
Abstract
Highlighting genomic profiles for geographically distinct subpopulations of the same breed may provide insights into adaptation mechanisms to different environments, reveal genomic regions divergently selected, and offer initial guidance to joint genomic analysis. Here, we characterized similarities and differences between the genomic patterns of Angus subpopulations, born and raised in Canada (N = 382) and Brazil (N = 566). Furthermore, we systematically scanned for selection signatures based on the detection of autozygosity islands common between the two subpopulations, and signals of divergent selection, via FST and varLD tests. The principal component analysis revealed a sub-structure with a close connection between the two subpopulations. The averages of genomic relationships, inbreeding coefficients, and linkage disequilibrium at varying genomic distances were rather similar across them, suggesting non-accentuated differences in overall genomic diversity. Autozygosity islands revealed selection signatures common to both subpopulations at chromosomes 13 (63.77–65.25 Mb) and 14 (22.81–23.57 Mb), which are notably known regions affecting growth traits. Nevertheless, further autozygosity islands along with FST and varLD tests unravel particular sites with accentuated population subdivision at BTAs 7 and 18 overlapping with known QTL and candidate genes of reproductive performance, thermoregulation, and resistance to infectious diseases. Our findings indicate overall genomic similarity between Angus subpopulations, with noticeable signals of divergent selection in genomic regions associated with the adaptation in different environments.
Similar content being viewed by others


Introduction
Angus is a taurine breed (Bos primigenius taurus) of moderate frame size, originated in the Highlands of Scotland. It has become adopted worldwide for beef production, due primarily to its distinguished carcass and meat quality highly attractive in currently demanding market1. The introduction of Angus in Canada and Brazil occurred during the 19th and early 20th centuries, respectively. In both countries, black and red coat color variants have been registered in a single herd book2,3. In Canada, purebred Angus is found throughout all provinces, being the most popular beef breed, in terms of number of new registered animals per year (56,003 in 2019)4,5. In Brazil, nearly 51% of 9.62 million beef cattle semen doses commercialized in 2018 were from Angus breed, having more than half of the semen straws imported from Canada and the U.S.6. Despite widely importing foreign genetic material, Brazil also performs national genetic evaluation including Angus purebred local records, which allows the identification of superior Brazilian sires. The progeny originated from locally selected animals tend to be more adaptable to tropical conditions and more robust to environmental changes7.
Combining international data of the same breed with the employment of proper adjustment factors has long been referred to as a promising strategy improving accuracies for cattle genetic analysis8,9,10, including the Angus breed11. Genomic evaluation also benefits from this data combining strategy as dense SNP genotypes will overcome the lack of common ancestry between subpopulations when the pedigree is not deep enough12. The genomic prediction relies on the linkage disequilibrium (LD) between the genotyped markers and quantitative trait loci (QTL), which may not persist between subpopulations, or may even be in reversed phase. Although it is expected that LD phases extend across large distances for subpopulations of the same breed in different countries13, this information is yet to be adequately assessed in Angus cattle.
The geographic division of Angus subpopulations may increase the diversity and sub-structure within the breed, albeit they originated from a unique domestication center. Since, differentiation in allelic frequencies, haplotypes diversity and LD tend to increase over time due to reduced common ancestry between subpopulations, different environmental pressures, and potential differences in selection schemes13,14. Furthermore, selection, either natural or human-oriented, intensifies the differentiation between subpopulations by driving particular genomic regions towards special patterns in each specific geographical location15.
Here, we characterize differences and similarities in the genomic profile of Angus from Canada and Brazil, by assessing the genomic level of relationship, inbreeding, linkage disequilibrium and persistence of linkage phase in both subpopulations. In addition, we scan for selection signatures that may shed light on the molecular basis of selected traits and divergent adaptation mechanisms on the two subpopulations. Although the current study has been focused on the population genomic aspects, our results are relevant to genomic predictions, especially to further collaborative analyses incorporating the studied herds.
Results
Population substructure and relationship
Here, we evaluated a sample of 948 Angus cattle (382 from Canada and 566 from Brazil) with genotypic information of 31,483 autosomal markers, spanning up to 2.5 GB of the bovine genome in an average spacing of 78.6Kb (ranging from 0.006Kb to 3,049Kb). The overall averages of observed heterozygosity (0.353 ± 0.012; 0.350 ± 0.009) and minor allele frequency (MAF: 0.268 ± 0.141; 0.263 ± 0.144) did not differ between Brazilian and Canadian subpopulations (Table 1). The genomic relationship between samples ranged from 0.5 to 0.81 and the average estimated for pairs of individuals from different subpopulations was 0.577 ± 0.021. The average genomic relationship within the Canadian subpopulation was slightly higher than the Brazilian average (Table 1).
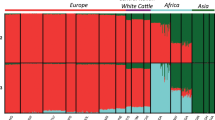
The proportion of variance in genomic relationship matrix (GRM) explained by the first and second principal components was 21.72% and 6.07%, respectively (Fig. 1). In line with the genomic relationship averages, principal component analysis (PCA) revealed greater diversity within the Brazilian samples than within Canadians and depicted two closely connected clusters representing the subpopulations. Although the Brazilian subpopulation included black and red coat colored samples, a clear extra population substructure was not observed within this subpopulation due to coat color variation (Fig. 1). It is noteworthy that the substructure that allowed distinguishing the two clusters representing the Angus Brazilian and Canadian subpopulations is not comparable with the stratification level involving different breeds (Supplementary Fig. S1). The level of differentiation between the two subpopulations, measured through the average of Wright’s FST statistics, was equal to 0.072 ± 0.021.

Principal component analysis of relationship matrix for Brazilian and Canadian Angus subpopulations. Blue and green circles represent black coat-color samples from Canada and Brazil, green triangles represent red samples from Brazil.
Averages of genomic inbreeding estimated using diagonal elements of GRM and runs of homozygosity (ROH) were similar in the two subpopulations (Table 1), being slightly higher in the Canadian. The correlation between these two metrics was equal to 0.9415, with FGRM estimates being overall higher than FROH (Table 1, Fig. 2). Table 2 summarizes the distribution of ROH lengths by subpopulations. The mean length of ROHs and the proportion of segments longer than 8 Mb were higher in the Brazilian subpopulation. All samples presented at least one ROH segment longer than 8 Mb. In total, 59 and 63% of samples in the Canadian and Brazilian subpopulations, respectively, presented at least one ROH segment longer than 16 Mb.

Genomic inbreeding estimated through the diagonal of the genomic relationship matrix (FGRM) and runs of homozygosity (FROH) in the Angus subpopulations. Green and blue dots represent Brazilian and Canadian samples.
Linkage disequilibrium
We used phased haplotypes to estimate linkage disequilibrium (LD), through r2 statistic. The number of SNPs, averages of SNP distances and r2 of adjacent SNPs per chromosome are presented in Supplementary Table S1. The r2 of all pairwise adjacent markers averaged 0.20 and 0.21 in the Brazilian and Canadian subpopulations, respectively (Table 3). The r2 average decreased according to the increase in distance classes of adjacent markers and increased according to the increase in the MAF threshold (Table 3). There was a smaller increase in r2 by rising the MAF threshold from 0 to 1%, than rising it from 1 to 5%.
With all combinations of syntenic SNPs, the overall average of r² kept higher than 0.2 for markers distanced up to 50Kb in both subpopulations (Fig. 3A). At distances around 1 Mb, the markers pairwise had r² averages equal to 0.06 and 0.07 in the Brazilian and Canadian subpopulations, respectively. Although the LD averages in the Canadian subpopulation were the largest for almost all distance classes, the slope of decay were consistent between the two subpopulations. The persistence of the phase between the two subpopulations remained high at a scale of nearly 0.50 up to a distance of 5 Mb (Fig. 3B) indicating agreement between linkage phases in the two subpopulations.

LD decay as a function of inter-marker distance (A) and correlation of linkage phase between Angus subpopulations for SNP pairs at varying distances (B).
Candidate regions under selection within and across subpopulation
The ROH scan revealed 3 and 2 regions with at least two consecutive SNPs with autozygosity score above the empirical threshold (99.9 percentile) in the Canadian and Brazilian subpopulations, respectively (Fig. 4, Supplementary Tables S2). The FST and varLD tests (between-population comparative methods) identified 5 regions each, with at least two consecutive windows exceeding the empirical threshold (Fig. 4, Supplementary Tables S3). The length of these regions ranged from 160 Kb to 1910Kb and presented some overlapping between them. Table 4 lists 6 selection signatures, which represent genomic regions concordant between the 99.9 percentile of at least two of the independent tests, hence more likely to be true positive signals16.

Genome-wide autozygosity, FST, and varLD scores. (A,B) Autozygosity scores per SNP within Brazilian and Canadian subpopulations. (C,D) FST and varLD scores averaged for 15 SNPs windows. Red lines indicate the cutoff of 99.9 percentile to ROH and varLD scores. Red dots represent FST windows in 99.9 percentile of their heterozygosity bins.
Two ROH islands common to both subpopulations were detected at BTA13:63.77–65.25 and BTA14:22.81–23.57 (Fig. 4A,B), surrounding candidate genes of stature, muscle development and lipid metabolisms, such as MYH7B (myosin heavy chain 7B), GDF5 (growth differentiation factor 5), PLAG1 (PLAG1 zinc finger) and XKR4 (XK related 4)17,18,19,20. A further ROH island exclusively detected in the Canadian subpopulation, BTA7:37.84–38.64, overlapped with a signal of divergent selection highlighted via FST (Table 4, Fig. 4A,C), indicating it as a region that has been targeted by selection only in the Canadian subpopulation. This genomic region encompassed several genes encoding integral components of membrane related to reproductive traits. The ARL10 (ADP-ribosylation factor-like 10) gene is suggested to play important role in bovine pre-implantation embryo development21 and the UNC5A (unc-5 netrin receptor A) gene previously showed association with reproductive traits in taurine breeds, such as the number of insemination per conception, fertility index and the interval between first and last insemination22.
Selection signatures revealed by the overlaps of the between-population differentiation methods (FST, and varLD), at BTA7:21.31–21.89 and BTA18:11.75–11.94, are putatively related to adaptation into adverse conditions across the different environments (Table 4, Fig. 4C,D). The selection signature BTA7:21.31–21.89 contained several adaptive immunity-related genes, including the IRF1 (interferon regulatory factor 1) and IL4 (Interleukin-4) genes, which control the differentiation of naive T helper (Th) cells and activation of cell-mediated or antibody-mediated immune responses23,24. In addition, the KIF3A (Kinesin family type 2 member 3 A) gene, mapped at this region, is a candidate to innate tick-resistance typical of some breeds25. Beyond immunity-related genes, the selection signature BTA18:11.75–11.94 comprised the COX4I1 (cytochrome c oxidase subunit 4I1) that is related to thermoregulatory efficiency. This gene represents a candidate gene to body temperature regulation, which was previously associated with cold tolerance26, and was up-regulated in the liver of cows experimentally submitted to cold-stress27.
A further selection signature identified via FST and varLD, at BTA18:13.76–15.40, was the longest signal detected in the current study and comprised the MC1R gene, major gene to coat color determination in Angus28,29. Interestingly, this signal was detected even when the selection signature scan was re-performed keeping only samples of black coat color in both subpopulations (Supplementary Fig. S2). Other genes of this region that are plausible of being causing this signal include DNAJA2 (DnaJ heat shock protein family member A2) that is involved in cellular responses to heat stress30, and some key players of immune responses, such as CYBA (cytochrome b-245 alpha chain) and CDK10 (cyclin dependent kinase 10)31,32,33.
The selection signatures common between Brazilian and Canadian subpopulation (BTA13:63.83–64.87 and BTA14:24.43–26.20) comprised 41 genes, whereas the regions representing signals of divergent selection (ROH islands exclusively detected in one of the two subpopulation, FST, and varLD signals) comprised 74 genes. Significant enrichment of particular terms or biological processes was not detected in any of these gene lists.
Discussion
Accentuated differences between the overall genomic diversity of two populations allow two assumptions, being (1) recent gene flow or adaptive genetic variation in the subpopulation of higher diversity, or (2) genetic drift, reduced effective population size or increased inbreeding in the population of lower diversity34,35. Although the genomic diversity patterns of the two subpopulations studied here were quite similar, the Brazilian samples showed higher heterozygosity, broader dispersion in PCA and lower averages of genomic relationship, inbreeding, and linkage disequilibrium than Canadian samples. The slightly lower diversity observed in the Canadian subpopulation is most likely due to the sample origin and its commercial purpose. The Canadian genotypes were obtained from a single experimental herd of reduced effective population size, in comparison with Brazilian samples that represented commercial herds, more opened to the eventual adoption of imported semen.
The observed heterozygosity within each Angus subpopulation was in agreement with averages varying from 0.357 to 0.399, reported in taurine breeds genotyped with high-density SNP panels34,36, but higher than the average of 0.28 previously reported to 42 Angus samples37. Presumably, this difference is due to the greater number of genotyped individuals studied here. Similar to previous results, the correlation between FGRM (with base allele frequency fixed to 0.5) and FROH was high, with greater estimates obtained to FGRM due to the non-distinction between IBS and IBD alleles as done by FROH38,39. Long ROHs (>8,000 Kb) were observed in both subpopulations, indicating relatively recent relatedness within both of them. Autozygous segments longer than 8,000 Kb and 16,000 Kb indicate nearly 6 and 3 generations, respectively, since the common ancestor that gives origin to both haplotype copies40.
The population structure depicted in PCA showed a great overlap between the clusters of the Brazilian and Canadian samples. One reason for this continued genomic connection resides in the recent common ancestry, also evidenced by the relative high LD phase persistence across the subpopulations. This recent common ancestry is most likely due to the importation of semen from Canada into Brazil, as well as the extensive importation of U.S. Angus sires into both countries (ASBIA 2018, http://www.asbia.org.br/wp-content/uploads/2018/10/INDEX-ASBIA-2017_completo.pdf).
The averages of r² considering all SNP pairs spaced up to 50Kb showed agreement with estimates previously reported for taurine breeds (r² ~ 0.25)41,42 and were higher than averages previously reported for indicine and taurine-indicine cross-bred cattle (r² ~ 0.18)41,43. The LD is a decisive parameter to define the required markers density for successful genomic prediction and genome-wide association studies10,44, being suggested that r² values of 0.2 are required to achieve high prediction accuracies (>0.8)45. Considering the bovine genome size of 2,875 Mb, and the averages of r² higher than 0.2 for adjacent SNPs spaced up to 100Kb in both Angus subpopulations, the minimum genotyping density required to these subpopulations would be 28,750 SNPs. The distribution of 31,483 SNPs common between two different commercial arrays maintained some gaps longer than 100Kb along chromosomes. Thus, the imputation of genotypes to the non-common SNPs would be a feasible strategy to enhance the marker density in both subpopulations, fulfilling long gaps and assuring great prediction accuracies in further studies. Furthermore, a single reference population genotyped with a higher density of markers could supply reference haplotypes to both subpopulations, due to the strong preserved phase between these two subpopulations.
The persistence of phase across populations may indicate allele-QTL preserved phases, being another very important issue to joint-genomic analysis. Porto-Neto et al.46 reported considerable gains in genomic prediction accuracies in five cattle traits using only SNPs of consistent linkage phase between two distinct populations. The linkage phase correlation between the Brazilian and Canadian subpopulations stayed high for genomic distances extended to 5 Mb, corresponding well with estimates of approximately 0.7 reported for subpopulations of a single breed42. This persistence was significantly higher than estimates involving different breeds (approximately 0.45)41. Therefore, combining the two subpopulations studied here to perform genomic prediction would result in gains in reliability, as it would enable increased reference population without the effect of SNPs in reversed phase masking the marker-QTL association.
The suitability of ROH islands for the selection signature scanning was reinforced here, due to several overlaps with QTL of traits undergoing selection and previously reported selection signatures of beef cattle. The two ROH islands found common between the Brazilian and Canadian samples suggested common selection events on BTA13 and BTA14, at genomic regions harboring genes known to be associated with stature in mammals19,47,48, thus putatively influential on growth traits undergoing selection in both subpopulations. These two signals were previously reported as selection signatures of Angus49 and were significantly associated with the body weight of Brahman50, in a study based on the reference UMD3.1. Reasonable candidate genes of BTA13 includes the GDF5 gene, which belongs to a family of bone morphogenetic proteins that stimulate bone formation and regulate growth51, and MHY7B that is expressed in the skeletal muscle and downregulated in double-muscled breeds20. The region of BTA14 comprising PLAG1 and XKR4 genes is well-known for its pleiotropic effect on economically important traits of beef cattle, such as backfat thickness, ribeye area, body and carcass weight5254,55, as well as the serum level of the growth-related hormones, IGF153.
An impressive signal of genomic differentiation between the subpopulations was identified at BTA7, corresponding to the ROH island exclusive of the Canadian subpopulation that partially coincided with FST results. The rs110428791 SNP, which is an intronic variant of gene UNC5A mapped at this selection signature, was previously associated with reproductive performance in taurine breeds, in a study based on the reference genome UMD3.122. In addition, homologous genes of RNF44 (ring finger protein 44), and UIMC1 (ubiquitin interaction motif containing 1), have been associated with reproductive risks and duration of reproductive life in women56,57. These findings suggest selection pressures in Canadian subpopulation with a balance between growth and reproductive traits.
The signal of divergent selection detected at BTA7:21.31–21.89 is most likely related to differences in the ability of quicker and strong response towards pathogen invasions across the subpopulations raised in different environments. This genomic region was previously identified as a QTL associated with resistance to viral load in Holstein cattle58, and it comprises genes that showed differential expression between susceptible and resistant hosts to cattle tick (KIF3A)25, mange infested and uninfested animals (IL4, IL5 and IL13)59, healthy and with mastitis cows (IL4)60. In addition, this selection signature was associated with the adaptation of taurine breeds to harsh environments61 and of indicine breeds to not so harsh, but still challenging environmental conditions62.
Two highlighted regions of BTA18 overlapped with selection signatures previously reported in Russian taurine breeds adapted to harsh weather61 and harbored candidate genes to immune and thermoregulatory efficiency. It is noteworthy that the longest selection signature (BTA18:13.76–15.40), encompassed the major gene for coat color variation in Angus, MC1R28. We were able to refuse the likelihood of this signal being related to the presence of red Angus samples (recessive homozygous) in the genotyped Brazilian subpopulation. However, we didn’t estimate frequencies of recessive allele among the samples of black coat color, since our genotyping information did not include the causal mutation for pigmentation in Angus, rs10968801329. Nevertheless, none of the two countries perform selection based on coat colors. Thus, it is not discarded the hypothesis of this signal being caused by other genes in this region, related to better performances on selected traits under challenging environmental conditions. Furthermore, this region has been indicated as QTL of resistance to infection in dairy cattle32,63, and revealed as a selection signature of cattle breeds with coat color phenotypes not determined by MC1R, such as Brown Swiss, Hanwoo and Nguni33,64,65. Further investigation adopting denser panels or even sequencing data could narrow down this region and empower the identification of the causal mutation(s) behind this signal.
Despite the potential of parallel selection in promoting differentiation of geographically isolated subpopulations of the same breed13, the Angus subpopulation from Canada and Brazil still present substantial genomic connection likely due to common ancestry (mainly influenced by the use of semen from common sires) and similarity between the selection schemes, primarily focused on growth-related traits in both countries7,66. Nevertheless, signatures of divergent selection pinpointed particular loci, related to reproductive performance, thermoregulation, and resistance to infectious diseases. Therefore, specific population subdivision might have been caused by both adaptation to distinct environments and particularities of selection criteria applied in each country, such as an additional emphasis on breeding indexes that combines fertility and growth traits in Canada67,68,69.
Methods
Genotyping information
Care and Use Committee approval was not required for the current study since it was performed with previously genotyped samples.
The genotypic data consisted of 382 and 566 Angus born and raised in Canada and Brazil, respectively. The Canadian subpopulation, previously described in Chen et al.70, consisted of steers born at the Onefour Research Substation of the Agriculture and Agri-Food Canada Research Centre (AAFC) at Lethbridge - AB, from 2004 to 2008. These samples were genotyped for approximately 54 K SNPs included in the BovineSNP50 (Illumina, San Diego, CA) panel. The Brazilian subpopulation comprised 352 heifers born in 2013 at Santa Helena farm at Uruguaiana - RS, and 214 influential Angus sires of Brazilian herds. Brazilian samples were genotyped for approximately 139 K SNPs included on GGP-150K - NEOGEN (GeneSeek, Lincoln, NE) panel. The genotyped Canadian subpopulation was exclusively composed of black coat color samples, whereas 206 out of 566 Brazilian samples were red Angus.
All the genotyped samples presented call-rate higher than 0.90. The SNPs from both arrays were remapped according to the new bovine assembly, ARS-UCD 1.271, using publicly available coordinates provided at https://www.animalgenome.org/repository/cattle/UMC_bovine_coordinates. SNPs presenting different positions among the different coordinate files aligned into the ARS-UCD 1.2 (N = 12) were filtered out from both arrays during the remapping process. Then, non-autosomal markers, markers presenting call-rate lower than 0.90 or showing a significant deviation from Hardy-Weinberg equilibrium (p < 10−5) were removed in both subpopulations datasets. Imputation of missing genotypes and haplotype phasing was performed per subpopulation using the Fimpute v372. Finally, only non-monomorphic markers, common between the two panels were kept to further analyses (n = 31,483).
Genomic relationships and inbreeding coefficients
Relatedness between all pairs of individuals was estimated using the GRM computed according to method 1 in VanRaden73, with base allele frequency fixed to 0.539,74. Genomic inbreeding coefficients based on GRM (FGRM) were estimated by subtracting one from its diagonal elements. Runs of homozygosity based inbreeding coefficients (FROH) were estimated as the sum of lengths of ROHs of each individual divided by the length of the genome covered by the SNPs75. The ROH discovery was conducted using PLINK v1.976, by sliding a 30 SNPs window in one SNP interval throughout each autosomal chromosome. One heterozygous genotype was permitted per window to account for occasional genotyping errors. The minimum length of an ROH segment was set to 1,000Kb, where a density of at least 1 SNP every 120Kb and no marker-intervals longer than 1,000Kb were required, following the recommended criteria of smaller density genotypes77.
SNP autozygosity scores were calculated per subpopulation and expressed as a proportion of animals in which each SNP appeared in an ROH. Genomic regions harboring at least two adjacent SNPs falling into the 99.9th percentile were assigned as ROH islands. The positions of the first and the last SNP of these regions were assumed as the start and end point of ROH islands.
To assess the influence of ROH detection using a fixed sliding window (approach implemented in PLINK software76) in our results, the ROH detection was repeated with an approach based on run detection in consecutive SNPs, implemented in the package detectRUNS78 of R software v3.4.479. This procedure revealed FROH values highly correlated (0.98) with those estimated in PLINK76, and essentially the same ROH islands (Supplementary Fig. S3, Tables S4).
Population substructure
Principal component analysis was applied to the GRM to investigate the population sub-structure. Additionally, we performed PCA after adding publicly available genotypes of Holstein80 and Nelore81 to compare the stratification level between the subpopulations of a single breed with the stratification across different breeds (Supplementary Fig. S1).
Analyses of linkage disequilibrium
Phased haplotypes were used to estimate the LD extent and persistence of phase, applying publicly available scripts at https:// www.msu.edu/~steibelj/JP_files/LD_estimate.htm. A measurement of LD was estimated for all syntenic SNP pairs by the genotype squared correlation (r2), as proposed by Hill and Robertson82. The decay of LD was then analyzed in each subpopulation using r² averages within bins according to the physical distances of SNP pairs, including 10 Kb intervals starting from 10 to 100 Kb, and the intervals 100–200 Kb, 200–500 Kb and 500–1000 Kb. To assess the effect of MAF on LD, the r2 was calculated with SNPs of minimum MAF higher than 0.01 (N = 30,657) and 0.05 (N = 28,048) in both subpopulations.
The consistency of the linkage phase for pairs of loci across subpopulations was analyzed through genotype correlation (rij). We estimated rij between all pairs of SNPs spaced for shorter intervals than 10 Mb in both subpopulations. Then, the persistence of phase was estimated within intervals of 100Kb, starting from 0.1 to 5 Mb, as Pearson’s correlation of rij for each subpopulation, as follows:
where Rm,n is the correlation of phase between rij in the subpopulation m (Brazilian) and n (Canadian); \({\bar{r}}_{(m)}\) and \({\bar{r}}_{(n)}\) are the averages of rij within interval p in the subpopulation m and n, respectively; and s(m) and s(n) are the standard deviation of rij in the subpopulation m and n, respectively83.
Contrasts between regional patterns of LD of the two subpopulations were performed through the statistic varLD84, implemented in varLD software85. The varLD method consists of building symmetric LD matrices in each subpopulation, with r2 of all pairs of SNPs within windows defined based on a fixed number of markers, sliding in one SNP interval. Next, eigenvalues of each symmetric matrix are computed per subpopulation, and raw varLD scores assigned as the sum of absolute differences between ranked eigenvalues of the homologous matrices. Finally, each raw varLD score is compared against the distribution with all windows values. Since one of the premises of varLD statistic is the establishment of windows encapsulating the same number of SNPs across the genome84, we choose to define windows of 15 SNPs (mean length of 1095.9 ± 389.9 Kb), instead of windows with fixed length size. Genomic regions with at least two consecutive windows falling in the 99.9th percentile of varLD were considered indicative of divergent selection. Preliminary analyses carried out using a range of SNP numbers (15, 20, 25 and 30) yielded consistent results related to the majority of signals, with the shortest boundaries of the detected signals associated with the windows of 15 SNPs (Supplementary Fig. S4).
Allelic frequency differentiation
The proportion of total variance in allele frequencies due to differences between subpopulations was quantified by applying the Wright’s fixation index (FST), estimated according to Cockerham and Weir86 using the “fsthet” package87 in R software, v3.4.479. FST and heterozygosity were averaged in sliding windows set equal to those of varLD procedure (15 SNPs, sliding in one SNP interval). To control false positive discoveries in FST test the empirical thresholds were defined based on the FST-heterozygosity outliers approach87. Windows were grouped into 20 bins according to heterozygosity and genomic regions were considered as putative signals of divergent selection if at least two sequential windows fell into the 99.9th percentile scores in its heterozygosity’s bin. Preliminary analyses carried out using windows defined based on physical size (1000 Kb, 750 Kb of overlapping) and SNP number (15 and 30) yielded consistent results (Supplementary Fig. S5).
Genes and QTL identification
Genomic regions identified into the top 99.9 percentile of more than one of the tests (ROH, varLD, and FST) were considered selection signatures, as they are more likely of being true positive signals16,88. Known QTL overlapping the selection signatures were assessed in the CattleQTL database - release 4089, with QTL positions lifted from the former reference genome UMD3.1.1 to the new reference ARS-UCD 1.2. The Ensembl cow gene - set 9990, which is based on the new ARS-UCD1.2 reference, was used for gene prospection.
Two lists of candidate genes were evaluated as over-representation of biological processes and molecular pathways, using the DAVID v.6.791 and PANTHER v.13.192. One list included 26 genes surrounded by ROH islands common for the two subpopulations and a further list included 73 genes of genomic regions divergently selected (ROH islands exclusively detected in one of the two subpopulations, FST and varLD signals).
Data availability
The datasets used and/or analyzed during the current study are available upon reasonable requests to the corresponding authors.
Change history
08 October 2020
An amendment to this paper has been published and can be accessed via a link at the top of the paper.
References
Buchanan, D. S. & Lenstra, J. A. Breeds of cattle. In The Genetics of Cattle (eds. Garrick, D. J. & Ruvinsky, A.) 641 (2015).
Herring, A. D. North American beef production. In Beef cattle production and trade (ed. Lewis Kahn, D. C.) 574 (2014).
Vasconcellos, L. P. et al. Genetic characterization of Aberdeen Angus cattle using molecular markers. Genet. Mol. Biol. 26, 133–137 (2003).
Canadian Angus Association. Canadian Angus Association - Annual Reports, Available at, https://cdnangus.ca/canadian-angus-tag-beef-program/beefprograms/, (Accessed: 9th March 2020) (2019).
Carruthers, C. R., Plante, Y. & Schmutz, S. M. Comparison of Angus cattle populations using gene variants and microsatellites. Can. J. Anim. Sci. 91, 81–85 (2011).
Index Asbia. Venda de sêmen Angus cresce 28% no Brasil - ASBIA - Associação Brasileira de Inseminação Artificial. Available at, http://www.asbia.org.br/venda-de-semen-angus-cresce-28-no-brasil/, (Accessed: 9th March 2020) (2019).
Cardoso, F. F. & Tempelman, R. J. Linear reaction norm models for genetic merit prediction of Angus cattle under genotype by environment interaction. J. Anim. Sci 90, 2130–2141 (2012).
Schaeffer, L. R. Model for international evaluation of dairy sires. Livest. Prod. Sci. 12, 105–115 (1985).
Schaeffer, L. R. Multiple-Country Comparison of Dairy Sires. J. Dairy Sci. 77, 2671–2678 (1994).
de Roos, A. P. W., Hayes, B. J. & Goddard, M. E. Reliability of Genomic Predictions Across Multiple Populations. Genetics 183, 1545–1553 (2009).
Meyer, K. Estimates of genetic parameters and breeding values for New Zealand and Australian Angus cattle. Aust. J. Agric. Res. 46, 1219 (1995).
de Haas, Y. et al. Improved accuracy of genomic prediction for dry matter intake of dairy cattle from combined European and Australian data sets. J. Dairy Sci. 95, 6103–6112 (2012).
de Roos, A. P. W., Hayes, B. J., Spelman, R. J. & Goddard, M. E. Linkage Disequilibrium and Persistence of Phase in Holstein-Friesian, Jersey and Angus Cattle. Genetics 179, 1503–1512 (2008).
Howard, J. T., Maltecca, C., Haile-Mariam, M., Hayes, B. J. & Pryce, J. E. Characterizing homozygosity across United States, New Zealand and Australian Jersey cow and bull populations. BMC Genomics 16, 187 (2015).
Akey, J. M., Zhang, G., Zhang, K., Jin, L. & Shriver, M. D. Interrogating a high-density SNP map for signatures of natural selection. Genome Res 12, 1805–14 (2002).
Wagh, K. et al. Lactase Persistence and Lipid Pathway Selection in the Maasai. 7, 1–12 (2012).
Randhawa, I. A. S., Khatkar, M. S., Thomson, P. C. & Raadsma, H. W. Composite selection signals can localize the trait specific genomic regions in multi-breed populations of cattle and sheep. BMC Genet. 15, 34 (2014).
Porto Neto, L. R., Bunch, R. J., Harrison, B. E. & Barendse, W. Variation in the XKR4 gene was significantly associated with subcutaneous rump fat thickness in indicine and composite cattle. Anim. Genet. 43, 785–789 (2012).
Utsunomiya, Y. T. et al. A PLAG1 mutation contributed to stature recovery in modern cattle. Sci. Rep 7, 1–15 (2017).
Cassar-Malek, I., Boby, C., Picard, B., Reverter, A. & Hudson, N. J. Molecular regulation of high muscle mass in developing Blonde d’Aquitaine cattle foetuses, https://doi.org/10.1242/bio.024950 (2017).
Jiang, Z. et al. Transcriptional profiles of bovine in vivo pre-implantation development. BMC Genomics 15, 756 (2014).
Höglund, J. K., Sahana, G., Guldbrandtsen, B. & Lund, M. S. Validation of associations for female fertility traits in Nordic Holstein, Nordic Red and Jersey dairy cattle. BMC Genet. 15, 8 (2014).
Lohoff, M. & Mak, T. W. Roles of interferon-regulatory factors in T-helper-cell differentiation. Nature Reviews Immunology 5, 125–135 (2005).
Zhang, R., Chen, K., Peng, L. & Xiong, H. Regulation of T helper cell differentiation by interferon regulatory factor family members. Immunologic Research 54, 169–176 (2012).
Franzin, A. M. et al. Immune and biochemical responses in skin differ between bovine hosts genetically susceptible and resistant to the cattle tick Rhipicephalus microplus. Parasit. Vectors 10, 51 (2017).
Howard, J. T. et al. Beef cattle body temperature during climatic stress: a genome-wide association study. Int. J. Biometeorol. 58, 1665–1672 (2014).
Skibiel, A. L., Zachut, M., do Amaral, B. C., Levin, Y. & Dahl, G. E. Liver proteomic analysis of postpartum Holstein cows exposed to heat stress or cooling conditions during the dry period. J. Dairy Sci. 101, 705–716 (2018).
Olson, T. A. Genetics of colour variation. in Genetics of Cattle (eds. Fries, R. F. & Ruvinsky, A.) 33–53 (CABI Publishing, 1999).
Boitard, S., Boussaha, M., Capitan, A., Rocha, D. & Servin, B. Uncovering Adaptation from Sequence Data: Lessons from Genome Resequencing of Four Cattle Breeds. Genetics 203, 433–450 (2016).
Sonna, L. A., Fujita, J., Gaffin, S. L. & Lilly, C. M. Highlighted topics Molecular Biology of Thermoregulation Invited Review: Effects of heat and cold stress on mammalian gene expression, https://doi.org/10.1152/japplphysiol.
Adams, N. M. et al. Transcription Factor IRF8 Orchestrates the Adaptive Natural Killer Cell Response Optimal NK cell proliferation & viral control Article Transcription Factor IRF8 Orchestrates the Adaptive Natural Killer Cell Response. Immunity 48, 1172–1182 (2018).
Chen, X., Cheng, Z., Zhang, S., Werling, D. & Wathes, D. C. Combining Genome Wide Association Studies and Differential Gene Expression Data Analyses Identifies Candidate Genes Affecting Mastitis Caused by Two Different Pathogens in the Dairy Cow. Open J. Anim. Sci. 05, 358–393 (2015).
Makina, S. O. et al. Genome-wide scan for selection signatures in six cattle breeds in South Africa. Genet. Sel. Evol. 47, 1–14 (2015).
Kelleher, M. M. et al. Inference of population structure of purebred dairy and beef cattle using high-density genotype data. animal 11, 15–23 (2017).
Vitti, J. J., Grossman, S. R. & Sabeti, P. C. Detecting natural selection in genomic data. Annu. Rev. Genet. 47, 97–120 (2013).
Signer-Hasler, H. et al. Population structure and genomic inbreeding in nine Swiss dairy cattle populations. Genet. Sel. Evol. 49, 83 (2017).
Edea, Z. et al. Genome-wide scan reveals divergent selection among taurine and zebu cattle populations from different regions. Anim. Genet., https://doi.org/10.1111/age.12724 (2018).
Forutan, M. et al. Inbreeding and runs of homozygosity before and after genomic selection in North American Holstein cattle. BMC Genomics 19, 98 (2018).
Bjelland, D. W., Weigel, K. A., Vukasinovic, N. & Nkrumah, J. D. Evaluation of inbreeding depression in Holstein cattle using whole-genome SNP markers and alternative measures of genomic inbreeding. J. Dairy Sci. 96, 4697–4706 (2013).
Howrigan, D. P., Simonson, M. A. & Keller, M. C. Detecting autozygosity through runs of homozygosity: A comparison of three autozygosity detection algorithms. BMC Genomics 12, 460 (2011).
Makina, S. O. et al. Extent of Linkage Disequilibrium and Effective Population Size in Four South African Sanga Cattle Breeds. Front. Genet 6, 337 (2015).
Biegelmeyer, P., Gulias-Gomes, C. C., Caetano, A. R., Steibel, J. P. & Cardoso, F. F. Linkage disequilibrium, persistence of phase and effective population size estimates in Hereford and Braford cattle. BMC Genet. 17, 32 (2016).
Espigolan, R. et al. Study of whole genome linkage disequilibrium in Nellore cattle. BMC Genomics 14, 305 (2013).
McKay, S. D. et al. Whole genome linkage disequilibrium maps in cattle. BMC Genet. 8, 74 (2007).
Hayes, B. J., Bowman, P. J., Chamberlain, A. J. & Goddard, M. E. Invited review: Genomic selection in dairy cattle: Progress and challenges. J. Dairy Sci. 92, 433–443.
Porto-Neto, L. R. et al. Genomic correlation: harnessing the benefit of combining two unrelated populations for genomic selection. Genet. Sel. Evol. 47, 84 (2015).
Bouwman, A. C. et al. Meta-analysis of genome-wide association studies for cattle stature identifies common genes that regulate body size in mammals. Nat. Genet. 50, 362–367 (2018).
Sanna, S. et al. Common variants in the GDF5-UQCC region are associated with variation in human height. Nat. Genet. 40, 198–203 (2008).
Zhao, F., McParland, S., Kearney, F., Du, L. & Berry, D. P. Detection of selection signatures in dairy and beef cattle using high-density genomic information. Genet. Sel. Evol. 47, 49 (2015).
Porto-Neto, L. R. et al. Genome-wide association for the outcome of fixed-time artificial insemination of Brahman heifers in Northern Australia. J. Anim. Sci 93, 5119–5127 (2015).
Van Der Eerden, B. C. J., Karperien, M. & Wit, J. M. Systemic and Local Regulation of the Growth Plate. Endocrine Reviews 24, 782–801 (2003).
Bolormaa, S. et al. A genome-wide association study of meat and carcass traits in Australian cattle. J. Anim. Sci 89, 2297–309 (2011).
Fortes, M. R. S. et al. Evidence for pleiotropism and recent selection in the PLAG1 region in Australian Beef cattle. Anim. Genet. 44, 636–47 (2013).
Fernandes Júnior, G. A. et al. Genome scan for postmortem carcass traits in Nellore cattle. J. Anim. Sci 94, 4087 (2016).
Cardoso, D. F. et al. Genome-wide scan reveals population stratification and footprints of recent selection in Nelore cattle. Genet. Sel. Evol. 50 (2018).
Day, F. R. et al. Large-scale genomic analyses link reproductive aging to hypothalamic signaling, breast cancer susceptibility and BRCA1-mediated DNA repair. Nat. Genet. 47, 1294–1303 (2015).
Stolk, L. et al. Meta-analyses identify 13 loci associated with age at menopause and highlight DNA repair and immune pathways. Nat. Genet. 44, 260–8 (2012).
Pant, S. D. et al. A principal component regression based genome wide analysis approach reveals the presence of a novel QTL on BTA7 for MAP resistance in holstein cattle. Genomics 95, 176–182 (2010).
Sarre, C. et al. Comparative immune responses against Psoroptes ovis in two cattle breeds with different susceptibility to mange. Vet. Res. 46, 131 (2015).
Bochniarz, M., Zdzisińska, B., Wawron, W., Szczubiał, M. & Dąbrowski, R. Milk and serum IL-4, IL-6, IL-10, and amyloid A concentrations in cows with subclinical mastitis caused by coagulase-negative staphylococci. J. Dairy Sci. 100, 9674–9680 (2017).
Yurchenko, A. A. et al. Scans for signatures of selection in Russian cattle breed genomes reveal new candidate genes for environmental adaptation and acclimation. Sci. Rep 8, 12984 (2018).
Carvalheiro, R. et al. Unraveling genetic sensitivity of beef cattle to environmental variation under tropical conditions. Genet. Sel. Evol. 51, 29 (2019).
Wijga, S. et al. Genomic associations with somatic cell score in first-lactation Holstein cows. J. Dairy Sci. 95, 899–908 (2012).
Porto-Neto, L. R. et al. Genomic divergence of zebu and taurine cattle identified through high-density SNP genotyping. BMC Genomics 14, 876 (2013).
Ramey, H. R. et al. Detection of selective sweeps in cattle using genome-wide SNP data. BMC Genomics 14, 382 (2013).
Lu, D. et al. Linkage disequilibrium in Angus, Charolais, and Crossbred beef cattle, https://doi.org/10.3389/fgene.2012.00152 (2012).
Cole, J. B. & VanRaden, P. M. Symposium review: Possibilities in an age of genomics: The future of selection indices1. J. Dairy Sci. 101, 3686–3701 (2018).
Spangler, M. Applied Reproductive Strategies in Beef Cattle (2016).
Campos, G. S. et al. Bioeconomic model and selection indices in Aberdeen Angus cattle. J. Anim. Breed. Genet. 131, 305–312 (2014).
Chen, L., Schenkel, F., Vinsky, M., Crews, D. H. & Li, C. Accuracy of predicting genomic breeding values for residual feed intake in Angus and Charolais beef cattle1. J. Anim. Sci 91, 4669–4678 (2013).
Rosen, B. D. et al. De novo assembly of the cattle reference genome with single-molecule sequencing. GigaScience 9, 3 (2020)
Sargolzaei, M., Chesnais, J. P. & Schenkel, F. S. A new approach for efficient genotype imputation using information from relatives. BMC Genomics 15, 478 (2014).
VanRaden, P. M. Efficient methods to compute genomic predictions. J. Dairy Sci. 91, 4414–23 (2008).
VanRaden, P. M., Olson, K. M., Wiggans, G. R., Cole, J. B. & Tooker, M. E. Genomic inbreeding and relationships among Holsteins, Jerseys, and Brown Swiss. J. Dairy Sci. 94, 5673–5682 (2011).
McQuillan, R. et al. Runs of Homozygosity in European Populations. Am. J. Hum. Genet. 83, 359–372 (2008).
Chang, C. C. et al. Second-generation PLINK: rising to the challenge of larger and richer datasets. Gigascience 4, 7 (2015).
Purfield, D. C., Berry, D. P., Mcparland, S. & Bradley, D. G. Runs of homozygosity and population history in cattle. BMC Genet. 13, 1 (2012).
Biscarini, F., Cozzi, P., Gaspa, G. & Marras, G. detectRUNS: an R package to detect runs of homozygosity and heterozygosity in diploid genomes. (2019).
R Core Team (2013). R: A language and environment for statistical computing. R Foundation for Statistical Computing; Vienna; Austria. URL, http://www.R-project.org/.
Sánchez-Molano, E., Bay, V., Smith, R. F., Oikonomou, G. & Banos, G. Quantitative Trait Loci Mapping for Lameness Associated Phenotypes in Holstein–Friesian Dairy Cattle. Front. Genet. 10 (2019).
Nascimento, A. V. D. et al. Genome-wide association study using haplotype alleles for the evaluation of reproductive traits in Nelore cattle. PLoS One 13, e0201876 (2018).
Hill, W. G. & Robertson, A. Linkage disequilibrium in finite populations. Theor. Appl. Genet. 38, 226–31 (1968).
Badke, Y. M., Bates, R. O., Ernst, C. W., Schwab, C. & Steibel, J. P. Estimation of linkage disequilibrium in four US pig breeds. BMC Genomics 13, 24 (2012).
Teo, Y. Y. et al. Genome-wide comparisons of variation in linkage disequilibrium. Genome Res. 19, 1849–1860 (2009).
Ong, R. T. H. & Teo, Y. Y. varLD: a program for quantifying variation in linkage disequilibrium patterns between populations. Bioinformatics 26, 1269–1270 (2010).
Cockerham, C. C. & Weir, B. S. Estimation of gene flow from F -statistics. Evolution (N. Y) 47, 855–863 (1993).
Flanagan, S. P. & Jones, A. G. Constraints on the FST–Heterozygosity Outlier Approach. J. Hered 108, 561–573 (2017).
Simianer, H., Ma, Y. & Qanbari, S. Statistical Problems in Livestock Population Genomics. In Proceedings of the World Congress on Genetics Applied to Livestock Production 202 (World Congress on Genetics Applied to Livestock Production, 2014).
Hu, Z.-L., Park, C. A. & Reecy, J. M. Building a livestock genetic and genomic information knowledgebase through integrative developments of Animal QTLdb and CorrDB. Nucleic Acids Res 47, D701–D710 (2019).
Cunningham, F. et al. Ensembl 2019. Nucleic Acids Res 47, D745–D751 (2019).
Huang, D. W., Sherman, B. T. & Lempicki, R. A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 4, 44–57 (2009).
Mi, H. & Thomas, P. PANTHER Pathway: An Ontology-Based Pathway Database Coupled with Data Analysis Tools. in. Methods in molecular biology (Clifton, N.J.) 563, 123–140 (2009).
Acknowledgements
The authors thank the first author grant #2016/22490-8, Sao Paulo Research Foundation (FAPESP), the support from AAFC Livestock Genetics & Genomics Program funds, AAFC A-base peer-reviewed research projects (RBPI-1139 and RBPI-1752) and the BCRC Beef Cluster project (FDE.05.09) for the collection of Canadian Angus data, and the support from CNPq Universal: 478780/2013-3, 480086/2013-3 and 474829/2013-8 for the collection of Brazilian Angus data. Also, thank the MSc. André V. do Nascimento and Camila Rosenberg for a critical review of the manuscript.
Author information
Authors and Affiliations
Contributions
L.G.A., H.T., R.C. and R.V.V. conceived and guided the study. C.L., L.G.A., R.C. and H.N.O. accomplished funding acquisition. D.F.C., D.C.B.S. and G.A.F.J.. performed the formal analysis. A.A.C.A., A.F.B.M., L.R.P.-N., C.L., T.B. and M.C.S.O. supported the data analysis and contributed to the interpretation of the results. D.F.C. composed the original draft. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Cardoso, D.F., Fernandes Júnior, G.A., Scalez, D.C.B. et al. Uncovering Sub-Structure and Genomic Profiles in Across-Countries Subpopulations of Angus Cattle. Sci Rep 10, 8770 (2020). https://doi.org/10.1038/s41598-020-65565-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-65565-1
This article is cited by
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
