and PSACT-treatment was determined according to a previous study51. Briefly, aliquots in BHI broth medium of bacterial suspensions (200 µL; 1.0 × 105 CFU/mL final concentration) were transferred into each well in 96-well microtiterplate. To maturation biofilm of A. actinomycetemcomitans then microtiterplate was incubated for 48 h in capnophilic condition, at 37 °C. After that, the medium was removed from each well, and planktonic cells were deducted by washing three times with PBS (pH 7.4). A. actinomycetemcomitans cells in biofilms were treated in aPDT-, SACT-, and PSACT-protocols as mentioned in the experimental design section.
After each treatment, biofilm killing/degradation was assessed with the colorimetric method based on CV assay52. The content of each well of microplates was removed and the planktonic cells were deducted were allowed to air dry at room temperature. Then, the wells were stained with CV dye (1%) for 20 min at 25 °C, and the dye was discarded and washed twice with PBS (pH 7.4). Ethanol (200 μL; 95%) was added into each well and was incubated for 15 min at 25 °C. Then, the content of each well quite was aspirated and the microtiterplate was allowed to air dry at room temperature. Thereafter, each well was filled with acetic acid (200 μL; 33%) and biofilms were quantified by measuring the optical densities (OD) value using a microplate reader machine (at 570 nm; Thermo Fisher Scientific, US). To assess the treatment efficiency of NPhs- and Cur-NPhs-mediated aPDT-, SACT-, and PSACT-efficiency on biofilm killing/degradation, the percentage of biofilm killing/degradation was determined by the following equation:
Assessment of metabolic activity of treatment groups using XTT reduction assay
The metabolic activity of treated A. actinomycetemcomitans was assessed using the XTT (2,3-bis [2-methyloxy-4-nitro-5-sulfophenyl]-2H-tetrazolium-5-carboxanilide) reduction assay, as described by Coraça-Hubér et al.53. Following each treatment described in the experimental design section, the content of wells was collected and centrifuged at 2000 rpm for 10 min. The supernatants were removed and resulting bacterial cell sediments were dissolved in 150 μL of XTT-menadione-PBS solution in microplate wells and incubated at 37 °C. After 3 h, 100 μL of the mixture was transferred to a new microplate and the optical intensity was measured at 492 nm using a microplate reader.
Determination of relative quantification of quorum-sensing-associated qseB and qseC genes expression
To determine the effects of treatment groups on QS genes expression in A. actinomycetemcomitans, the treated A. actinomycetemcomitans was used to extract RNA using GeneAll Hybrid-R RNA purification kit following the manufacturer’s instructions. The extracted RNA was quantified spectrophotometrically using a NanoDrop spectrophotometer Thermo Fisher Scientific, US) and the quality was confirmed on the agarose gel. Total RNA was pretreated with RNase-free DNase I treatment according to the manufacturer's protocol to remove any residual chromosomal DNA. The first-strand cDNA was synthesized from the total RNA using a Revert Aid First Strand cDNA Synthesis Kit according to the manufacturer’s protocol. Quantitative real-time polymerase chain reaction (qRT-PCR) was then performed to quantify gene transcription of qseB and qseC. The nucleotide sequences of primers used in this study were shown in Table 1. The specificity of the primers was evaluated on agarose gel electrophoresis. Reaction plates were processed on the Line-GeneK Real-Time PCR Detection System and Software (Bioer Technology, Hangzhou, China) using SYBR Green master mixes with the following cycling parameters: initial denaturation at 95 °C/2 min, followed by 40 cycles at 94 °C/10 s, 60 °C/10 s, and 72 °C/10 s. Finally, the expression level of qseB and qseC genes was analyzed based on the previous study54. 16S rRNA was used as an internal control to normalize RNA concentration.
Determination of relative quantification of biofilm-associated rcpA gene expression
The evaluation of rcpA expression was performed based on the previous study42. Total RNA extraction, cDNA synthesis, and qRT-PCR was done as described above (Section Determination of relative quantification of quorum-sensing-associated qseB and qseC genes expression).
Statistical analyses
All experiments were performed in triplicate and expressed as mean ± standard deviation (SD). All quantitative data were analyzed with one-way analysis of variance (ANOVA) and Tukey test using the SPSS software version 23.0. The significance level was set at P < 0.05. Fold differences in RNA expression were determined by the 2−ΔΔCt method and the changes greater than or equal to two-fold were considered significant54.
References
Machado, V. et al. Prevalence and extent of chronic periodontitis and its risk factors in a Portuguese subpopulation: A retrospective cross-sectional study and analysis of Clinical Attachment Loss. PeerJ 6, 5258–5263 (2018).
Nazir, M. A. Prevalence of periodontal disease, its association with systemic diseases and prevention. Int. J. Health Sci. (Qassim) 11(2), 72–80 (2017).
Castano, A. P., Demidova, T. N. & Hamblin, M. R. Mechanisms in photodynamic therapy: Part one photosensitizers, photochemistry and cellular localization. Photodiagn. Photodyn. Ther. 1(4), 279–293 (2004).
Allison, R. R. & Moghissi, K. Photodynamic therapy (PDT): PDT mechanisms. Clin. Endosc. 46(1), 24–29 (2013).
Belibasakis, G. N. et al. Virulence and pathogenicity properties of Aggregatibacteractinomycetemcomitans. Pathogens 8(4), 222 (2019).
Pourhajibagher, M. et al. Monitoring gene expression of rcpA from Aggregatibacter actinomycetemcomitans versus antimicrobial photodynamic therapy by relative quantitative real-time PCR. Photodiagn. Photodyn. Ther. 19, 51–55 (2017).
Ventola, C. L. The antibiotic resistance crisis: Part 1: Causes and threats. P T 40(4), 277–283 (2015).
Pourhajibagher, M. et al. Photo-sonodynamic antimicrobial chemotherapy via chitosan nanoparticles-indocyanine green against polymicrobial periopathogenic biofilms: Ex vivo study on dental implants. Photodiagn. Photodyn. Ther. 31, 101834 (2020).
Bailey, M. et al. Physical mechanisms of the therapeutic effect of ultrasound (A review). Acoust. Phys. 49, 369–388 (2003).
McHale, A. P. et al. Sonodynamic therapy: Concept, mechanism and application to cancer treatment. Adv. Exp. Med. Biol. 880, 429–450 (2016).
Rosenthal, I., Sostaric, J. Z. & Riesz, P. Sonodynamic therapy—A review of the synergistic effects of drugs and ultrasound. Ultrason. Sonochem. 11, 349–363 (2004).
Braham, P. et al. Antimicrobial photodynamic therapy may promote periodontal healing through multiple mechanisms. J. Periodontol. 80(11), 1790–1798 (2009).
Cieplik, F. et al. Antimicrobial photodynamic therapy—What we know and what we don’t. Crit. Rev. Microbiol. 44(5), 571–589 (2018).
Huang, L. et al. Type I and Type II mechanisms of antimicrobial photodynamic therapy: An in vitro study on Gram-negative and Gram-positive bacteria. Lasers Surg. Med. 44(6), 490–499 (2012).
Bolhari, B. et al. Ex vivo assessment of synergic effect of chlorhexidine for enhancing antimicrobial photodynamic therapy efficiency on expression patterns of biofilm-associated genes of Enterococcusfaecalis. Photodiagn. Photodyn. Ther. 22, 227–232 (2018).
Tortamano, A. C. A. C. et al. Antimicrobial photodynamic therapy mediated by methylene blue in surfactant vehicle on periodontopathogens. Photodiagn. Photodyn. Ther. 31, 101784 (2020).
Sukumar, K. et al. Evaluation of combined efficacy of photodynamic therapy using indocyanine green photosensitizer and non-surgical periodontal therapy on clinical and microbial parameters in the management of chronic periodontitis subjects: A randomized split-mouth design. Photodiagn. Photodyn. Ther. 31, 101949 (2020).
Pan, H., Wang, D. & Zhang, F. In vitro antimicrobial effect of curcumin-based photodynamic therapy on Porphyromonasgingivalis and Aggregatibacteractinomycetemcomitans. Photodiagn. Photodyn. Ther. 31, 102055 (2020).
Joshi, K. et al. Clinical effectiveness of indocyanine green mediated antimicrobial photodynamic therapy as an adjunct to scaling root planing in treatment of chronic periodontitis—A randomized controlled clinical trial. Photodiagn. Photodyn. Ther. 29, 101591 (2020).
Garcia, V. G. et al. Effect of the concentration of phenothiazine photosensitizers in antimicrobial photodynamic therapy on bone loss and the immune inflammatory response of induced periodontitis in rats. J. Periodont. Res. 49(5), 584–594 (2014).
Park, D. et al. Antimicrobial photodynamic therapy efficacy against specific pathogenic periodontitis bacterial species. Photodiagn. Photodyn. Ther. 30, 101688 (2020).
Saitawee, D. et al. Photodynamic therapy of Curcumalonga extract stimulated with blue light against Aggregatibacteractinomycetemcomitans. Photodiagn. Photodyn. Ther. 22, 101–105 (2018).
Kocaadam, B. & Şanlier, N. Curcumin, an active component of turmeric (Curcumalonga), and its effects on health. Crit. Rev. Food Sci. Nutr. 57(13), 2889–2895 (2017).
Dagar, P., Dahiya, P. & Bhambi, M. Recent advances in curcumin nanoformulations. Nano Sci. Nano Technol. Indian J. 8, 458–474 (2014).
Ghanbarzadeh, B., Babazadeh, A. & Hamishehkar, H. Nano-phytosome as a potential food-grade delivery system. Food Biosci. 15, 126–135 (2016).
Purpura, M. et al. Analysis of different innovative formulations of curcumin for improved relative oral bioavailability in human subjects. Eur. J. Nutr. 57(3), 929–938 (2018).
Mirzaei, H. et al. Phytosomal curcumin: A review of pharmacokinetic, experimental and clinical studies. Biomed. Pharmacother. 85, 102–112 (2017).
Appendino, G. et al. Potential role of curcumin phytosome (Meriva) in controlling the evolution of diabetic microangiopathy. A pilot study. Panminerva Med. 53(3 Suppl 1), 43 (2011).
Aldini, R. et al. Curcumalonga extract exerts a myorelaxant effect on the ileum and colon in a mouse experimental colitis model, independent of the anti-inflammatory effect. PLoS ONE 7(9), e44650 (2012).
Steigerwalt, R. et al. Meriva®, a lecithinized curcumin delivery system, in diabetic microangiopathy and retinopathy. Panminerva Med. 54(4), 11 (2012).
Allegri, P., Mastromarino, A. & Neri, P. Management of chronic anterior uveitis relapses: Efficacy of oral phospholipidic curcumin treatment. Long-term follow-up. Clin. Ophthalmol. 4, 1201 (2010).
Sahebkar, A. A systematic review and meta-analysis of randomized controlled trials investigating the effects of curcumin on blood lipid levels. Clin. Nutr. 33(3), 406–414 (2014).
Cao, Y. et al. Non-antibiotic antimicrobial agents to combat biofilm-forming bacteria. J. Glob. Antimicrob. Resist. 21, 445–451 (2019).
Sharma, D., Misba, L. & Khan, A. U. Antibiotics versus biofilm: An emerging battleground in microbial communities. Antimicrob. Resist. Infect. Control. 8(1), 1 (2019).
Pourhajibagher, M. et al. Monitoring of virulence factors and metabolic activity in Aggregatibacter actinomycetemcomitans cells surviving antimicrobial photodynamic therapy via nano-chitosan encapsulated indocyanine green. Front. Phys. 6, 124 (2018).
Pourhajibagher, M. et al. Inhibitory effects of antimicrobial photodynamic therapy with curcumin on biofilm-associated gene expression profile of Aggregatibacteractinomycetemcomitans. J. Dent. (Tehran, Iran) 15(3), 169–175 (2018).
Kaplan, J. B., Meyenhofer, M. F. & Fine, D. H. Biofilm growth and detachment of Actinobacillusactinomycetemcomitans. J. Bacteriol. 185(4), 1399–1404 (2003).
Rocha, M. P. et al. Photodynamic therapy with curcumin in the reduction of enterococcus faecalis biofilm in bone cavity: Microbiological and spectral fluorescense analysis. Photodiagn. Photodyn. Ther. 33, 1–7 (2020).
Pourhajibagher, M. et al. Modulation of virulence in Acinetobacterbaumannii cells surviving photodynamic treatment with toluidine blue. Photodiagn. Photodyn. Ther. 15, 202–212 (2016).
Mahdizade-Ari, M., Pourhajibagher, M. & Bahador, A. Changes of microbial cell survival, metabolic activity, efflux capacity, and quorum sensing ability of Aggregatibacteractinomycetemcomitans due to antimicrobial photodynamic therapy-induced bystander effects. Photodiagn. Photodyn. Ther. 26, 287–294 (2019).
Juárez-Rodríguez, M. D., Torres-Escobar, A. & Demuth, D. R. ygiW and qseBC are co-expressed in Aggregatibacteractinomycetemcomitans and regulate biofilm growth. Microbiology 159(Pt 6), 989 (2013).
Novak, E. A. et al. Autoinducer-2 and QseC control biofilm formation and in vivo virulence of Aggregatibacteractinomycetemcomitans. Infect. Immun. 78(7), 2919–2926 (2010).
Weigel, W. A. & Demuth, D. R. Qse BC, a two-component bacterial adrenergic receptor and global regulator of virulence in Enterobacteriaceae and Pasteurellaceae. Mol. Oral Microbiol. 31(5), 379–397 (2016).
Pourhajibagher, M. et al. Real-time quantitative reverse transcription-PCR analysis of expression stability of Aggregatibacteractinomycetemcomitans fimbria-associated gene in response to photodynamic therapy. Photodiagn. Photodyn. Ther. 18, 78–82 (2017).
Azeez, N. A., Deepa, V. S. & Sivapriya, V. Phytosomes: Emergent promising nano vesicular drug delivery system for targeted tumor therapy. Adv. Nat. Sci. 9(3), 1–6 (2018).
Allam, A. N., Komeil, I. A. & Abdallah, O. Y. Curcumin phytosomal softgel formulation: Development, optimization and physicochemical characterization. Acta Pharm. 65(3), 285–297 (2015).
Hasan, M. et al. The positive role of curcumin-loaded salmon nanoliposomes on the culture of primary cortical neurons. Mar. Drugs 16(7), 218 (2018).
Goulart, R. D. et al. Photodynamic therapy in planktonic and biofilm cultures of Aggregatibacteractinomycetemcomitans. Photomed. Laser Surg. 28(S1), 53–60 (2010).
Nakonechny, F. et al. Sonodynamic excitation of Rose Bengal for eradication of Gram-positive and Gram-negative bacteria. BioMed Res. Int. 2013, 1–7 (2013).
Gomes, F. et al. Evaluation of antimicrobial activity of certain combinations of antibiotics against in vitro Staphylococcusepidermidis biofilms. Indian J. Med. Res. 135(4), 542 (2012).
Partoazar, A. et al. Antibiofilm activity of natural zeolite supported NanoZnO: Inhibition of Esp gene expression of Enterococcusfaecalis. Nanomedicine 14(6), 675–687 (2019).
Pourhajibagher, M. et al. Assessment of biofilm formation among Acinetobacter baumannii strains isolated from burned patients. Der Pharm. Lett. 8, 225–229 (2016).
Coraça Hubér, D. C. et al. Evaluation of MBEC™-HTP biofilm model for studies of implant associated infections. J. Orthop. Res. 30(7), 1176–1180 (2012).
Livak, K. J. & Schmittgen, T. D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 25, 402–408 (2011).
Funding
This research was supported by Dental Research Center, Dentistry Research Institute, Tehran University of Medical Science and Health Services Grant No. 98-3-134-43563.
Author information
Authors and Affiliations
Contributions
M.P. designed the experiments, performed the experiments, analyzed the data, authored or reviewed drafts of the paper, prepared figures and/or tables, and approved the final draft. A.B. conceived and designed the experiments, analyzed the data, authored or reviewed drafts of the paper, and approved the final draft. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Pourhajibagher, M., Bahador, A. Attenuation of Aggregatibacter actinomycetemcomitans virulence using curcumin-decorated nanophytosomes-mediated photo-sonoantimicrobial chemotherapy. Sci Rep 11, 6012 (2021). https://doi.org/10.1038/s41598-021-85437-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-021-85437-6
This article is cited by
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
