
Abstract
The nuclear-encoded DAG-like (DAL) gene family plays critical roles in organelle C-to-U RNA editing in Arabidopsis thaliana. However, the origin, diversification and functional divergence of DAL genes remain unclear. Here, we analyzed the genomes of diverse plant species and found that: DAL genes are specific to spermatophytes, all DAL genes share a conserved gene structure and protein similarity with the inhibitor I9 domain of subtilisin genes found in ferns and mosses, suggesting that DAL genes likely arose from I9-containing proproteases via exon shuffling. Based on phylogenetic inference, DAL genes can be divided into five subfamilies, each composed of putatively orthologous and paralogous genes from different species, suggesting that all DAL genes originated from a common ancestor in early seed plants. Significant type I functional divergence was observed in 6 of 10 pairwise comparisons, indicating that shifting functional constraints have contributed to the evolution of DAL genes. This inference is supported by the finding that functionally divergent amino acids between subfamilies are predominantly located in the DAL domain, a critical part of the RNA editosome. Overall, these findings shed light on the origin of DAL genes in spermatophytes and outline functionally important residues involved in the complexity of the RNA editosome.
Similar content being viewed by others

Introduction
C-to-U RNA editing (deamination of cytidine to uridine) is an essential step of RNA maturation in chloroplasts and mitochondria of land plants from bryophytes to angiosperms1, 2. U-to-C RNA editing is also observed in ferns and mosses3, 4. More than 400 editing sites in mitochondria and 30–40 editing events in chloroplasts are typically found in flowering plants5,6,7. RNA editing occurring in plant organelle mRNAs can restore functionally conserved amino acids at the post-transcriptional level, create functional proteins and play important roles in efficient splicing of introns and processing of precursor tRNA molecules7,8,9. In plants, some mutants with impaired RNA editing at specific nucleotide sites cause deleterious phenotypes and even lethality. The site specificity of the cytidines to be edited in a plant organelle is determined by a crucial cis-acting regulatory sequence10,11,12,13,14 and the RNA editosome that will bind to it. The RNA editosome is composed of nuclear-encoded trans-acting factors that recognize the cis-element and perform RNA editing15. Recent extensive genetics studies have revealed that these trans-factors enlisted in the RNA editosome include DYW-type pentatricopeptide repeat (PPR) proteins1, RNA-Editing Factor Interacting Protein (RIP) family or Multiple Organelle RNA Editing Factor (MORF) family proteins16, 17, RNA-recognition motif (RRM)-containing proteins18,19,20, protoporphyrinogen IX oxidase 1 (PPO1)21 and organelle zinc-finger 1 (OZ1)22. PPR proteins are characterized by tandem 35-amino acid PPR motifs23. The DYW-PPR proteins each recognize one or a few editing sites that have similar cis-elements and thereby bind directly to the cis-acting sequences. The DYW domain of DYW-PPR proteins has a sequence similar to the active sites of known cytidine deaminases and editing enzymes24, and may be responsible for deamination of cytidine to uridine.
The RIP/MORF gene family, which controls multiple organelle RNA editing sites, was identified in Arabidopsis thaliana and designated the RIP gene family and the MORF gene family by two research groups16, 17. Here, we adopt the name, the DAG-like (DAL) gene family, based on the first identified member (DAG) of the gene family in Antirrhinum majus 25, 26. Arabidopsis DAL proteins are all targeted to mitochondria or chloroplasts and required for RNA editing at all sites in both organelles16, 17. The mutation of DAL genes in plants results in abnormal development of plants, even lethality16, 17, 25,26,27. Yeast two-hybrid analysis confirmed that DAL/RIP/MORF proteins can interact selectively with diverse PPR proteins by the binding of the DAL domain to PPR motifs17, and moreover, DAL proteins can connect to form hetero- and homodimers16. A variation of the DAL gene (ORRM1) was identified and functionally analyzed; it harbors a pair of truncated RIP domains (RIP-RIP) at its N terminus and an RRM domain at its C terminus28. ORRM1 is an essential plastid editing factor that can interact selectively with PPR proteins via its RIP-RIP domain, and the ORRM1 protein can also bind to sequences near at least some of its RNA targets in vitro 28. Furthermore, the RRM domain can rescue the editing defect in orrm1 protoplasts independent of RIP domains, and three other RRM-containing proteins were identified because of their roles in organelle RNA editing, suggesting that the RRM domain participates in the RNA editosome18,19,20. Together, DAL proteins may be connectors between the site-specific PPRs and the as-yet-unknown deaminase or other components in the RNA editosome, such as RRM-containing proteins, PPO121 and OZ122.
Compared with the RNA editosomes responsible for C-to-U or A-to-I (deamination of adenosine to inosine) RNA editing in mammals, the plant organelle RNA editosomes have more diverse components15. In addition to the interpretation that more RNA-edited sites in plant organelles require more trans-acting editing factors, the diverse composition of the organelle RNA editosome in plants probably overcomes the deficiency in RNA editing caused by the mutation of PPR protein or changes in the cis-acting sequences of edited sites15, especially for those edited sites in plant mitochondrial genomes which evolve much more quickly. Thus, the origin, classification and evolution analysis of trans-acting factors is important for understanding the evolution and molecular mechanism of the RNA editosome in plants. In the RNA editosome, DYW-PPR genes undergo purifying selection at sites targeted for RNA editing because they are important for recognizing cis-element sequences1, 29, 30. However, the functional evolution and origin of the DAL gene family is unknown.
In this study, we identified the DAL proteins in various plant lineages, including green algae, moss, ferns, gymnosperms and flowering plants, to investigate functional divergence and origin of the DAL gene family in plants. The result indicated DAL genes are specific to spermatophytes other than to lower plants. Plant DAL genes shared a strong conserved gene structure and appear to have evolved from the I9-containing proprotease via exon shuffling. Functional divergence analysis revealed that there was significant functional divergence between different DAL clades which may be associated with differences in the roles different DAL genes play in RNA editing and RNA metabolism. The evolutionary and functional divergence analysis of the DAL genes in plants presented here provides useful information for further probing the molecular mechanism by which DAL proteins contribute to the RNA editosome.
Results
Identification and sequence analysis of DAL genes in maize
To identify putative DAL genes in the maize genome, we searched the maize genome annotation data with known plant DAL proteins as a query. In total, we obtained 7 putative DAL genes in maize named ZmDAL1—ZmDAL7 based on their order on the chromosomes (Fig. 1a and Supplementary Table S1). ZmDALs were distributed on 5 of 10 maize chromosomes, and chromosomes 9 and 10 both had two ZmDAL genes (Supplementary Fig. S1). The gene model of ZmDAL1 was reannotated correctly by analyzing the similarity between ZmDAL genes and their orthologs (Supplementary Fig. S2). The veracity of each gene model of ZmDAL genes was assessed using reverse transcription polymerase chain reaction (RT-PCR) assays with the gene-specific primers listed in Supplementary Table S2, as 4 of 7 ZmDAL genes had more than one transcript for each ZmDAL gene in the MaizeGDB database (http://www.maizegdb.org/). The RT-PCR results indicated that seven ZmDAL genes were expressed in maize seedlings and only a single transcript was found for each ZmDAL gene (Supplementary Fig. S3). All identified maize DAL genes encoded proteins ranging from 215 (ZmDAL1) to 412 amino acids (aa) (ZmDAL2), and their isoelectric points (Ip) were similar (>8.0).

Maize DAL proteins and their conserved motifs. (a) Multiple sequence alignment of maize DAL proteins was carried out using ClustalW2.0, and the NJ tree was built using MEGA v5.0. The chloroplast and mitochondrial transit peptides of maize DAL proteins were predicted using Predotar (http://urgi.versailles.inra.fr/predotar/predotar.html) and (/) TargetP (http://www.cbs.dtu.dk/services/TargetP/). M, mitochondria; C, chloroplast. The DAL proteins marked by an asterisk were observed in the plastid proteome. (b) Alignment of conserved DAL domains in maize DAL proteins was conducted using ClustalW2.0 and displayed with GeneDoc. The secondary structure of the DAL domain was inferred using MINNOU (http://minnou.cchmc.org/). (c,d) Putative motifs were explored using the MEME server with the parameters of between 10 and 50 aa in length and sharing of each motif among all ZmDAL proteins.
No known motif was found in the maize DAL proteins by screening the PFAM and INTERPRO databases, except the MORF box (called the DAL domain in this study) which had been identified previously16, 17. Novel putative motifs were explored using the MEME server with different motif lengths. By selecting a motif length between 10 and 50 aa, we identified 4 conserved motifs, and all 4 motifs were located in the DAL domain (Fig. 1b,c,d), suggesting that the DAL domain is a conserved sequence among Arabidopsis and maize DAL proteins. To obtain an intact motif containing the DAL domain, we enlarged the MEME motif length and identified one motif containing 114 aa (Supplementary Fig. S4). Like their homologs in Arabidopsis 16, maize DAL proteins were predicted using TargetP and Predotar to enter mitochondria or chloroplasts. Of them ZmDAL1 and ZmDAL6 were also detected in the plastid nucleoid proteome by searching the maize organelle proteomics database (PPDB, http://ppdb.tc.cornell.edu/) (Fig. 1a).
The gene structures of the ZmDAL genes were constructed by aligning the extracted genomic sequences to predicted cDNA sequences of maize DAL genes. This showed that ZmDAL genes have a conserved gene structure (Fig. 2); each of the ZmDAL genes has 3 introns with the intron phases 2, 1 and 1 separating DAL domain-encoding exons 1, 2, 3 and 4 (Fig. 2b). Motifs 1 and 2 are encoded by exon 1; motif 3 is encoded by exons 2 and 3; and motif 4 is located in exon 4 (Fig. 2c and d). Furthermore, the length of exons 2 (98 basepairs, bp) and 3 (66 bp) is conserved among all five ZmDAL genes, even though the size of the introns between the exons varies between different ZmDAL genes.

Conserved gene structures of maize DAL genes. The gene structures of ZmDAL genes were built using GSDraw (http://wheat.pw.usda.gov/piece/GSDraw.php) through both alignment of DNA obtained from MaizeGDB (http://www.maizegdb.org/) and coding sequences (CDS) of ZmDAL genes.
Identification and phylogenetic analysis of plant DAL genes
To mine more DAL domain-encoding genes in plants, we used the HMMER 3.0 package31 to build a hidden Markov model (HMM) file (dal.hmm, Supplementary Data File S1) with 17 DAL domain sequences of those DAL proteins from A. majus, maize and Arabidopsis (Supplemental Data File S2). We then used the dal.hmm algorithm to query the genomes of a variety of plants representing the major evolutionary lineages, including Chlamydomonas reinhardtii, Physcomitrella patens, Selaginella moellendorffii, Picea abies, Brachypodium distachyon, Oryza sativa Japonica, Zea mays, Sorghum bicolor, Aquilegia coerulea, Vitis vinifera, A. thaliana, Arabidopsis lyrata and Populus trichocarpa. The result showed that putative DAL genes were only identified in seed plants but not in lower plants (C. reinhardtii, P. patens and S. moellendorffii) (Fig. 3). The numbers of DAL genes of higher plants used here are comparable, ranging from 6 (in A. coerulea) to 11 (in A. lyrata). In total, 79 DAL genes were identified in 10 plant genomes (Supplementary Table S3). In addition, we identified ORRM1-like genes in this study that were also specific to seed plants, and these genes encoded two tandem truncated DAL domains at the N terminus and one RNA recognition motif (RRM) at the C terminus, except the MA_10436715g0010 protein found in P. abies, which had no C-terminal RRM domain (Supplementary Fig. S5).

Distribution of DAL genes in the plant kingdom. (a) A schematic diagram of plant evolution tree was constructed according to the plant tree shown in PGDD (http://chibba.agtec.uga.edu/duplication/). (b) RNA editing sites (organelle C-to-U RNA editing), DYW-type PPR genes, DAG-like genes, ORRM genes, and Inhibitor I9 genes were identified in the plants listed on the left. The checkmark in the box denotes that the above genes can be found in the corresponding genomes, and the cross in the box indicates none of above genes are found in these genomes.
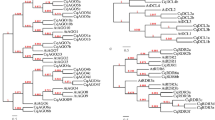
To investigate the phylogenetic relationship among plant DAL genes, an unrooted neighbor-joining (NJ) tree containing all 79 DAL proteins was generated based on the conserved DAL domain alignment (Figs 4 and S5). On the basis of the phylogeny, the DAL gene family in plants was subdivided into five groups, named group I to group V (Fig. 4). In the NJ tree shown in Fig. 4, DAL genes of each group were all from diverse plant species. In groups I, II and V, species-specific gene duplication events occurred after the lineages diverged, resulting in the inclusion of more than one DAL gene per species (Fig. 4). Since the DAL genes were found to be specific to spermatophytes, we can infer that the ancestral DAL gene appeared after the divergence of seed plants and ferns.

Phylogenetic relationship of plant DAL proteins. The neighbor-joining (NJ) phylogenetic tree of 79 plant DAL genes was constructed based on the multiple protein sequence alignment of the conserved regions (Supplemental Data File S3) according to the Poisson model. Bootstrap values >60% are indicated at each node, and the scale bar denotes the substitute rate per site. The species names are displayed before plant DAL genes.
The exon-intron organization analysis of 79 plant DAL genes indicated that plant DAL genes all share a conserved gene structure, with the 2-1-1 intron phase pattern separating DAL domain-encoding exons, as observed in maize DAL genes, except Al_477997 and MA_489006g0010, which have intron phase patterns 0-1-1 and 0-2-2, respectively (Supplementary Fig. S6).
The source of plant DAL genes
Putative genes or gene fragments homologous to DAL genes were identified in lower plants to identify the origin of DAL genes in higher plants by lowering the HMMER search threshold (E-value of full sequence <0.01). Peptidase S8 propeptide/proteinase inhibitor I9 domain of subtilisins were identified as putative homologs of DAL proteins in P. patens and S. moellendorffii but not in C. reinhardtii (Supplementary Table S4). The proteinase inhibitor I9 domain is the propeptide of the serine peptidase family S8A (subtilisin family) and is responsible for the modulation of folding and activity of these proenzymes32. In addition to the protein similarity of the inhibitor I9 domain and the DAL domain, inhibitor I9 domain-encoding genes or gene fragments have conserved gene structure with DAL genes, including the 2-1-1 intron phase pattern and the 98-bp exon (Figs 5 and S7), which suggests that DAL genes probably originated from inhibitor I9 domain-encoding DNA sequences. The combination of inhibitor I9 domain-encoded exons and other exons, such as RRM-encoding exons, could be responsible for the appearance of DAL genes and ORRM1-like genes in higher plants.

Alignment of inhibitor I9 domains and the DAL domain. A multiple protein sequence alignment was performed using hmmalign in the HMMER 3.0 package28 and was displayed using GeneDoc (http://www.nrbsc.org/gfx/genedoc/ebinet.htm). The DAL domain of the ZmDAL6 protein was used to represent plant DAL domains. The codons harboring intron splicing sites denoted by red dashed lines were from Pp1s121_135V6.4 (the upper ones) and ZmDAL6 (the basal ones).
Functional divergence evaluation between plant DAL subfamilies
As reported previously, Arabidopsis DAL genes play inequable roles in RNA editing for different RNA sites by binding diverse DYW-PPR proteins27. To investigate the different functional constraints between these members, we conducted a maximum likelihood test of functional divergence using DIVERGE v3.033. The unrooted NJ tree was generated with complete amino acid sequences of plant DAL proteins excluding those of P. abies and V. vinifera (Supplementary Fig. S8). Two types of functional divergence (type I and type II) between gene clusters of the DAL gene family in flowering plants were examined. The theta (θ) ML values were calculated, and the results demonstrated that the coefficients of type I (θ I) for 6 of 10 pairwise comparisons between DAL subfamilies were significantly greater than zero (Bonferroni corrected P < 0.05), and only one pairwise subfamily comparisons showed significant divergence with the coefficients of type II (θ II) test (Bonferroni corrected P < 0.05) (Table 1). Functional divergence-related sites were identified based on the posterior probabilities with a cut-off value of 0.85, and most were located in the DAL domain (Supplementary Fig. S9). These observations indicate that there were significant site-specific shifted selective constraints on most members of the DAL gene family. Furthermore, we also observed that the values of θ I were much larger than the estimates of θ II in each pairwise comparison (Table 1), indicating that type I functional divergence predominantly contributed to the diversified evolution of plant DAL genes. In addition, we checked the functional divergence of intragroup members of the DAL genes, such as Sub.Ia vs. Sub.Ib (Supplementary Fig. S8), but there was no significant functional divergence in any intragroup comparison (Supplementary Table S5), suggesting that intragroup DAL genes might play similar conserved roles in different plant lineages.
GC content of DAL genes in monocots and dicots
The GC content, an important genomic feature, plays a critical role in determining the physical properties of DNA molecules and genome regulation by providing substrates for DNA methylation34. The base composition analysis of plant DAL genes revealed that the DAL genes of monocots (grass) have a higher GC content than those of dicots (Fig. 6a). To investigate DNA methylation of the GC-rich DAL genes, CpG islands of ZmDAL6 and At1g11430 as representatives were predicted, and there were two CpG island regions identified in ZmDAL6 but not in At1g11430 (Fig. 6b). We analyzed the DNA methylation of the first CpG island of ZmDAL6, which was located at the exon 1-harboring region, using bisulfite sequencing and observed that 7 cytosines within a 19-bp region (nucleotides 1,222–1,240 of the ZmDAL6 DNA sequence in Fig. 6c) were methylated, including three CHH sites, two CHG sites and one CG site (Fig. 6c). In the 607-bp CpG island, however, most cytosines were not substantially modified by DNA methylation. Moreover, the codon base composition of the DAL genes was analyzed, and the result showed that the GC content of each base of one codon in monocot DAL genes was higher than that of dicot DAL genes (Student t-test, P < 0.01). For each group, only the codon second base (GC2) of group III DAL genes showed no significant difference in GC content between dicots and monocots. The largest difference of GC content was found in the codon third base (GC3) of groups I and IV between dicots and monocots (Fig. 6d), and in these two groups, more than 75% of the monocot DAL GC3 bases were guanine or cytosine nucleotides. Given that no significant functional divergence was observed between intragroup DAL genes, we inferred that the higher GC content of DAL genes in monocots could be caused by GC-biased gene conversion because the base composition of DAL genes was consistent with the high GC content of monocot genomes34,35,36.

GC content and DNA methylation analysis of DAL genes in higher plants. (a,b) GC content and CpG islands of plant DAL genes were identified and displayed using CpGplot of EMBOSS (http://www.ebi.ac.uk/Tools/emboss/) with a window size of 100 bp and the following set options: Observed/Expected ratio >0.60, Percent C+ Percent G >50.00 and Length >200. The DNA sequences of the DAL genes used here contain 1000 bp before the start codon and 1000 bp after the stop codon. Two CpG islands within the 607-bp (nucleotides 817–1423) and 359-bp (nucleotides 817–1423) regions (817–1423) were observed and delimited by two close red lines, respectively. (c) The DNA methylation of the 607-bp CpG island was analyzed using bisulfite sequencing with the primers listed in Supplementary Table S2, and the data were displayed using Kismeth software (http://katahdin.mssm.edu/kismeth/revpage.pl). (d) Codon base composition of DAL genes in flowering plants. The significant differences of codon base composition between dicot and monocot DAL genes were statistically analyzed according to Student’s t-test. *P < 0.05; **P < 0.01. GC1, the GC contents at the first base of one codon; GC2, the GC contents at the second base; GC3, the GC contents at the third base.
Expression analysis of ZmDAL genes by RT-PCR
Given the important roles of DAL genes in plant organelle RNA editing, the preferential expression patterns of ZmDAL genes were analyzed. To analyze the expression pattern of ZmDAL genes, a NimbleGen maize microarray data (ZM37)37 was performed for 60 tissues representing 11 major organ systems and various developmental stages of the B73 maize inbred line using ZmDAL probes. The median log signal values for all 7 ZmDAL genes were extracted. Four ZmDAL genes (ZmDAL2, ZmDAL3, ZmDAL4, and ZmDAL5) showed a constitutive expression pattern in 60 different tissues, with a CV value <5% (Supplementary Fig. S10). ZmDAL1 had a much higher expression level in the leaves than in other tissues, while ZmDAL6 showed a lower expression level in the roots and first internode compared with that in other tissues or organs. The predominant expression levels of ZmDAL7 were observed in the developing seed, embryo and endosperm. Of these DAL genes, ZmDAL1 and ZmDAL6 were predicted to localize to chloroplasts and were preferentially expressed in the leaves, despite different expression patterns of the two genes (Supplementary Fig. S10). The expression pattern was similar for paralogous genes (ZmDAL3 and ZmDAL4), indicating they were formed by segmental duplication and retained their function (Supplementary Fig. S1).
To confirm the organ-specific expression of ZmDAL genes shown by microarray data, RT-PCR was performed with total RNA isolated from the roots, leaves, ears, and immature tassels. The RT-PCR analysis revealed that the results for ZmDAL2, ZmDAL3, ZmDAL4, and ZmDAL5 match with the DNA chip data but that the other ZmDAL genes do not (Fig. 7). However, the expression levels of ZmDAL2, ZmDAL3, ZmDAL4, and ZmDAL5 were abundant in the ears and tassels, where more biological energy from mitochondria is required38. ZmDAL1 was expressed little in the four tissues. ZmDAL7 showed higher expression levels in the leaves, ears, and tassels in comparison to the roots. ZmDAL6 was predominately expressed in tassels but showed little or no expression in roots, leaves and ears, according to the RT-PCR analysis (Fig. 7).

RT-PCR analysis of maize DAL genes in the four different tissues of maize. The total RNA of four tissues including seedling roots, seedling leaves, 5-cm immature ears and non-emerged immature tassels was isolated and used to perform the semi-quantitative RT-PCR of ZmDAL genes. Actin1 was used for internal controls to normalize the RNA contents in each sample. Primers used are shown in Supplementary Table S2.
Discussion
RNA editing of a single nucleotide, such as C-to-U and A-to-I substitution, requires trans-factors to recognize the nucleotide to be edited and remove the amino group. In mammals, A-to-I RNA editing is catalyzed by a family of enzymes called adenosine deaminases that act on RNA (ADARs)39, while C-to-U editing of apolipoprotein B mRNAs is performed by the zinc-dependent RNA-editing enzyme apolipoprotein B editing catalytic subunit 1 (APOBEC-1), which interacts with APOBEC-1 complementation factor (ACF) for site-specific editing40. In plant organelles, C-to-U RNA editing is mediated by site-specific DYW-PPR proteins and by several other nuclear-encoded factors, including DAL proteins, PPO121 and OZ122. However, the precise roles of each component of the plant RNA editosome in the complicated editing machinery are not yet well known. In this study, we performed a comprehensive analysis of DAL genes in plants and uncovered their seed plant-specific distribution, origin and the potential role of functional divergence.
Using a custom-built HMM file derived from multiple DAL domain alignments, we screened a plant annotation database for putative DAL genes and found that DAL genes were specific to seed plants. The absence of DAL genes in lower plants, in which there are thousands of organelle RNA-edited sites41, 42, indicates that the RNA editosome differs between higher and lower plants2, 16, 17. Given that the presence of DYW-PPR genes in plants is associated with C-to-U RNA editing events42 and that DAL proteins interact with PPR proteins15, PPR genes may not be necessary for the emergence of DAL genes in plants (Fig. 3), although DAL proteins can interact with P. patens PPR proteins43. Furthermore, the homologs of OZ1, ORRM and PPO1 proteins, which have been proven to interact with DAL proteins, were found in P. patens and S. moellendorffii 15 (Fig. 3), and in particular, putative PPO1 proteins in P. patens (Pp1s28_304V6.1) and S. moellendorffii (Sm_82264) all have the 22-aa regions that are essential for their interaction with DAL proteins21, suggesting that these additional RNA editing factors are also not required for the evolution/function of DAL genes, although there is no evidence for the roles of these proteins in RNA editing in lower plants. However, we cannot exclude the possibility that unidentified alternative genes aside from those of the DAL gene family may play similar roles in RNA editing in lower plants. In addition to the regulation of RNA editing via bridging multiple subunits in the RNA editosome, DAL proteins probably have other functions required in higher plants.
When we searched DAL proteins with the dal.hmm information as a query, propeptide inhibitor I9 domains of plant subtilisins were found to show high similarity to part of the DAL domain. In addition, the inhibitor I9 domain-encoding genes and DAL genes have a conserved gene structure, including the intron phases 2, 1, and 1 and the 98-bp exon (Supplementary Fig. S7), and inhibitor I9 genes were present prior to the divergence of seed plants from ferns (Fig. 3). Therefore, it is inferred that the DAL genes originated from inhibitor I9 genes by combining I9 domain-encoding exons with another unidentified sequence via exon shuffling. Additional evidence for this hypothesis stems from the fact that the ORRM1-like genes encode two tandem DAL domains followed by an RRM domain at the C terminus28, which could have arisen from the combination of two I9 domain-encoding exons and an extra RRM domain-encoding sequence. In addition to the protein sequence similarity and conserved gene structure between the DAL genes and I9 domain-encoding genes, they also have similar molecular function, as the DAL domain mediates the protein-protein interaction of DAL proteins with other RNA editing factors15, while the I9 domain inhibits proenzymes by hiding substrate-binding domains32.
Plant DAL genes were assigned to five distinct subfamilies based on their phylogenetic relationships (Fig. 4). In Arabidopsis, DAL/RIP genes play nonequivalent roles in RNA editing. The major factor is RIP1 (At3g15000), which belongs to group II and controls 75% of RNA-edited sites in mitochondria and 20% of RNA-edited sites in chloroplasts27. RIP2 (At2g33430), RIP3 (At3g06790), RIP9 (At1g11430) and RIP8 (At4g20020) have moderate effects on RNA editing and are in groups I, III, IV and V, respectively (Fig. 4). As minor genes, RIP4 (At5g44780), RIP5 (At1g32580), RIP6 (At2g35240) and RIP7 (At1g72530) each shares clades with the above major or moderate factors, suggesting that these DAL genes have recently duplicated and have evolved to only control few RNA editing sites. We analyzed the functional divergence between DAL groups to identify the sites distinguishing the different DAL members. Five groups resulted in 10 pairwise comparisons; of these comparisons, 6 showed significant type I functional divergence, and one showed significant type II functional divergence (Table 1). In addition, we analyzed the functional divergence between intragroup members. These intragroup DAL genes were divided into two subgroups: a dicot subgroup (a) and a monocot subgroup (b) (Supplementary Fig. S8). No significant functional divergence was observed between intragroup members of DAL genes, although the GC contents of DAL genes in monocots and dicots were different. The sites involved in functional divergence between DAL groups were predominantly localized to the DAL domains, and relaxed selection on these sites would serve to increase the complexity for determination of RNA editing because DAL proteins act on RNA editing in the form of heterodimers in addition to homodimers. Also, these sites probably account for the interaction of each DAL with different PPR proteins in the RNA editosome. Therefore, it is understandable that different dimers formed with homogenous or heterogeneous DAL proteins, which confer RNA editing to corresponding sites, have increased the diversity of RNA editing regulation in higher plants. However, further studies on the biochemical character of DAL proteins and the crystal structure of the RNA editosome are required to parse the roles of DAL proteins with the functionally diverged sites. In addition, the putative effects of DAL proteins on other RNA processing events in addition to RNA editing should be further investigated.
Materials and Methods
Identification and sequence analysis of putative DAG-like genes in plants
Known MORF/RIP genes At4g20020, At2g33430, At3g06790, At5g44780, At1g32580, At2g35240, At1g72530, At3g15000, and At1g11430 from A. thaliana 16, 17, 26, 44 and DAG (NCBI Protein ID: Q38732) from A. majus 25 were used to query the maize filtered gene set (ZmB73_5b_FGS_translations.fasta downloaded from www.maizesequence.org) using a local BLASTP program with an E-value <1e-10 and a bit score >100. ZmDAL protein sequences were analyzed using ExPASy tools available at http://us.expasy.org/tools/. Multiple sequence alignments of ZmDAL proteins and the above known DAL proteins were performed using ClustalW (http://www.ebi.ac.uk/Tools/msa/clustalw2/)45. To mine the conserved domain, the alignment results (Supplementary Data File S1) were used to build a protein HMM file, dubbed dal.hmm (Supplementary Data File S2) by the hmmbuild program in HMMER 3.0 package31.
To investigate the evolution of DAL genes in the plant kingdom, the dal.hmm information was used as a query to search the genome annotation data of the following representative species from Phytozome v8.0 (http://www.phytozome.net/), except those of P. abies, which were from ConGenIE (http://congenie.org/), in HMMER 3.0 package: C. reinhardtii, P. patens, S. moellendorffii, P. abies, B. distachyon, O. sativa Japonica, S. bicolor, A. formosa, V. vinifera, A. lyrata and P. trichocarpa. Protein hits with an e-value <1e-10 and sequence score of “best 1 domain” >100 were collected. The homologs of the OZ1, PPO1 and ORRM genes in the above plants were identified using a local BLASTP program with the protein sequences of known Arabidopsis OZ1, PPO1, ORRM2, ORRM3 and ORRM4 proteins as queries15.
The PFAM (http://pfam.sanger.ac.uk/) and INTERPRO (http://www.ebi.ac.uk/interpro/) databases were screened to detect known motifs in ZmDAL proteins and the DAL proteins of other plants. The MEME program (http://meme.nbcr.net/meme/cgi-bin/meme.cgi) was used to investigate the putative conserved motifs among these ZmDAL proteins with the following parameters: length between 10 and 50 aa, maximum number of motifs to find = 5, and one per sequence. To obtain the intact conserved DAL domain, different limits for length of each motif were taken that were between 100 and 120 aa.
Gene structures of plant DAL genes
The DNA and transcript sequences of ZmDAL genes obtained from the maize sequence annotation database MaizeGDB (http://www.maizegdb.org/) were used to analyze the gene structures of ZmDAL genes. Several ZmDAL genes had more than one gene model annotated in MaizeGDB. To confirm the putative alternative splicing transcripts, transcript-specific primers (Supplementary Table S2) were designed to amplify corresponding DNA isolated from B73 seedlings and cDNA derived from B73 seedling RNA. Gene structures producing validated transcripts were drawn and displayed using the online GSDraw program of the PIECE server (http://wheat.pw.usda.gov/piece/GSDraw.php)46. Conserved DAL domains were also displayed using the GSDraw program. The gene structures of DAL genes from other plant species were obtained from the Phytozome v8.0 annotation database (http://www.phytozome.net/) and displayed using the GSDraw program46.
Subcellular localization prediction of ZmDAL proteins
Two in silico programs, Predotar47 and TargetP48, were used to predict the putative organelle localization of ZmDAL proteins. The maize organelle proteomics database (PPDB, http://ppdb.tc.cornell.edu/)49 was screened to detect the accumulation of ZmDAL proteins.
Phylogenetic dendrogram
The multiple sequence alignment analysis of conserved DAL domains collected from 79 DAL proteins identified in maize and in other higher plants was carried out using MUSCLE v3.8.3150, and the resulting alignment was used to build the NJ distance phylogenetic tree using MEGA v5.051 by applying the Poisson substitution model, 1000 bootstrap samples, and pairwise deletion for gaps/missing data. The tree was displayed using FigTree v1.4.0 (http://tree.bio.ed.ac.uk/software/figtree/).
Functional divergence analysis
To investigate the functional alteration of duplicated DAL genes in plants, the GU99 method within DIVERGR v3.033 was used to calculate the coefficients of type I and type II functional divergence (θ I and θ II, respectively) between two groups after gene duplication and to predict functionally divergent amino acids based on their different evolutionary rates. Within two duplicated groups of a gene family, type I functional divergence helps identify the relaxation of functional constraint in one group relative to that of another, while type II identifies shifting patterns of functional constraint.
GC content and DNA methylation analysis with bisulfite sequencing
The entire DNA sequences of plant DAL genes together with 1 kb of upstream and downstream flanking sequences were used for calculation of GC content and prediction of CpG islands in the EMBOSS CpGplot online server (http://www.ebi.ac.uk/Tools/seqstats/emboss_cpgplot/). To confirm the DNA methylation of ZmDAL DNA sequences, leaf DNA of B73 seedlings was isolated and treated with bisulfate using the EpiTect® bisulfite kit (QIAGEN, USA) according to the manufacturer’s instructions. The primers for detection of DNA methylation were designed using MethPrimer (http://www.urogene.org/methprimer/)52 and modified using Primer3web (http://primer3.ut.ee/). PCR products were cloned into a pGEM-T vector (Promega, USA) and subsequently sequenced using an ABI3730 DNA sequencer (Shanghai Sunny Bio., China). DNA methylation states via bisulfite sequencing were analyzed and displayed using Kismeth software (http://katahdin.mssm.edu/kismeth/revpage.pl)53.
Expression analysis of ZmDAL genes in different tissues
To investigate the spatiotemporal expression patterns of ZmDAL genes, the log2-transformed and RMA-normalized data for ZmDAL genes were downloaded from PLEXdb (http://www.plexdb.org/)54, and cluster analysis of these expression data were performed using Cluster v3.055 with the hierarchic method. A heat map was produced using Java TreeView v1.1.556. The coefficient of variation (CV) was calculated according to the following equation to estimate the expression variation of each ZmDAL gene in different tissues: CV = sd/mean, where sd indicates the standard deviation of a gene in different tissues and the mean represents the average expression level.
Semi-quantitative reverse transcription PCR (semiq-RT-PCR)
Total RNA was isolated from different tissues of the B73 inbred lines, including seedling roots, leaves, 5-cm ears and immature tassels, using the Trizol® reagent (Invitrogen, USA) according to the manufacturer’s protocol. First-strand cDNA was produced from 1 μg of total RNA (25 μl reaction volume) using M-MLV reverse transcriptase (Invitrogen, USA) at 37 °C for 1 h. All gene-specific primers were designed as shown in Supplementary Data Table S2. Specific primers for the maize Actin1 gene (GenBank ID: NM_001155179) were used as an internal control. Reactions were performed with Taq Polymerase (Dalian Takara Biotechnology, China) on a Bio-Rad Thermal Cycler (Bio-Rad, USA) using the following procedure: 5 min at 94 °C to start; 32 cycles of 30 s at 94 °C, 30 s at 58 °C and 1 min at 72 °C; and a final extension step of 72 °C for 10 min to complete the reaction, and the Actin1 transcript was amplified with 28 PCR cycles. Each PCR pattern was performed in triplicate, mixtures without a template were employed as negative controls, and the maize Actin1 amplicon served as an internal control for each gene investigated.
References
Fujii, S. & Small, I. The evolution of RNA editing and pentatricopeptide repeat genes. New Phytol. 191, 37–47 (2011).
Takenaka, M., Zehrmann, A., Verbitskiy, D., Härtel, B. & Brennicke, A. RNA editing in plants and its evolution. Annu. Rev. Genet. 47, 335–352 (2013).
Kugita, M., Yamamoto, Y., Fujikawa, T., Matsumoto, T. & Yoshinaga, K. RNA editing in hornwort chloroplasts makes more than half the genes functional. Nucleic Acids Res. 31(9), 2417–2423 (2003).
Grewe, F., Viehoever, P., Weisshaar, B. & Knoop, V. A trans-splicing group I intron and tRNA-hyperediting in the mitochondrial genome of the lycophyte Isoetes engelmannii. Nucleic Acids Res. 37(15), 5093–5104 (2009).
Giegé, P. & Brennicke, A. RNA editing in Arabidopsis mitochondria effects 441 C to U changes in ORFs. Proc. Natl. Acad. Sci. USA 96, 15324–15329 (1999).
Tsudzuki, T., Wakasugi, T. & Sugiura, M. Comparative analysis of RNA editing sites in higher plant chloroplasts. J. Mol. Evol. 53, 327–332 (2001).
Chateigner-Boutin, A. L. & Small, I. Plant RNA editing. RNA Biol. 7, 213–219 (2001).
Bock, R. Sense from nonsense: How the genetic information of chloroplastsis altered by RNA editing. Biochimie 82(6–7), 549–557 (2000).
Marchfelder, A. & Binder, S. Plastid and plant mitochondrial RNA processing and RNA stability. In: Daniell, H. & Chase, C. D. (Eds), Molecular Biology and Biotechnology of Plant Organelles. Springer, Dordrecht, pp. 261–294 (2004).
Bock, R., Hermann, M. & Kössel, H. In vivo dissection of cis-acting determinants for plastid RNA editing. EMBO J. 15, 5052–5059 (1996).
Chaudhuri, S. & Maliga, P. Sequences directing C to U editing of the plastid psbL mRNA are located within a 22 nucleotide segment spanning the editing site. EMBO J. 15, 5958–5964 (1996).
Farré, J. C., Leon, G., Jordana, X. & Araya, A. cis recognition elements in plant mitochondrion RNA editing. Mol. Cell. Biol. 21, 6731–6737 (2001).
Takenaka, M., Neuwirt, J. & Brennicke, A. Complex cis-elements determine an RNA editing site in pea mitochondria. Nucleic Acids Res. 32, 4137–4144 (2004).
Neuwirt, J., Takenaka, M., van der Merwe, J. A. & Brennicke, A. An in vitro RNA editing system from cauliflower mitochondria: Editing site recognition parameters can vary in different plant species. RNA 11, 1563–1570 (2005).
Sun, T., Bentolila, S. & Hanson, M. R. The unexpected diversity of plant organelle RNA editosomes. Trends Plant Sci. 21(11), 962–973 (2016).
Takenaka, M. et al. Multiple organellar RNA editing factor (MORF) family proteins are required for RNA editing in mitochondria and plastids of plants. Proc. Natl. Acad. Sci. USA 109(13), 5104–5109 (2012).
Bentolila, S. et al. RIP1, a member of an Arabidopsis protein family, interacts with the protein RARE1 and broadly affects RNA editing. Proc. Natl. Acad. Sci. USA 109(22), E1453–1461 (2012).
Shi, X., Hanson, M. R. & Bentolila, S. Two RNA recognition motif-containing proteins are plant mitochondrial editing factors. Nucleic Acids Res. 43(7), 3814–3825 (2015).
Shi, X., Bentolila, S. & Hanson, M. R. Organelle RNA recognition motif-containing (ORRM) proteins are plastid and mitochondrial editing factors in Arabidopsis. Plant Signal. Behav. 11(5), e1167299 (2016).
Shi, X., Germain, A., Hanson, M. R. & Bentolila, S. RNA recognition motif-containing protein ORRM4 broadly affects mitochondrial RNA editing and impacts plant development and flowering. Plant Physiol. 170(1), 294–309 (2016).
Zhang, F. et al. Tetrapyrrole biosynthetic enzyme protoporphyrinogen IX oxidase 1 is required for plastid RNA editing. Proc. Natl. Acad. Sci. USA 111(5), 2023–2028 (2014).
Sun, T. et al. A zinc finger motif-containing protein is essential for chloroplast RNA editing. PLoS Genet. 11(3), e1005028 (2016).
Lurin, C. et al. Genome-wide analysis of Arabidopsis pentatricopeptide repeat proteins reveals their essential role in organelle biogenesis. Plant Cell 16, 2089–2103 (2004).
Salone, V. et al. A hypothesis on the identification of the editing enzyme in plant organelles. FEBS Lett. 581, 4132–4138 (2007).
Chatterjee, M. et al. DAG, a gene required for chloroplast differentiation and palisade development in Antirrhinum majus. EMBO J. 15(16), 4194–4207 (1996).
Bisanz, C. et al. The Arabidopsis nuclear DAL gene encodes a chloroplast protein which is required for the maturation of the plastid ribosomal RNAs and is essential for chloroplast differentiation. Plant Mol. Biol. 51(5), 651–663 (2003).
Bentolila, S., Oh, J., Hanson, M. R. & Bukowski, R. Comprehensive high-resolution analysis of the role of an Arabidopsis gene family in RNA editing. PLoS Genet. 9(6), e1003584 (2013).
Sun, T. et al. An RNA recognition motif-containing protein is required for plastid RNA editing in Arabidopsis and maize. Proc. Natl. Acad. Sci. USA 110(12), E1169–1178 (2013).
Hayes, M. L., Giang, K. & Mulligan, R. M. Molecular evolution of pentatricopeptide repeat genes reveals truncation in species lacking an editing target and structural domains under distinct selective pressures. BMC Evol. Biol. 12, 66 (2012).
O’Toole, N. et al. On the expansion of the pentatricopeptide repeat gene family in plants. Mol. Biol. Evol. 25(6), 1120–1128 (2008).
Eddy, S. R. Accelerated Profile HMM Searches. PLoS Comput. Biol. 7(10), e1002195 (2011).
Li, Y., Hu, Z., Jordan, F. & Inouye, M. Functional analysis of the propeptide of subtilisin E as an intramolecular chaperone for protein folding. Refolding and inhibitory abilities of propeptide mutants. J. Biol. Chem. 270(42), 25127–25132 (1995).
Gu, X. et al. An update of DIVERGE software for functional divergence analysis of protein family. Mol. Biol. Evol. 30(7), 1713–1719 (2013).
Šmarda, P. & Bureš, P. The variation of base composition in plant genomes. In: Wendel, J. F., Greilhuber, J., Dolezel, J. & Leitch, I. J. (eds) Plant genome diversity volume 1. Springer, Vienna, pp 209–235 (2012).
Šmarda, P. Ecological and evolutionary significance of genomic GC content diversity in monocots. Proc. Natl. Acad. Sci. USA 111(39), E4096–4102 (2014).
Singh, R., Ming, R. & Yu, Q. Comparative analysis of GC content variations in plant genomes. Tropical Plant Biol. 9(3), 136–149 (2016).
Sekhon, R. S. et al. Genome-wide atlas of transcription during maize development. Plant J. 66(4), 553–563 (2011).
Mackenziea, S. & McIntoshb, L. Higher plant mitochondria. Plant Cell 11(4), 571–585 (1999).
Nishikura, K. Functions and regulation of RNA editing by ADAR deaminases. Annu. Rev. Biochem. 79, 321–349 (2010).
Keegan, L. P., Gallo, A. & O’Connell, M. A. The many roles of an RNA editor. Nat. Rev. Genet. 2(11), 869–878 (2001).
Oldenkott, B., Yamaguchi, K., Tsuji-Tsukinoki, S., Knie, N. & Knoop, V. Chloroplast RNA editing going extreme: more than 3400 events of C-to-U editing in the chloroplast transcriptome of the lycophyte Selaginella uncinata. RNA 20(10), 1499–1506 (2014).
Rüdinger, M., Polsakiewicz, M. & Knoop, V. Organellar RNA editing and plant-specific extensions of pentatricopeptide repeat proteins in jungermanniid but not in marchantiid liverworts. Mol. Biol. Evol. 25(7), 1405–1414 (2008).
Schallenberg-Rüdinger, M., Kindgren, P., Zehrmann, A., Small, I. & Knoop, V. A DYW-protein knockout in Physcomitrella affects two closely spaced mitochondrial editing sites and causes a severe developmental phenotype. Plant J. 76(3), 420–432 (2013).
Babiychuk, E., Fuangthong, M., Van Montagu, M., Inzé, D. & Kushnir, S. Efficient gene tagging in Arabidopsis thaliana using a gene trap approach. Proc. Natl. Acad. Sci. USA 94(23), 12722–12727 (1997).
Larkin, M. A. et al. Clustal W and Clustal X version 2.0. Bioinformatics 23(21), 2947–2948 (2007).
Wang, Y. et al. PIECE: a database for plant gene structure comparison and evolution. Nucleic Acids Re. 41(Database issue): D1159–1166 (2013).
Small, I., Peeters, N., Legeai, F. & Lurin, C. Predotar: A tool for rapidly screening proteomes for N-terminal targeting sequences. Proteomics 4(6), 1581–1590 (2004).
Emanuelsson, O., Brunak, S., von Heijne, G. & Nielsen, H. 2007. Locating proteins in the cell using TargetP, SignalP and related tools. Nat. Protoc. 2(4), 953–971 (2007).
Sun, Q. et al. PPDB, the Plant Proteomics Database at Cornell. Nucleic Acids Res. 37(Database issue), D969–974 (2009).
Edgar, R. C. MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinformatics 5, 113 (2004).
Tamura, K. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 28(10), 2731–2739 (2011).
Li, L. C. & Dahiya, R. MethPrimer: designing primers for methylation PCRs. Bioinformatics 18(11), 1427–1431 (2002).
Gruntman, E. et al. Kismeth: analyzer of plant methylation states through bisulfite sequencing. BMC Bioinformatics 9, 371 (2008).
Dash, S., Van Hemert, J., Hong, L., Wise, R. P. & Dickerson, J. A. PLEXdb: gene expression resources for plants and plant pathogens. Nucleic Acids Res. 40(Database issue), D1194–1201 (2012).
Eisen, M. B., Spellman, P. T., Brown, P. O. & Botstein, D. Cluster analysis and display of genome-wide expression patterns. Proc. Natl. Acad. Sci. USA 95(25), 14863–14868 (1998).
Saldanha, A. J. Java Treeview–extensible visualization of microarray data. Bioinformatics 20(17), 3246–3248 (2004).
Acknowledgements
This work was supported by the National Natural Science Foundation of China (31571675, 91335205, 31171565 and 31500253) and by the BAAFS Innovation Team of Corn Germplasm Innovation and Breeding of New Varieties (JNKYT201603). We are grateful to Dr. Jingqi Zhou (Fudan University) for helpful assistance to functional divergence analysis with DIVERGE v3.0.
Author information
Authors and Affiliations
Contributions
Conceived and designed the experiments: Yx.Z., F.Q. and Jr. Z. Performed the experiments: M.C., Jh.Z., Y.L., H.X. and K.Z. Analyzed the data: Yx.Z., M.L., R.Z., H.X., B.Y. and W.S. Wrote the paper: Yx.Z. and Yl.Z. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Luo, M., Cai, M., Zhang, J. et al. Functional divergence and origin of the DAG-like gene family in plants. Sci Rep 7, 5688 (2017). https://doi.org/10.1038/s41598-017-05961-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-05961-2
This article is cited by
-
Maize—Azospirillum brasilense interaction: accessing maize’s miRNA expression under the effect of an inhibitor of indole-3-acetic acid production by the plant
Brazilian Journal of Microbiology (2024)
-
Genome-wide identification and expression analysis of peach multiple organellar RNA editing factors reveals the roles of RNA editing in plant immunity
BMC Plant Biology (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
