
Abstract
Borrelia miyamotoi, a member of the tick-borne relapsing fever spirochetes, shows a serum-resistant phenotype in vitro. This ability of B. miyamotoi may contribute to bacterial evasion of the host innate immune system. To investigate the molecular mechanism of serum-resistance, we constructed a membrane protein-encoding gene library of B. miyamotoi using Borrelia garinii strain HT59G, which shows a transformable and serum-susceptible phenotype. By screening the library, we found that bom1093 and bom1515 of B. miyamotoi provided a serum-resistant phenotype to the recipient B. garinii. These B. miyamotoi genes are predicted to encode P35-like antigen genes and are conserved among relapsing fever borreliae. Functional analysis revealed that BOM1093 bound to serum vitronectin and that the C-terminal region of BOM1093 was involved in the vitronectin-binding property. Importantly, the B. garinii transformant was not serum-resistant when the C terminus-truncated BOM1093 was expressed. We also observed that the depletion of vitronectin from human serum enhances the bactericidal activity of BOM1093 expressing B. garinii, and the survival rate of BOM1093 expressing B. garinii in vitronectin-depleted serum is enhanced by the addition of purified vitronectin. Our data suggests that B. miyamotoi utilize BOM1093-mediated binding to vitronectin as a mechanism of serum resistance.
Similar content being viewed by others


Introduction
Borrelia miyamotoi was first discovered in Hokkaido, Japan in 19951. Although B. miyamotoi is classified to relapsing fever (RF) borreliae, it was discovered in the hard-bodied tick, Ixodes persulcatus1. B. miyamotoi has also been detected from Ixodes scapularis and Ixodes pacificus in North America2,3,4 and Ixodes ricinus in Europe5,6. The first cases of B. miyamotoi infection in humans were reported in Russia and were referred to as “Emerging RF”7. Following the initial report, several cases of B. miyamotoi infection have been confirmed in humans in the United States, Europe, and Asia8,9,10,11,12,13.
Emerging RF (recently renamed B. miyamotoi disease, or BMD) is a systemic illness causing fever, headache, myalgia, arthralgia, elevated liver enzymes, neutropenia, and thrombocytopenia7,14, and several cases of meningitis have been reported8,9,10,15. Spirochetemia has been reported in cases of BMD, and survival of spirochetes in the bloodstream may be important in establishing systemic infection. Resistance to human complement was demonstrated for B. miyamotoi in 201416, and the complement binding and inhibitory protein A (CbiA) has been identified as a serum-resistance factor in B. miyamotoi17. However, the mechanisms utilized by the BMD pathogen, B. miyamotoi, are not fully understood.
Although genetic approaches such as mutagenesis and complementation have been employed to study genes of genus Borrelia over the last few decades, these processes have not been established for B. miyamotoi. Röttgerding et al., however, successfully identified and characterized the contribution of CbiA to serum resistance of B. miyamotoi in vitro using a surrogate strain, B. garinii G117. We, therefore, employed a similar surrogate system by first establishing a transformable/serum susceptible Borrelia strain to use in our investigation. Using this strain, we attempted to comprehensively screen genes involved in serum resistance of B. miyamotoi and found that a vitronectin (Vn)-binding protein contributed to serum resistance of B. miyamotoi in vitro. Vn is a serum glycoprotein that circulates in the bloodstream and has roles in many biological processes including the regulation of the terminal pathway of complement in which it inhibits the C5b7 complex formation and C9 polymerization18,19. Our data suggests B. miyamotoi may utilize Vn-binding to evade the complement system in human serum.
Results
Identification of serum-sensitive B. garinii HT59G which shows a transformable phenotype
We first sought to evaluate the susceptibility of Borrelia strains to human serum in detail using strains isolated from different biological and geographical samples. For this purpose, 17 Borrelia strains of B. garinii and B. bavariensis were examined for serum-sensitivity by determining the survival rate following treatment with 40% Normal human serum (NHS) or Heat-inactivated human serum (HIS) for 16 h (Figure 1). Of these 17 strains, nine strains (B. bavariensis strains J-14, J-16, J-20t, J-32, J-39, J-40, J-41 and B. garinii strains J-21, J-37) obtained from the skin of Lyme disease patients, two B. garinii strains (strains VSBM and VSBP) isolated from cerebrospinal fluid (CSF) of patients, and one B. garinii (strain NT25) isolated from a tick exhibited a serum-resistant phenotype. One B. garinii strain (strain VSDA) isolated from patient CSF and four strains of B. garinii (strains Fis01, Far01, Far02, and HT59) isolated from ticks were serum-sensitive. These serum-sensitive strains were selected as candidate hosts for gene library construction of B. miyamotoi. To investigate the transformability of these B. garinii strains, the shuttle vector pBSV2 was electroporated into each serum-susceptible B. garinii strain. Among the five strains tested, transformants were obtained only from B. garinii strain HT59. We therefore picked 10 single colonies of strain HT59 and established 10 clones. Of these 10 clones, clone G also showed a transformable phenotype. When B. garinii strain HT59G was transformed with plasmid pBSV2, an average of 15 transformants was obtained per 1 µg of plasmid DNA (Table 1).

Serum susceptibility of Borrelia strains used in this study. Spirochetes were incubated in either 40% normal human serum (NHS) or heat-inactivated serum (HIS) for 5 days at 37 °C. Cell viability was assessed using microscopic counts of cells in 10 fields under ×300 magnification. The figure depicts the means, and error bars represent the positive and negative errors of the mean of triplicates from one representative experiment. Borrelia species names abbreviated as follows: Borrelia bavariensis (B. bav), B. garinii (B. gar), B. burgdorferi, (B. bur), and B. miyamotoi (B. miy).
Construction of plasmid archives for B. garinii HT59G transformation
At the time of this study, the genome of B. miyamotoi strain MYK3 was not available. Therefore, candidate genes encoding membrane proteins were selected from the genome sequence of B. miyamotoi strain FR64b, which is published in GenBank (Acc. Nos. CP004218–CP004266). From this database, 649 open reading frames (ORFs) that were predicted to be non-chromosomal encoding were extracted. Of these 649 ORFs, 90 ORFs were predicted to be displayed on the bacterial surface of B. miyamotoi using SignalP or LipoP analysis. For each of these 90 ORFs, specific PCR primers were used for DNA amplification. All ORF PCR products were detected from template genomic DNA of B. miyamotoi strain MYK3. The shuttle vector was created for the 90 ORFs by combining linearized pBSV2 and each PCR fragment using the In-Fusion procedure according to the manufacturer’s instructions (see “Materials and methods”****). Of these 90 ORFs, 84 ORFs were isolated from E. coli DH5α. Of these 84 ORFs, transformation of B. garinii HT59G was successful with 76 ORFs (coverage ratio: 84.4%).
bom1093 and bom1515 provide serum-resistance phenotype to recipient Borrelia
In order to investigate serum resistance of the 76 transformants, we subjected them to a screening test. After culturing for 5 days with 40% NHS, survival of the bacteria was observed using a dark-field microscope. The negative control (B. garinii HT59G/pBSV2) was completely destroyed in the presence of NHS. In contrast, B. garinii HT59G/pCspZ, with introduced Complement Regulator-Acquiring Surface Protein-2 gene (cspZ) of B. burgdorferi strain 297, a gene encoding a factor H (FH)-binding protein, used as a survival control, showed growth. Of the 76 transformants examined, two (B. garinii HT59G/pBOM1093 and HT59G/pBOM1515) exhibited a serum-resistant phenotype in a bactericidal assay. These transformants were further subjected to a quantitative serum-sensitivity assay. The transformed strains treated with 40% NHS or HIS for up to 16 h were plated on a 1% soft agar overlay on BSK-M plates with kanamycin. The B. garinii HT59G/pBSV2 and B. garinii HT59G/pCspZ strains were used as controls for serum susceptibility and serum resistance to NHS, respectively. The survival ratio was calculated using colony-forming units (CFUs) of NHS-treated cells and HIS-treated cells. In this study, B. garinii HT59G/pCspZ and B. garinii HT59G/pBSV2 showed survival ratios of 91% and 0.1%, respectively (Fig. 2). Moreover, B. garinii HT59G/pBOM1093 and B. garinii HT59G/pBOM1515 showed 54.6% and 37.5% survival ratios, respectively. Amino acid sequences of BOM1093 and BOM1515 from the strain B. miyamotoi MYK3 showed high similarity (94% identity) with each other, and the BOM1093 sequence of B. miyamotoi strain MYK3 was identical to BOM1093 of the strain FR64b (Acc. No. NZ_CP004225).

Quantification of serum susceptibility for Borrelia garinii transformants. Each transformant was incubated in either 40% NHS or HIS for 16 h at 34 °C. The colony forming unit (CFU) represents the number of colonies formed. The mock strain (B. garinii HT59G/pBSV2) and serum-resistant transformant (B. garinii HT59G/CspZ) were used as controls. The figure depicts the average CFU, and error bars represent the positive and negative errors of the mean of triplicates. *p value (≤ 0.01) was calculated by Student’s t test.
Proteinase K accessibility of BOM1093
In silico analyses (SignalP and LipoP) predicted BOM1093 to be a membrane protein. To further investigate the localization of the BOM1093 protein, a Proteinase K accessibility test was performed. Proteinase K treatment was performed for B. garinii HT59G/pBOM1093. As shown in Fig. 3, both P83/100 and OspA were digested by proteinase K treatment in a dose-dependent manner. BOM1093 was also accessible to proteinase K. In contrast, flagellin protein, which reacted with monoclonal antibody H9724, was resistant to proteinase K treatment.

Protease treatment affects surface exposed protein, BOM1093. B. garinii HT59G/pBOM1093 cells were incubated with the indicated concentrations of proteinase K. After 1 h of incubation, the cells were lysed by SDS-PAGE buffer, and each protein lysate was separated by 12.5% SDS-PAGE. (A) Total proteins were detected by Coomassie Brilliant Blue (CBB) staining, (B) BOM1093 was detected with anti-BOM1093 rabbit serum (dilution 1:1000), (C) P83/100 was detected with MAb958 (dilution 1:1000), and (D) OspA was detected with MAb H5332 (dilution 1:1000). (E) Flagellin was detected with MAb H9724 (dilution 1:1000) by western blotting.
Human serum Vn co-precipitated with His-tagged BOM1093
Serum resistance phenotype of Borrelia is due to its ability to bind complement regulator (for e.g., FH). Therefore, we examined the binding of B. garinii HT59G/pBOM1093 to complement regulators FH, factor H-like protein 1 (FHL-1), factor I, properdin, carboxy peptidase N, C4b binding protein (C4BP), C1 inhibitor, complement factor H-related protein 1, clusterin and vitronectin (Vn). B. garinii HT59G/pBOM1093 (6xHis-tagged on C terminus) incubated in 20% NHS was tested by a pull-down assay using Ni–NTA magnetic beads. The only complement regulator that bound to BOM1093 was Vn, which is reported to inhibit the terminal pathway of the complement system (e.g. C9 polymerization). In contrast, Vn was not detected when B. garinii HT59G/pBSV2 was used as a prey antigen (Fig. 4). Based on this data, we conducted further analyses for Vn and BOM1093.

Human serum Vn is co-precipitated with BOM1093. B. garinii HT59G expressed 6xHis-tagged BOM1093 (~ 108 cells/tube) were incubated with 20% NHS for 1 h at 37 °C and washed 3 times with Tris–HCl buffer (pH 7.0) containing 5 mM Pefabloc SC. The 6xHis-tagged BOM1093 from sonicated cells was captured using Ni–NTA magnetic beads. The captured proteins were separated on a 10% SDS-PAGE gel, transferred to PVDF membrane, and Vn was detected by western blotting. B. garinii HT59G/pBSV2 was used as a Mock control.
B. garinii HT59G/pBOM1093 binds purified-recombinant Vn in a dose-dependent manner
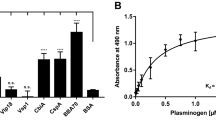
To confirm the results of the pull-down assay, we performed an ELISA assay to examine the binding of purified recombinant Vn to B. garinii HT59G/pBOM1093. A Vn-binding ELISA assay found dose-dependent and saturation binding of recombinant Vn for B. garinii HT59G/pBOM1093. In contrast, Vn binding was not detected when B. garinii HT59G/pBSV2 was tested. The results are shown in Fig. 5. The KD value for the interaction was estimated to be 10.8 nM (95% Confidential interval range 8.5–13.0 nM).

Vn-binding to B. garinii transformants using recombinant Vn. Vn-binding to B. garinii HT59G/BOM1093 was quantified by ELISA and depicted as the ratio of O.D. 450 nm to the background absorbance at 620 nm. Recombinant human Vn (6.25–400 nM) was used for this study. Values are means ± SD of three independent experiments. Values were compared and analyzed between B. garinii HT59G/BOM1093 and B. garinii HT59G/BOM1093. **p value (≤ 0.001) was calculated by Student’s t test.
C-terminal region of BOM1093 is predicted to contribute to serum resistance
Next, we attempted to determine the region of BOM1093 that contributes to serum resistance. Briefly, the effect of truncation of BOM1093 on serum susceptibility was examined. In this study, four truncated mutants of BOM1093 were created by gradually deleting different nucleotide sequences from the bom1093 gene; BOM10931–208 (bom1093∆209–308), BOM10931–158 (bom1093∆159–308), BOM10931–108 (bom1093∆109–308), and BOM10931–58 (bom1093∆59–308) were generated by PCR. These truncated genes were concatenated with pflaB on pBSV2 and were then introduced into B. garinii HT59G. The four mutants used in this study are schematically represented in Fig. 6A. All four truncated mutants survived equally well in the HIS (Fig. 6B). As previously mentioned, B. garinii HT59G/pBOM1093 showed a serum-resistant phenotype with a survival rate of 54%, and the mock control (B. garinii HT59G/pBSV2) demonstrated a survival rate of less than 1% (Fig. 6B). All the truncated mutants (B. garinii HT59G/pBOM10931–208, HT59G/pBOM10931–158, HT59G/pBOM10931–108, and HT59G/pBOM10931–58) were observed to be serum-susceptible phenotypes (all with a survival ratio less than 2.3%) with dramatically decreased survival compared to B. garinii HT59G/pBOM1093. These results suggest that the C-terminal region of BOM1093 between 209 and 308 is important for converting B. garinii HT59G/pBOM1093 to a serum-susceptible phenotype.

The effect of C-terminal truncation of BOM1093 on serum susceptibility. Schematic picture of C-terminal truncation of BOM1093 (left) and serum susceptibility of each mutant (right) are shown. *Ratio of serum resistance (%) was calculated by dividing the CFUs of NHS by CFUs of HIS, respectively.
C-terminal region of BOM1093 is required for Vn-binding
To define the Vn-binding facilities of BOM1093, C-terminal-truncation mutants were subjected to a pull-down assay. Although intact 6xHis tagged-BOM1093 co-precipitated with Vn, none of the truncated proteins (6xHis tagged-BOM10931–208, 6xHis tagged-BOM10931–158, 6xHis tagged-BOM10931–108, and 6xHis tagged-BOM10931–58) co-precipitated with Vn (Fig. 7A,B). Moreover, an ELISA assay revealed that B. garinii HT59G/pBOM10931–208, in which the C-terminal residues 209–308 were deleted, showed reduced binding compared to B. garinii HT59G/pBOM1093 (Fig. 7B). Furthermore, Vn-binding was not detected in the other mutants due to deletion of the C terminus of BOM1093. These results suggest that the 209–308 region of the C-terminal amino acids of the BOM1093 protein is essential in enabling B. garinii HT59G/pBOM1093 to bind to Vn in vitro.

Decrease of serum Vn binding in C-terminal truncation mutants. B. garinii expressed BOM1093 or C-terminal truncation mutants were used in this study. B. garinii HT59G/pBSV2 was used as Mock control. (A) Detection of co-precipitated serum Vn by western blotting. M.W. indicated molecular weight marker. (B) The amount of co-precipitated Vn was quantified by ELISA.
Depletion of Vn enhanced bactericidal activity of serum-resistant B. garinii HT59G/pBOM1093
It is well characterized that NHS-derived Vn inhibits the complement system. Therefore, removal of Vn from NHS enhances bactericidal activity. In this study, we prepared human serum depleted of Vn (HSΔVn) and used it in the serum susceptibility assay of B. garinii transformants. In this study, Vn depletion was confirmed by western blotting with clusterin (CLU), as a positive control (Fig. 8A). The Vn-depleted serum was subjected to bactericidal assays using B. garinii HT59G/pBOM1093 and the mock control (B. garinii HT59G/pBSV2). B. garinii HT59G/pBOM1093 showed a significant increase in serum susceptibility when incubated with HSΔVn (Fig. 8B). However, when 1 mM of purified recombinant Vn was added to the HSΔVn, serum-resistance was observed for B. garinii HT59G/pBOM1093.

Vn-depleted human serum has bactericidal activity against serum-resistant B. garinii HT59G/pBOM1093. (A) Serum Vn was depleted for NHS. Vn depletion from human serum was repeated three times (HSΔVn 1st, HSΔVn 2nd, and HSΔVn 3rd). Levels of Vn and clusterin (CLU) in tested sera were determined by western blotting. (B) Survival ratio was calculated for B. garinii HT59G/pBOM1093. ΔVn indicates the HSΔVn third sample (HSΔVn 3rd). ΔVn/Vn indicates the tested serum, which was supplemented with 1 mM of recombinant Vn to the HSΔVn 3rd. **p ≤ 0.01.
Discussion
In previous studies, several transformable Borrelia strains have been used as surrogate strains for serum susceptibility analysis. B. burgdorferi strain B313, which is a derivative of strain B31, is one of the transformable and serum susceptible strains20. The strain is convenient for genetic analysis because it forms colonies on a semi-solid BSK agar plate. B. garinii strains G1 and 50.97 have also been used for genetic analyses of serum susceptibility because these strains are transformable and susceptible to human serum21,22. However, transformants of B. garinii strains were isolated using a limiting dilution technique in liquid BSK medium. This process for isolating clonal transformants is an intricate procedure that requires several weeks. Furthermore, more than 10 µg of plasmid DNA is required for efficient transformation for these strains. In this study, we established another transformable and serum-susceptible B. garinii HT59G. Transformation efficacy of the strain was 15 transformants/µg plasmid DNA when pBSV2 plasmid was used, and transformants were isolated from semi solid agar plates. These results suggest that the B. garinii strain HT59G used in our study may be a useful and convenient tool to investigate serum resistance mechanisms of borreliae.
Bacterial pathogens display proteins on their surface, some of which bind complement regulators and inhibit the host complement system. Several reports have indicated that borreliae have the ability to evade the innate immune system. Lyme disease borreliae produce the protein p43 which binds C4BP to the bacterial surface, thereby regulating the classical and lectin pathways through degradation of C4b23. Most Lyme disease borreliae produce several complement regulator-acquiring surface proteins (CRASPs): CRASP-1 to CRASP-5 (CspA, CspZ, and Erps). CspA and CspZ are ligands for FH and/or FHL-1. These proteins can also bind C3b and then promote C3b degradation on the bacterial surface to regulate the complement pathway24. Erps (ErpP, ErpC, and ErpA) can also bind to FH and contribute to serum resistance of Lyme disease borreliae; however, the biological significance of these interactions remains unclear. RF borreliae also express complement regulator-binding proteins on their surface25. BHA007 (B. hermsii) and CihC (B. recurrentis and B. duttonii) can bind C4BP. CihC is also known as C1-INH-binding protein. The binding of BHA007/CihC to C1-INH and C4BP on the bacterial surface prevents the formation of C1 and mannan/mannose-binding lectin-associated serine protease complexes and induces the cleavage of C4b, respectively, resulting in inhibition of the classical and lectin pathways. FhbA of B. hermsii, BpcA of B. parkeri, and HcpA of B. recurrentis bind FH (and/or FHL-1), which inhibits the alternative pathway through C3b cleavage. BBK32 of Lyme disease borreliae, known for both fibronectin26 and glycosaminoglycan binding27, was recently reported to be a C1r-binding protein. By binding to the inactive form of C1r, BBK32 blocks the formation of the active C1 complex thereby inhibiting the classical pathway28. CspA, BGA66 and BGA71 of Lyme disease borreliae bind C7, C8, and C9, and may inhibit the terminal cascade of the complement pathway29,30. A CD59-like protein of B. burgdorferi that binds to C9 has also been suspected; however, the borrelial factor was not identified31. These interactions contribute to the inhibition of the membrane attack-complex, thereby preventing bacteriolysis. Complement regulation by Vn binding, however, has not been reported in Borrelia.
Vn was discovered in 196732 and initially called S-protein, but was later renamed by Hayman et al.33. Vn binds to the membrane attachment site of the C5b-7 complex, thereby blocking insertion into the target membrane and inhibiting C9 polymerization and attachment18,34. Bacterial pathogens thereby evade bactericidal action by the complement system through binding host Vn to their surfaces35. Vn-dependent serum resistance has been well studied for several pathogens. The serum resistance mechanism by Vn binding has been reported in LcpA of Leptospira species, Lpd of Pseudomonas aeruginosa, PE of non-typable Haemophilus influenzae, and UspA2 of Moraxella catarrhalis36,37,38,39. In this study, we revealed that B. miyamotoi BOM1093 acts as a virulence factor contributing to human serum resistance by binding to serum Vn.
In previous studies, B. miyamotoi has shown a serum-resistant phenotype16,40, and only CbiA (locus tag BOM1283, Acc. No. AHH05826) has been identified as a factor responsible for this phenotype17. Briefly, it was shown that CbiA binds to FH and interacts to complement components (C3, C3b, C4, C4b, C5, and C9), thereby potentially blocking the alternative, classical, and terminal pathways of the complement system. However, it has not been shown whether this multi-functional protein is also capable of binding Vn. To our knowledge, this is the first study to report that Vn-binding to a borrelial factor promoted serum resistance of B. miyamotoi.
BOM1093 was identified as the Vn-binding protein in this study. Using Protein Basic Local Alignment Search Tool (BLASTP) analysis, BOM1093 and BOM1515 proteins were identified as related to antigen P3541 in Lyme disease borreliae. In addition, the amino acid residue of BOM1093, which was suggested to be an important region for Vn binding, had a homology of 94.0% identity with the sequence of BOM1515. BOM1515 is also expected to confer serum resistance to B. miyamotoi by Vn-binding. From BLASTP analysis, we found that BOM1093 was conserved in RF borreliae including hard-tick-borne RF borreliae. Sequence similarity of BOM1093 ranged from 59 to 100% in B. miyamotoi and from 56 to 68% in RF borreliae. The bom1093 lineage possessed by RF borreliae may have a function similar to that of B. miyamotoi bom1093. In this study, we conclude that the C-terminal region of BOM1093 is involved in Vn-binding. However, it is also possible that the C-terminal region of BOM1093 is required for structural stability of the protein and that its removal results in a protein with disrupted structure and function. To resolve this question, further investigation is required.
Conclusion
In conclusion, using a newly established transformable B. garinii strain, we revealed that B. miyamotoi has the ability to bind Vn through the membrane protein BOM1093. We hypothesize that Vn-binding may contribute to pathogenicity of B. miyamotoi in humans by allowing it to evade the serum complement system. This is the first study to report that Vn-binding is associated with serum resistance of Borrelia.
Materials and methods
Bacterial strains and culture conditions
Borrelial strains used in this study are listed in Table 2. B. garinii strains J-21, J-37, VSBM, VSBP, VSDA, Fis01, Far01, Far02, HT59, NT25, Borrelia bavariensis strains J-14, J-16, J-20t, J-32, J-39, J-40, J-41, Borrelia burgdorferi 297, and B. miyamotoi MYK3 were used in this study. B. garinii HT59G was isolated from strain HT59 by subsurface colony formation42. These Borrelia strains were grown at 34 °C in Barbour–Stoenner–Kelly (BSK)-M medium43 supplemented with 7% rabbit serum with or without selectable antibiotics. For antibiotic selection of shuttle vector transformants, kanamycin (200 µg/ml) was used. Escherichia coli DH5α was used for the preparation of plasmids for electroporation into B. garinii HT59G.
Preparation of NHS and HIS
NHS from a human blood donor without history of a borrelial infection was used. The serum was confirmed serologically negative by the absence of IgG and IgM antibodies against Borrelia spp. A serum diagnostic test was performed for Lyme disease by immunoblotting using a commercial kit, recomLine Borrelia IgM/IgG (Mikrogen GmbH, Neuried, Germany). We obtained ethical approval for the use of human serum (The details are provided in the Medical Ethics section). HIS was prepared by incubating NHS at 56 °C for 30 min.
Screening assay for serum sensitivity
The serum sensitivity of each borrelial strain was assessed using cells harvested from mid-log phase cultures. The cells (~ 107 cells/ml) were incubated in 40% NHS or HIS at 37 °C. After incubation for 16 h, cell viability was assessed using dark-field microscopic counts of moving cells in 10 fields under 300× magnification. Data are presented as percent survival, calculated as follows: (average number of moving cells/numbers of morphologically collapsed cells per ten 300× magnification fields) × 100. In each assay, the B. burgdorferi strain 297 and B. garinii VSDA were used as survival control and serum-sensitive control, respectively35,49.
Electroporation of Borrelia strains
Electroporation of serum-sensitive Borrelia strains was performed as described previously50,51 with minor modifications. Briefly, Borrelia strains were grown in BSK-M medium and harvested at the late-log phase (5 × 107–1 × 108 cells/ml), which were subsequently prepared for electroporation by washing once with phosphate-buffered saline (PBS) (FUJIFILM Wako Pure Chemical Corp., Osaka, Japan) and twice with electroporation solution [EPS: 0.27 M sucrose, 10% [v/v] glycerol] by centrifugation at 5000×g for 15 min at 4 °C. Freshly prepared competent cells (approximately 109 cells in 50 µl of electroporation solution) were transformed with 0.3–2.0 µg of plasmid DNA. After electroporation at 2.5 kV, 25 μF, and 200 Ω, bacterial cells were immediately resuspended in 1 ml of prewarmed BSK-M medium and incubated for 20–24 h at 34 °C. The cultures were then plated as a 1% soft agar overlay42 on BSK-M plates with kanamycin (200 µg/ml) and incubated for 2 weeks at 34 °C. To determine transformation efficiency (transformants/µg DNA), plasmid pBSV2 was used52. The colonies were picked up from the BSK-M plates and were cultured until mid-log phase. The presence of the kanamycin resistance gene (kanR) of post-transformation Borrelia strains was confirmed by kanR-PCR using the primers mentioned in the Supplemental Table S1.
In silico analysis of ORFs
The plasmid sequences of B. miyamotoi strain FR64b (Acc. Nos. CP004218–CP004266) were subjected to in silico analyses. From these sequences, 649 ORFs (locus-tag nos. BOM 0875–BOM 1523) were extracted and translated into amino acid sequences. These 649 polypeptides were analyzed using SignalP 4.1 (http://www.cbs.dtu.dk/services/SignalP/)53 and LipoP 1.0 (http://www.cbs.dtu.dk/services/LipoP/)54 to predict the existence and location of signal peptide cleavage sites, and to predict the existence of lipoprotein signal peptides, respectively.
Plasmid construction and transformation of recipient B. garinii
The outline of plasmid construction is summarized in Fig. 9. The shuttle vector pBSV2 was used for transformation of B. garinii HT59G. Plasmid pBSV2 was originally constructed by Stewart et al.52. The flagellin gene (flaB) promoter (pflaB) was used for gene expression of the borrelial gene in the recipient Borrelia strain. DNA fragment of pflaB on plasmid pTM6155 was amplified by PCR using a set of primers: pTM61_pflaB_R and pTM61_pflaB_F + tag (Supplemental Table S1). Plasmids for borrelial transformation were constructed using the In-Fusion HD Cloning System (Clontech Laboratories, Mountain View, CA, USA). Briefly, plasmid pBSV2 was linearized by digestion with restriction enzymes (Hind III and Xba I, Takara Bio, Shiga, Japan). DNA fragments of each ORF of B. miyamotoi were amplified by PCR using specific primers for each ORF. Concatenation of pflaB, each amplified fragment from the genomic DNA of B. miyamotoi strain MYK3 and linearized pBSV2 was performed using the In-Fusion system according to manufacturer’s instructions. The primers used for amplification of DNA fragments from B. miyamotoi are listed in the Supplemental Table S2. The constructed plasmid was propagated using E. coli DH5α and purified with the Qiaprep Spin Miniprep kit (QIAGEN, Calif, USA). The purified plasmids were subjected to nucleotide sequencing to ensure no mutations introduced during the cloning process. The oligonucleotide primer pair (T7 and AS-T, Supplemental Table S1), which amplified the DNA fragment of multi-cloning site of pBSV2, was used for PCR amplification and sequencing. Each plasmid was used to transform B. garinii HT59G. The transformed B. garinii HT59G was picked up from the BSK-M plate containing 200 µg/ml kanamycin. Transformation with plasmid was confirmed by kanR-PCR and PCR using DNA primers T7 and AS-T.

Schematic model of plasmid construction and transformation of recipient Borrelia. These were abbreviated in this figure as follows; promoter region of flaB gene (pflaB), upstream 5′-DNA primer (USP), and downstream 3′-DNA primer (DSP), respectively.
Serum resistance assay of transformed B. garinii HT59G clones
For initial screening, each B. garinii HT59G transformant (~ 107 cells/ml) was incubated with rabbit serum-free BSK-M medium with 40% NHS for 5 days at 34 °C, and the survival of transformants was assessed using dark-field microscopy. For calculation of serum resistance, the transformants (~ 106 cells/ml), which showed a survival phenotype, were exposed to 40% NHS or HIS for 16 h at 34 °C. The cells were plated as a 1% soft agar overlay on BSK-M plates with kanamycin (200 µg/ml) and incubated at 34 °C for 2 weeks; the number of surviving cells was counted and calculated as CFUs49.
Proteinase K accessibility of BOM1093
B. garinii HT59G/pBOM1093 was grown ***at 34 °C and collected by centrifugation at 2000×g for 15 min at 4 °C and washed twice with PBS containing 5 mM MgCl2. The cells (8.3 × 107 cells/tube) were resuspended in 0.5 ml of either PBS or PBS with proteinase K (to a final concentration of 200 µg/ml). All the samples were incubated for 1 h at 20 °C. Reactions were terminated by the addition of Pefabloc SC (Roche Diagnostics, Mannheim, Germany) to a final concentration of 100 mM. Cells were again pelleted by centrifugation (5000×g for 10 min at 4 °C), washed twice with PBS containing 5 mM MgCl2 and 20 mM Pefabloc SC and resuspended in Laemmli sample buffer (62.5 mM Tris pH6.8, 2% SDS, 10% glycerol, 0.01% bromophenol blue, and 0.5% 2-mercaptoethanol) to carry out sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE)56. The samples corresponding to 1.6 × 106 whole cell equivalents were separated on a 12.5% SDS-PAGE gel, transferred to a Sequi-Blot PVDF membrane (Bio-Rad Laboratories, Hercules, CA, USA), and western blotting was performed as previously described57. Monoclonal antibodies (mAb) against borrelial OspA (H5332)58, flagellin (H9724)59, p100 (Mab958) (Merck, Darmstadt, Germany), or anti-BOM1093 rabbit serum (prepared in this study) were used. To detect these antibodies, Horseradish peroxidase (HRP)-conjugated goat anti-mouse IgG (Jackson ImmunoResearch, West Grove, PA, USA) or rabbit IgG (Merck) were used. HRP-conjugated antibodies were detected by chemiluminescence using the electrogenerated chemiluminescence (ECL) Prime detection reagent (GE Healthcare Bioscience, Piscataway, NJ, USA).
Pull down assay
Borrelia garinii HT59G cells expressed as 6xHis-tagged to C-terminal of BOM1093 (108 cells/reaction) were incubated with NHS (20%) for 1 h at 37 °C and washed 3 times with TBS buffer (20 mM Tris–HCl pH 7.0, 0.25 M NaCl) containing 5 mM Pefabloc SC. Hexa-His-tagged BOM1093 obtained from sonicated cells was purified using HisPur Ni–NTA Magnetic Beads (Thermo Fisher Scientific, Waltham, MA, USA).
The sample was separated by 10% SDS-PAGE, transferred to a PVDF membrane, and analyzed using either mAb or polyclonal antibody (pAb) (1:1000) to detect complement regulators for 1 h followed by the HRP-conjugated monoclonal antibody (1:5000) for 1 h. The blot was developed using the ECL Prime detection reagent. For the detection of complement regulators, antibodies were used as follows; Anti-Vn, Anti-clusterin, Anti-FHL-1, Anti-factor I and Anti-complement factor H-related protein 1 mAbs were purchased from R&D Systems (Minneapolis, MN, USA), Anti-C4BP mAb was from Santa Cruz Biotechnology (Dallas, TX, USA), Anti-properdin mAb was from Abcam, Anti-carboxy peptidase N mAb was from Bioss (Woburn, MA, USA), Anti-FH pAb was from Merck, Anti-C1 inhibitor pAb was from Complement Technology (Tyler, TX, USA). To detect these antibodies, HRP-conjugated goat anti-mouse IgG (Jackson ImmunoResearch, West Grove, PA, USA), HRP-conjugated anti-rabbit IgG (Merck) and HRP-conjugated rabbit anti-goat IgG (Abcam) were used.
Quantitation of binding of Vn to transformants by ELISA
Vn-binding to B. garinii HT59G/pBOM1093 was evaluated by ELISA using a 96-well Maxisorp plate (Thermo Fisher Scientific). The wells were coated with 0.5 µg/well of sonicated B. garinii HT59G/pBOM1093 or B. garinii HT59G/pBSV2 in TBS buffer and the plate was incubated at 4 °C overnight. Next, the plate was washed three times with TBS buffer and treated with blocking buffer (TBS buffer with 1% skim milk) at room temperature for 1 h. After soaking in TBS buffer, the plates were incubated for 2 h at room temperature with recombinant human Vn (Sigma-Aldrich, St. Louis, MO, US) (6.25–400 nM). The plate was then washed 3 times with washing buffer (TBS buffer with 0.05% Tween 20) and incubated with mouse anti-human Vn IgG (1:1000 dilution with blocking buffer) at 37 °C for 1 h. After washing, HRP-conjugated goat anti-mouse IgG (1:5000) was incubated at 37 °C for 1 h. To detect the HRP-labeled secondary antibodies, a 3,3′,5,5′-tetramethylbenzidine solution (TMB; Nakarai Tesque, Kyoto, Japan) was added to the wells and allowed to react for 10 min before the absorbance was measured at 450/620 nm. The values of B. garinii HT59G/pBSV2 was subtracted from the values of B. garinii HT59G/BOM1093 and the KD was calculated using Prism Ver. 6 (GraphPad Software, San Diago, CA, USA).
Depletion of Vn from NHS
Vn-depleted human serum (HSΔVn), which retained complement activity, was generated according to the method reported by Hallstrom60. A total of 250 µg of protein A/G-sepharose (Abcam) was incubated with 200 µg of anti-Vn polyclonal rabbit serum (Gene Tex, CA, USA) in PBS overnight at 4 °C with mild shaking. Unbound anti-Vn antibody was removed by washing three times with PBS. Thereafter, NHS was added thrice and the mixture was incubated each time for 20 min at 4 °C. The Vn-depleted serum was analyzed for the presence of Vn by ELISA and western blotting. For ELISA, anti-human Vn monoclonal antibody (1:2000) and HRP-conjugated goat anti-mouse IgG monoclonal antibody (1:10,000) were used. The signal was detected using the TMB reagent. For western blotting, the HSΔVn was separated by 10% SDS-PAGE. For detection of Vn, anti-human Vn monoclonal antibody (1:2000) was used. As an internal control, serum clusterin was assessed in this study. Anti-human clusterin monoclonal antibody (1:2000) (Quidel, CA, USA) and HRP-conjugated goat anti-mouse IgG monoclonal antibody (1:10,000) were used to detect serum clusterin. The signal was detected by a chemiluminescence-based technique using ECL Prime detection reagent.
Evaluation of the effect of Vn depletion activity of NHS
The B. garinii HT59G transformants (~ 106 cells/ml) were reacted to HSΔVn with or without 1 mM of recombinant human Vn (Sigma-Aldrich, USA). Briefly, the Vn-depleted serum (the third HSΔVn sample; HSΔVn3rd) was incubated in bactericidal assays at a final concentration of 10% (v/v) for 4 h at 34 °C. The cells were plated as a 1% soft agar overlay on BSK-M plates with kanamycin (200 µg/ml) and incubated for 10–14 days at 34 °C.
Medical ethics
The normal human serum (NHS) was obtained from healthy Japanese blood donors. The blood collection was carried out in accordance with international guideline and regulations (Declaration of Helsinki, 1964). All experimental protocols used human serum, the procedure of blood collection, and documented informed consent were approved by the Ethical Committee of the National Institute of Infectious Diseases for medical research using human subjects (Approval No. 791 on June 26, 2017). All volunteers provided informed consent.
Statistical analysis
Results were assessed using the Student's t test for paired data. A value of p ≤ 0.01*** was considered statistically significant.
Data availability
Materials established in this study are available from the corresponding author on reasonable request.
References
Fukunaga, M. et al. Genetic and phenotypic analysis of Borrelia miyamotoi sp. nov., isolated from the ixodid tick Ixodes persulcatus, the vector for Lyme disease in Japan. Int. J. Syst. Bacteriol. 45, 804–810 (1995).
Scoles, G. A., Papero, M., Beati, L. & Fish, D. A. Relapsing fever group spirochete transmitted by Ixodes scapularis ticks. Vector Borne Zoonot. Dis. 1, 21–34 (2001).
Mun, J., Eisen, R. J., Eisen, L. & Lane, R. S. Detection of a Borrelia miyamotoi sensu lato relapsing-fever group spirochete from Ixodes pacificus in California. J. Med. Entomol. 43, 120–123 (2006).
Salkeld, D. J., Cinkovich, S. & Nieto, N. C. Tick-borne pathogens in northwestern California, USA. Emerg. Infect. Dis. 20, 493–494 (2014).
Fraenkel, C. J., Garpmo, U. & Berglund, J. Determination of novel Borrelia genospecies in Swedish Ixodes ricinus ticks. J. Clin. Microbiol. 40, 3308–3312 (2002).
Richter, D., Schlee, D. B. & Matuschka, F. R. Relapsing fever-like spirochetes infecting European vector tick of Lyme disease agent. Emerg. Infect. Dis. 9, 697–701 (2003).
Platonov, A. E. et al. Humans infected with relapsing fever spirochete Borrelia miyamotoi, Russia. Emerg. Infect. Dis. 17, 1816–1823 (2011).
Krause, P. J., Fish, D., Narasimhan, S. & Barbour, A. G. Borrelia miyamotoi infection in nature and in humans. Clin. Microbiol. Infect. 21, 631–639 (2015).
Henningsson, A. J. et al. Two cases of Borrelia miyamotoi meningitis, Sweden, 2018. Emerg. Infect. Dis. 25, 1965–1968 (2019).
Boden, K., Lobenstein, S., Hermann, B., Margos, G. & Fingerle, V. Borrelia miyamotoi-associated neuroborreliosis in immunocompromised person. Emerg. Infect. Dis. 22, 1617–1620 (2016).
Tobudic, S. et al. Human Borrelia miyamotoi infection, Austria. Emerg. Infect. Dis. 26, 2201–2204 (2020).
Sato, K. et al. Human infections with Borrelia miyamotoi, Japan. Emerg. Infect. Dis. 20, 1391–1393 (2014).
Jiang, B. G. et al. Borrelia miyamotoi infections in humans and ticks, northeastern China. Emerg. Infect. Dis. 24, 236–241 (2018).
Molloy, P. J. et al. Borrelia miyamotoi disease in the northeastern United States: A case series. Ann. Intern. Med. 163, 91–98 (2015).
Gugliotta, J. L., Goethert, H. K., Berardi, V. P. & Telford, S. R. 3rd. Meningoencephalitis from Borrelia miyamotoi in an immunocompromised patient. N. Engl. J. Med. 368, 240–245 (2013).
Wagemakers, A., Oei, A., Fikrig, M. M., Miellet, W. R. & Hovius, J. W. The relapsing fever spirochete Borrelia miyamotoi is cultivable in a modified Kelly-Pettenkofer medium, and is resistant to human complement. Parasit. Vectors 7, 418 (2014).
Röttgerding, F. et al. Immune evasion of Borrelia miyamotoi: CbiA, a novel outer surface protein exhibiting complement binding and inactivating properties. Sci. Rep. 7, 303 (2017).
Podack, E. R., Preissner, K. T. & Müller-Eberhard, H. J. Inhibition of C9 polymerization within the SC5b-9 complex of complement by S-protein. Acta. Pathol. Microbiol. Immunol. Scand. Suppl. 284, 89–96 (1984).
Singh, B., Su, Y. C. & Riesbeck, K. Vitronectin in bacterial pathogenesis: A host protein used in complement escape and cellular invasion. Mol. Microbiol. 79, 545–560 (2010).
Hartmann, K. et al. Functional characterization of BbCRASP-2, a distinct outer membrane protein of Borrelia burgdorferi that binds host complement regulators factor H and FHL-1. Mol. Microbiol. 61, 1220–1236 (2006).
Siegel, C. et al. Complement factor H-related proteins CFHR2 and CFHR5 represent novel ligands for the infection-associated CRASP proteins of Borrelia burgdorferi. PLoS One 5, e13519 (2010).
Alitalo, A. et al. Expression of complement factor H binding immunoevasion proteins in Borrelia garinii isolated from patients with neuroborreliosis. Eur. J. Immunol. 35, 3043–3053 (2005).
Pietikainen, J., Meri, T., Blom, A. M. & Meri, S. Binding of the complement inhibitor C4b-binding protein to Lyme disease borreliae. Mol. Immunol. 47, 1299–1305 (2010).
Kraiczy, P. & Stevenson, B. Complement regulator-acquiring surface proteins of Borrelia burgdorferi: Structure, function and regulation of gene expression. Ticks Tick Borne Dis. 4, 26–34 (2013).
Röttgerding, F. & Kraiczy, P. Immune evasion strategies of relapsing fever spirochetes. Front. Immunol. 11, 1560 (2020).
Probert, W. S. & Johnson, B. J. B. Identification of a 47 kDa fibronectin-binding protein expressed by Borrelia burgdorferi isolate B31. Mol. Microbiol. 30, 1003–1015 (1998).
Fischer, J. R., LeBlanc, K. T. & Leong, J. M. Fibronectin binding protein BBK32 of the Lyme disease spirochete promotes bacterial attachment to glycosaminoglycans. Infect. Immun. 74, 435–441 (2006).
Garcia, B. L., Zhi, H., Wager, B., Hook, M. & Skare, J. T. Borrelia burgdorferi BBK32 inhibits the classical pathway by blocking Activation of the C1 complement complex. PLoS Pathog. 12, e1005404 (2016).
Hallström, T. et al. CspA from Borrelia burgdorferi inhibits the terminal complement pathway. mBio 4, 20 (2013).
Hammerschmidt, C. et al. BGA66 and BGA71 facilitate complement resistance of Borrelia bavariensis by inhibiting assembly of the membrane attack complex. Mol. Microbiol. 99, 407–424 (2016).
Pausa, M. et al. Serum-resistant strains of Borrelia burgdorferi evade complement-mediated killing by expressing a CD59-like complement inhibitory molecule. J. Immunol. 170, 3214–3222 (2003).
Holmes, R. Preparation from human serum of an alpha-one protein which induces the immediate growth of unadapted cells in vitro. J Cell Biol. 32, 297–308 (1967).
Hayman, E. G., Pierschbacher, M. D., Ohgren, Y. & Ruoslahti, E. Serum spreading factor (vitronectin) is present at the cell surface and in tissues. Proc. Natl. Acad. Sci. U S A 80, 4003–4007 (1983).
Podack, E. R., Kolb, W. P. & Müller-Eberhard, H. J. The SC5b-7 complex: Formation, isolation, properties, and subunit composition. J. Immunol. 119, 2024–2029 (1977).
Patarakul, K., Cole, M. F. & Hughes, C. A. Complement resistance in Borrelia burgdorferi strain 297: Outer membrane proteins prevent MAC formation at lysis susceptible sites. Microb. Pathog. 27, 25–41 (1999).
Da Silva, L. B. et al. Pathogenic Leptospira species acquire factor H and vitronectin via the surface protein LcpA. Infect. Immun. 83, 888–897 (2015).
Hallström, T., Uhde, M., Mörgelin, M., Skerka, C. & Zipfel, P. F. Pseudomonas aeruginosa CRASP-2 is a surface protein that uses the human terminal complement regulator vitronectin for protection against complement mediated attack. Mol. Immunol. 47, 2837 (2010).
Hallström, T., Blom, A. M., Zipfel, P. F. & Riesbeck, K. Nontypeable Haemophilus influenzae protein E binds vitronectin and is important for serum resistance. J. Immunol. 183, 2593–2601 (2009).
Attia, A. S., Ram, S., Rice, P. A. & Hansen, E. J. Binding of vitronectin by the Moraxella catarrhalis UspA2 protein interferes with late stages of the complement cascade. Infect. Immun. 74, 1597–1611 (2006).
Teegler, A., Herzberger, P., Margos, G., Fingerle, V. & Kraiczy, P. The relapsing fever spirochete Borrelia miyamotoi resists complement-mediated killing by human serum. Ticks Tick Borne Dis. 5, 898–901 (2014).
Casjens, S. et al. A bacterial genome in flux: The twelve linear and nine circular extrachromosomal DNAs in an infectious isolate of the Lyme disease spirochete Borrelia burgdorferi. Mol. Microbiol. 35, 490–516 (2000).
Norris, S. J., Howell, J. K., Garza, S. A., Ferdows, M. S. & Barbour, A. G. High- and low-infectivity phenotypes of clonal populations of in vitro-cultured Borrelia burgdorferi. Infect. Immun. 63, 2206–2212 (1995).
Takano, A. et al. Tick Surveillance for Relapsing Fever Spirochete Borrelia miyamotoi in Hokkaido, Japan. PLoS ONE 9, e104532 (2014).
Takano, A. et al. Multilocus sequence typing implicates rodents as the main reservoir host of human-pathogenic Borrelia garinii in Japan. J. Clin. Microbiol. 49, 2035–2039 (2011).
Fukunaga, M., Sohnaka, M., Takahashi, Y., Nakao, M. & Miyamoto, K. Antigenic and genetic characterization of Borrelia species isolated from Ixodes persulcatus in Hokkaido, Japan. J. Clin. Microbiol. 31, 1388–1391 (1993).
Péter, O., Bretz, A. G., Postic, D. & Dayer, E. Association of distinct species of Borrelia burgdorferi sensu lato with neuroborreliosis in Switzerland. Clin. Microbiol. Infect. 3, 423–431 (1997).
Bunikis, J. et al. Molecular polymorphism of the Lyme disease agent Borrelia garinii in northern Europe is influenced by a novel enzootic Borrelia focus in the North Atlantic. J. Clin. Microbiol. 34, 364–368 (1996).
Steere, A. C. et al. The spirochetal etiology of Lyme disease. N. Engl. J. Med. 31, 733–740 (1983).
van Dam, A. P. et al. Complement-mediated serum sensitivity among spirochetes that cause Lyme disease. Infect. Immun. 65, 1228–1236 (1997).
Lawrenz, M. B., Kawabata, H., Purser, J. E. & Norris, S. J. Decreased electroporation efficiency in Borrelia burgdorferi containing linear plasmids lp25 and lp56: Impact on transformation of infectious B. burgdorferi. Infect. Immun. 70, 4798–4804 (2002).
Kawabata, H., Norris, S. J. & Watanabe, H. BBE02 disruption mutants of Borrelia burgdorferi B31 have a highly transformable, infectious phenotype. Infect. Immun. 72, 7147–7154 (2004).
Stewart, P. E., Thalken, R., Bono, J. L. & Rosa, P. Isolation of a circular plasmid region sufficient for autonomous replication and transformation of infectious Borrelia burgdorferi. Mol. Microbiol. 39, 714–721 (2001).
Nielsen, H. Predicting secretory proteins with SignalP. In Protein Function Prediction: Methods and Protocols (ed. Kihara, D.) 59–73 (Springer, 2017).
Rahman, O., Cummings, S. P., Harrington, D. J. & Sutcliffe, I. C. Methods for the bioinformatic identification of bacterial lipoproteins encoded in the genomes of Gram-positive bacteria. World J. Microbiol. Biotechnol. 24, 2377–2382 (2008).
Moriarty, T. J. et al. Real-time high resolution 3D imaging of the Lyme disease spirochete adhering to and escaping from the vasculature of a living host. PLoS Pathog. 4, e1000090 (2008).
Laemmli, U. K. Cleavage of Structural Proteins during the Assembly of the Head of Bacteriophage T4. Nature 227, 680-685 (1970).
Masuzawa, T. et al. Characterization of monoclonal antibodies for identification of Borrelia japonica, isolates from Ixodes ovatus. Microbiol. Immunol. 38, 393–398 (1994).
Barbour, A. G., Tessier, S. L. & Todd, W. J. Lyme disease spirochetes and ixodid tick spirochetes share a common surface antigenic determinant defined by a monoclonal antibody. Infect. Immun. 41, 795–804 (1983).
Barbour, A. G., Hayes, S. F., Heiland, R. A., Schrumpf, M. E. & Tessier, S. L. A Borrelia-specific monoclonal antibody binds to a flagellar epitope. Infect. Immun. 52, 549–554 (1986).
Hallström, T. et al. Pseudomonas aeruginosa Uses Dihydrolipoamide Dehydrogenase (Lpd) to Bind to the Human Terminal Pathway Regulators Vitronectin and Clusterin to Inhibit Terminal Pathway Complement Attack. PLoS One 10, e0137630 (2015).
Acknowledgements
We are grateful to P. E. Stewart and P. A. Rosa (National Institute of Allergy and Infectious Diseases Rocky Mountain Laboratories), T. Moriarty and G. Chaconas (The University of Calgary), R. Lienhard (National Reference Centre for tick-transmitted diseases), M. Nakao (Asahikawa Medical University), and T. Masuzawa (Chiba Institute of Science) for providing DNA plasmids, Borrelia strains, and monoclonal antibodies and M. Ato and and J. Mitobe (National Institute of Infectious Disease) for collection of blood from volunteers. Plasmids pBSV2 and pTM61 were graciously provided by P. E. Stewart and P. A. Rosa, and T. Moriarty and G. Chaconas (The University of Calgary), respectively. The monoclonal antibodies against OspA (MAb H5332) and flagellin (MAb H9724) were kindly provided by T. Masuzawa. Borrelia strains J-21, J-37, HT59, NT25, J-14, J-16, J-20t, J-32, J-39, J-40, and J-41 were provided from M. Nakao. Borrelia burgdorferi 297 were provided from T. Masuzawa. Borrelia strains VSBM, VSDA, Fis01, Far01, Far02 were kindly provided from R. Lienhard. This research was supported by AMED under Grant numbers JP20wm0225016 and JP20fk0108068 (H.K.), and JSPS KAKENHI Grant numbers JP18K07132 and JP15K08487 (H.K.).
Author information
Authors and Affiliations
Contributions
K.S. and H.K. designed the study and wrote the manuscript. K.S., Y.K., and H.K. constructed the plasmid library and transformation of Borrelia. K.S. performed all the biochemical experiments. T.S. and M.K. provided genome analysis data for Lyme disease borreliae. T.H., A.T., and Gaowa provided unpublished B. miyamotoi genome data. K.R.T. and M.O. provided helpful advice and discussion. All the authors agreed to the final version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Sato, K., Kumagai, Y., Sekizuka, T. et al. Vitronectin binding protein, BOM1093, confers serum resistance on Borrelia miyamotoi. Sci Rep 11, 5462 (2021). https://doi.org/10.1038/s41598-021-85069-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-021-85069-w
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
