
Abstract
RNA interference is a highly conserved mechanism wherein several types of non-coding small RNAs regulate gene expression at the transcriptional or post-transcriptional level, modulating plant growth, development, antiviral defence, and stress responses. Argonaute (AGO), DCL (Dicer-like), and RNA-dependent RNA polymerase (RDR) are key proteins in this process. Here, these three protein families were identified in Chenopodium quinoa. Further, their phylogenetic relationships with Arabidopsis, their domains, three-dimensional structure modelling, subcellular localization, and functional annotation and expression were analysed. Whole-genome sequence analysis predicted 21 CqAGO, eight CqDCL, and 11 CqRDR genes in quinoa. All three protein families clustered into phylogenetic clades corresponding to those of Arabidopsis, including three AGO clades, four DCL clades, and four RDR clades, suggesting evolutionary conservation. Domain and protein structure analyses of the three gene families showed almost complete homogeneity among members of the same group. Gene ontology annotation revealed that the predicted gene families might be directly involved in RNAi and other important pathways. Largely, these gene families showed significant tissue-specific expression patterns, RNA-sequencing (RNA-seq) data revealed that 20 CqAGO, seven CqDCL, and ten CqRDR genes tended to have preferential expression in inflorescences. Most of them being downregulated in response to drought, cold, salt and low phosphate stress. To our knowledge, this is the first study to elucidate these key protein families involved in the RNAi pathway in quinoa, which are significant for understanding the mechanisms underlying stress responses in this plant.
Similar content being viewed by others


Introduction
RNA interference (RNAi), also known as RNA silencing, is an extremely important and highly conserved gene-expression regulatory mechanism widely distributed among eukaryotes. RNAi is mediated by small non-coding RNAs that regulate gene expression at the transcriptional and post-transcriptional levels by specifically identifying complementary RNA targets, and protecting cells against harmful exogenous and endogenous genetic elements1,2. Thus, RNAi plays an important role in the regulation of plant development, epigenetic modification, genome stability maintenance, and abiotic and biotic stress responses3,4,5. Argonaute (AGO), Dicer-like (DCL), and RNA-dependent RNA polymerase (RDR) are key proteins of the RNAi pathway6.
RNAi is initially triggered by double-stranded RNA (dsRNA) or partially double-stranded stem–loop RNA that is cleaved by DCL into 21–24-nt small RNA (sRNA) duplexes7, which are then incorporated with AGO protein to form the pre-RNA-induced silencing complex (pre-RISC) that requires the molecular chaperone Heat shock protein 70 (Hsp70)/90 (Hsp90)8. The duplex is melted by the action of the N-domain of AGO and only the guide RNA strand remains in the complex to form a mature RISC9,10. RISC binds to complementary mRNA guided by single-stranded sRNA to inhibit translation during post-transcriptional gene silencing (PTGS) or mediates DNA methylation and heterochromatin formation during transcriptional gene silencing (TGS), resulting in specific gene silencing11,12. RDR recognizes aberrant RNA and catalysing phosphodiester bond formation between ribonucleotides to synthesize other dsRNA, providing a new substrate to DCL, which can enhance RNAi signals or initiate a new round of RNAi13.
To date, several studies have shown that the sizes of AGO, DCL, and RDR gene families vary among species. For example, Arabidopsis14, rice15, maize16, millet17, grapevine18, tomato19, wheat20, soybean21, pepper22, cucumber23, barley24, sugarcane25, sweet orange6, and tea26 genomes encode ten, 19, 18, 19, 13, 15, 39, 21, 12, seven, 11, 21, eight, and 18 AGO genes; four, five, five, eight, four, seven, seven, seven, four, five, five, four, four, and five DCL genes; six, eight, five, 11, five, six, 16, seven, six, eight, seven, 11, four, and nine RDR genes, respectively. These studies have also shown that these gene families are highly conserved in plants, although little is known about the corresponding genes in quinoa.
Quinoa is a tetraploid dicotyledonous species with a cultivation history of over 7000 years. Its seeds can be consumed as entire grains or ground into flour, and its leaves and stems can be used as animal feed27. Quinoa is high in nutritional value and contains a variety of essential amino acids, fats, dietary fibre, vitamins, and minerals, among other valuable nutrients28. In addition, quinoa contains a large number of secondary metabolites, such as steroids, flavonoids, and triterpene saponins29, which have anti-microbial27, anti-diabetic30, anti-inflammatory31, and immunomodulatory activities32. Moreover, quinoa is resistant to salinity, frost, and drought, and can be planted in marginal environments. Therefore, quinoa has garnered increasingly widespread attention, and the year 2013 was declared ‘The International Year of Quinoa’ by the United Nations33.
Although quinoa exhibits excellent resistance to stress, the mechanisms at play are not well understood. Studies have shown that when plants are subjected to biotic or abiotic stress, the sRNAs involved in the RNAi pathway play an important role in the regulation of gene expression34,35. Here, we systematically studied the AGO, DCL, and RDR gene families in quinoa through whole genome analysis. We identified the evolutionary relationship of these gene families with those of Arabidopsis, and analysed the secondary domains, three-dimensional (3D) structure, subcellular localization, and functional annotation of the identified AGO, DCL, and RDR genes. The results reported herein provide further insights into the molecular mechanism of RNAi and will help understand the mechanisms underlying stress resistance in quinoa.
Results
Screening of AGO, DCL, and RDR genes in quinoa
To identify quinoa AGO, DCL, and RDR genes, the Chenopodium quinoa v1.0 database was searched for the transcripts of each gene family that contained the characteristic domains. The assigned names, primary transcript ID, chromosome localization, description of main transcripts, CDS, and peptide lengths are shown in Tables 1 and 2. A total of 25 AGO, 12 DCL, and 12 RDR genes were initially recognized, and after considering the lack or overlap of the functional domain and insufficient length of the amino acid (aa) sequence, 21 AGO, eight DCL, and 11 RDR genes were ultimately identified. The gene IDs of AtAGOs, AtDCLs, and AtRDRs are shown in Table S1.
A total of 21 CqAGO homologues were localized on nine chromosomes and mostly concentrated on chromosomes 07, 11, 15, and 18. Chromosomes 07, 11, and 15 harbor three CqAGO genes, and chromosome 18 harbors four CqAGO genes (Fig. S1). The length of the CDS ranged from 1953 to 3165 bp (Table 1). Most CqAGO genes possessed 18–23 introns, whereas CqAGO2/3, CqAGO7a, and CqAGO7b contained two introns each, where they were localized in the AtAGO2/3/7 clade (Figs. 1A and 2). Two CqDCL genes were detected on chromosomes 01 and 10, one CqDCL gene was localized to chromosomes 02, 03, 05, and 12 (Fig. S1). CDS length varied from 2976 to 7065 bp, produced by CqDCL10b and CqDCL1a, with coding proteins of 992 and 2355 aa, respectively. The number of introns varied from 0 to 42 in CqDCLs (Fig. 3A). CqRDR genes were mainly present on chromosomes 01, 02, and 04 (Fig. S1), and the length of the CDS ranged from 1677 to 3624 bp (Table 2). There were significant differences in the number of introns among CqRDR members, and the intron numbers of CqRDR1, 2a, 2b, 6a, and 6b, which were localized in the AtRDR1/2/6 clade, were concentrated in the range from 1 to 3. In the AtRDR3 clade, CqRDR3c possessed only 13 introns, whereas the other CqRDRs contained a significantly higher number of introns, i.e., 17–20 (Figs. 1C and 3B).

Phylogenetic analysis. (A) Relationship between AtAGO and CqAGO proteins. (B) Relationship between AtDCL and CqDCL proteins. (C) Relationship between AtRDR and CqRDR proteins. Branch length was ignored, and branch support values are displayed. The scale bar at the bottom left corner represents the branch length.

Conserved domains of CqAGO proteins identified by SMART and Pfam, generated using IBS (left). The protein domains include N domain, DUF1785, PAZ, L2, MID, and PIWI. Introns in CqAGO genes are shown on the right.

Domain structure of CqDCLs and CqRDRs (left). (A) Conserved domains of CqDCL proteins identified by SMART and Pfam, and generated using IBS. The protein domains include DEXDc, HELICc, Dicer-dimer, PAZ, RIBOc, and DSRM. (B) Conserved domains of CqRDR proteins identified by SMART and Pfam, and generated by IBS. The protein domains include RRM and RdRP. Introns in CqDCL and CqRDR genes are shown on the right.
Phylogenetic tree and domain analysis of CqAGO, CqDCL, and CqRDR proteins
To determine the potential function of critical proteins in the RNAi pathway, we predicted the domains of 21 CqAGOs, eight CqDCLs, and 11 CqRDRs using SMART. Detailed prediction data, and corresponding confidence values of the domains of CqAGO (Table S2 and Fig. 2), CqDCL (Table S3 and Fig. 3A), and CqRDR (Table S4 and Fig. 3B) were obtained from SMART/Pfam, and visually analysed. Visual analysis of protein domains revealed similarities and differences in the position of typical conserved domains among members of each protein family. We found that CqAGO, CqDCL, and CqRDR had more copies than those of Arabidopsis, which further indicates that they may be functionally more diverse.
Phylogenetic analysis showed that the AGO protein sequences of Arabidopsis can be divided into three clades: AtAGO1/5/10, AtAGO2/3/7, and AtAGO4/6/8/9. We observed that CqAGO proteins were placed in all these clades; there were the following three CqAGOs: CqAGO2/3, 7a, and 7b within the AtAGO2/3/7 clade, CqAGO7a and CqAGO7b were grouped with the AtAGO7 clade, although they were localized on different chromosomes, and the sequence similarity between them was as high as 98.2% at the aa level. Furthermore, CqAGO7a and CqAGO7b shared 70% sequence similarity with AtAGO7. The AtAGO1/5/10 and AtAGO4/6/8/9 clades were highly diverse in quinoa, with nine CqAGO proteins. CqAGO1a, 1b, 5a, 5b, 5c, 5d, 5e, 10a, and 10b clustered into the AtAGO1/5/10 clade, whereas CqAGO4a, 4b, 4c, 4d, 4e, 4f, 6a, 6b, and 8/9 clustered into the AtAGO4/6/8/9 clade (Fig. 1A).
Consistent with other eukaryotic AGO proteins, four typical characteristic domains, including N domain, PIWI/Argonaute/Zwille (PAZ), middle (Mid), and p-body-induced wimpy tests (PIWI) domains, were found in several CqAGO proteins (Fig. 2), and the order of functional domains was consistent with AtAGO proteins. All CqAGO proteins had the PAZ and PIWI domains. Most predicted CqAGOs identified a variable N-t domain, which is composed of an N domain and a DUF1785 domain; CqAGO2/3 and CqAGO6a contained only the DUF1785 domain, whereas CqAGO4a, CqAGO5b and CqAGO5e contained only the N domain. In addition, all CqAGO proteins in the AtAGO2/3/7 clade did not contain the MID domain, whereas all CqAGOs in the AtAGO1/5/10 clade were predicted to contain the MID domain. AGOs in the same clade had high structural similarity, indicating that they may exhibit high functional similarity. MSA analysis of the PIWI domain of CqAGO proteins exhibited a conserved QF-V (Q = glutamine, F = phenylalanine, V = valine) motif and the metal-chelating residue motif DEDD/H (D = aspartic acid, E = glutamate, D = aspartic acid, and H = histidine) required for cleavage activity, except for CqAGO2/3 and CqAGO5e; the first D in CqAGO2/3 was replaced by N (asparagine), and CqAGO5e lacked the D/H residue of the catalytic tetrad (Fig. 4A). Moreover, Arabidopsis H798 (H798 of AtAGO1) is a very important aa residue, and most CqAGOs in the AtAGO1/5/10 and AtAGO2/3/7 clades retained H residues. H was replaced by N in CqAGO2/3. Furthermore, almost all H residues were replaced by P (proline) in the AtAGO4/6/8/9 clade, and was only replaced by S (serine) in CqAGO4a (Fig. 4A). Residues Y (tyrosine), K (lysine), Q, and K which are related to 5′- phosphate binding in sRNA, were completely conserved in all CqAGOs, except for CqAGO2/3, CqAGO4f, and CqAGO5e. Additionally, CqAGO2/3 lacked the conserved residue Q, while CqAGO5e only retained the residue Y, and the second K was replaced by H in CqAGO4f (Fig. 4B). The N residue, preferentially bound to 5′U 21-nt sRNA36, was conserved in CqAGO1a, 1b, 4a, 4b, 4c, 4d, 4e, 4f, 8/9, 10a, and 10b (Fig. 4B).

Functionally conserved amino acids of CqAGO proteins. (A) DEDD/H tetrad (blue arrows), H798 (red arrow) and QF-V motif within PIWI domains. (B) 5′-terminal nucleotide selection N (red arrow), 5′-phosphate-binding selection YKQK (blue arrows).
Compared to Arabidopsis, the CqDCL families were more diverse, with the number of members exceeding those of Arabidopsis (four AtDCL members from four clades). Each member of the AtDCL expanded into two copies in quinoa (Fig. 1B). Analysis of the CqDCL proteins revealed that most CqDCL proteins consist of DEAD-like helicase superfamily (DEXDc), helicase superfamily C-terminal (HELICc), Dicer dimerization (Dicer-dimer or DUF283), PAZ, Ribonuclease III family (RIBOc), and double-stranded RNA-binding motif (DSRM) domains. CqDCL1a, CqDCL1b, CqDCL4a, and CqDCL4b contained all characteristic domains. CqDCL2a and CqDCL2b contained only one DSRM domain, whereas CqDCL3a and CqDCL3b contained only the PAZ and RIBOc domains (Fig. 3A). MSA analysis of CqDCL proteins showed that L/IPSI/L/VM/I(X)11LK/R in the core region of the connecting helix is relatively conserved. Except for CqDCL2a and CqDCL2b in the AtDCL2 clade, the NLL motif of the PAZ loop was responsible for connection with dsRNA in other CqDCLs were conserved (Fig. 5A). RIBOc has two domains: RNase IIIA and RNase IIIB. The TEKCHER motif of RNase IIIA and the HPSYN loop of RNase IIIB in AtDCL4 may interact with dsRNA37, but limited conservation was observed in quinoa; only the HPSYN loops in CqDCL4a and CqDCL4b were fully conserved. In addition, the catalytic aa residues N and K in the RIBOc domain were highly conserved (Fig. 5B,C).

Functionally conserved amino acids in CqDCL proteins. (A) NLL motif and connector helix core L/IPSI/L/VM/I(X)11LK/R. (B) RNase III A TEKCHER motif, N, and K residues. (C) RNase III B HPSYN motif, N, and K residues.
According to phylogenetic analysis, AtRDR proteins were grouped into four clades: RDR1, RDR2, RDR3, and RDR6. There were six CqRDR proteins (CqRDR3a, 3b, 3c, 3d, 3e, and 3f) grouped together with RDR3, and the RDR1, RDR2, RDR6 clade contained one, two, two CqRDR proteins, respectively (Fig. 1C). CqRDR6a and CqRDR6b, belonging to the AtRDR6 clade, shared 96% sequence similarity. Structural analysis of the CqRDR proteins showed that the RNA-dependent RNA polymerase (RdRP) domain was present in all predicted CqRDRs; CqRDR1 and CqRDR2a had an RNA-recognition motif (RRM) domain (Fig. 3B). Moreover, CqRDR1, 2a, 2b, 6a, 6b, 3a, and 3b possessed canonical DLDGD, whereas CqRDR3c, CqRDR3d, CqRDR3e, and CqRDR3f contained DYDGD (Fig. 6).

Functionally conserved DL/YDGD motif in CqRDR proteins.
3D modelling of CqAGO proteins
SWISS-MODEL is the best software for protein 3D model prediction38. We used SWISS-MODEL to obtain a 3D model of CqAGO and verified the predicted structure using four different measures.
QMEAN and GMQE are two different measures for evaluating models in SWISS-MODEL. QMEAN is based on a single model that is used to derive the absolute quality mass of each residue and the entire structure. The QMEAN z-score of a high-quality model should be between − 4.0 and 0. In turn, GMQE combines the attributes of target-template alignment and template structure, with values between 0 and 1. The larger the score, the more reliable the quality of the predicted structure. This study also used PROCHECK, ERRAT, Verify 3D, and WHATCHECK to evaluate the quality of the model, with higher values indicating a better model in each case. The results are shown in Table S5. Figures 7 and S2 demonstrate the models of CqAGOs and the corresponding AtAGOs in the same clade. The predicted structure of CqAGOs was similar to that of the corresponding AtAGOs, suggesting a high degree of functional conservation.

3D structure predictions for the AtAGO1/5/10 and AtAGO2/3/7 clades, as predicted using SWISS-MODEL. PAZ (yellow), PIWI (blue), and MID (red) domains as predicted using SMART and Pfam are displayed. DEDD/H is marked by magenta spheres.
Function prediction and subcellular localization
To better understand the biological functions of CqDCL, CqAGO, and CqRDR, Expasy was used to perform gene ontology (GO) annotations. The GO annotations for CqDCL and CqRDR were relatively complete. In the CqAGO family, CqAGO5a had the most comprehensive annotations, it participates in miRNA binding (GO:0035198), miRNA loading onto RISC involved in gene silencing by miRNA (GO:0035280), and miRNA-mediated inhibition of translation (GO:0035278), all of which are closely related to the RNAi pathway. CqAGO1a is involved in miRNA binding (GO:0035198) and gene silencing by miRNA (GO:0035195). Eight CqRDR genes play a role in the production of small interfering RNA (siRNA) involved in chromatin silencing by small RNA (GO:0070919) and the production of siRNA involved in RNA interference (GO:0030422). Moreover, 12 genes (eight AGOs and four RDRs) are involved in nucleic acid binding (GO:0003676), four genes (three DCLs and one RDR) are involved in RNA binding (GO:0003723), and CqAGO7a is involved in RNA interference (GO:0016246) (Tables S6–8). Most CqAGOs, CqDCLs, and CqRDRs showed some annotations related to RNAi, indicating that these genes are closely related to the RNAi pathway in quinoa.
Most CqAGOs are located in the nucleus, except for CqAGO4a (predicted to localize in the cytosol), CqAGO5a (predicted to localize in the mitochondrion and chloroplast), and CqAGO6a and CqAGO6b (predicted to localize in the cytosol and mitochondrion). All CqDCLs are localized in the nucleus, CqDCL1a, CqDCL1b, CqDCL3a, and CqDCL4b are also localized in the cytosol, and CqDCL2b is also localized in the membrane. As for CqRDRs, CqRDR3a, CqRDR3c, CqRDR3f, and CqRDR6b are localized in the cytosol and chloroplast, CqRDR2a, CqRDR3d, and CqRDR 3e are localized only in the cytosol, CqRDR1 and CqRDR3b are localized only in the nucleus, CqRDR6a is localized in the nucleus and cytosol, and CqRDR2b is localized in the nucleus and mitochondrion (Table S9).
Expression profiles of CqAGOs, CqDCLs, and CqRDRs
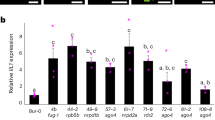
The RNA-seq results showed that eight CqDCLs were expressed in dry seeds, one-week-old seedlings, stems, leaves, and inflorescences from six-week-old plants. CqAGO4b, CqAGO4e, and CqRDR2b were not expressed in dry seeds, while CqAGO4b, and CqAGO5e were not detected in leaves. Most of the CqAGOs, CqDCLs and CqRDRs had the highest expression levels in inflorescences. Only CqAGO10b and CqDCL4b had the highest expression in internode stems, and CqRDR3c had the highest expression in seedlings (Fig. 8A). In addition, most CqAGO, CqDCL and CqRDR genes responded to drought, heat, salt and low phosphorus stresses, being up- or downregulated. The expression levels of CqAGO5a, CqAGO5b, CqAGO7a, CqDCL2a, CqDCL4a, and CqDCL4b all showed a downward trend under the four stresses in the two tissues (Fig. 8B). According to the result of RNA-seq, 11 candidate genes were screened. Because of the high homology of CqAGO10a and CqAGO10b, we could not design primers with high specificity. Thus, five, three, and one genes from CqAGOs, CqDCLs, and CqRDRs, respectively, were chosen to analysed by electrophoresis. The electrophoresis results showed that all the tested genes were expressed in five tissues, with high expression in the stems, leaves, and inflorescences, and relatively low expression in seedlings and dry seeds (Fig. 8C,D). These findings were consistent with the RNA-seq results.

(A) Expression profiles of CqAGOs, CqDCLs, and CqRDRs in different tissues, including dry seeds; one-week-old seedlings; stems, leaves, and inflorescences of six-week-old plants. (B) Expression profiles of CqAGOs, CqDCLs, and CqRDRs under low-phosphate, heat, drought, and salt stresses in root and shoot, respectively. The log2 normalized value of original TPM data are represented in both figures. The colour bar at the right of the heat map represents relative expression values. Electrophoresis analysis in different tissues of CqAGOs (C) and CqDCLs, CqRDR (D). β-actin was used as a control for each tissue types.
Discussion
In this study, we investigated the phylogenetic relationship, structure, and functions of CqAGO, CqDCL, and CqRDR proteins. Our results showed that, compared to Arabidopsis, CqAGO, CqDCL, and CqRDR exhibited more copies and may thus have higher functional diversification. We discuss the phylogenetic relationships and predict structural domains in detail.
The AGO protein is the main executive element of RISC and the main effector of RNAi. In Arabidopsis, AtAGO4, 6, and 9 in the AGO4/6/8/9 clade are mainly bound to 24-nt siRNA and are responsible for RNA-directed DNA methylation39,40. In turn, AGO proteins in the AGO1/5/10 clade participate in the regulation of plant development and stress responses. For example, AGO1 binds to miR156 in seedlings, is involved in shoot development41,42, and can also bind to chromatin in response to hormones and biotic and abiotic stress conditions43,44; AGO5 interacts with miR156 to control flowering time45; and AGO10 regulates the development of shoot apex meristem46. Additionally, AtAGO1, 2, 3, and 7 play an important role in plant adaptation to salt stress46, and AtAGO7 plays a critical role in the transition from the juvenile to the adult stage during plant growth47. In addition, AGO1, AGO2, AGO3, AGO4, and AGO5 in Arabidopsis are involved in the antiviral defence response48,49.
Structural visualization can provide an understanding of the differences between CqAGO and AtAGO. As demonstrated by the secondary and 3D structures, AGO generally has several important domains: N domain, L2, PAZ, MID, and PIWI. The 3D model of the AGO protein shows a bilobal protein, wherein the N domain, the linker region, and the PAZ domain form the N-terminal lobe, while the MID and PIWI domains constitute the C-terminal lobe50, with a cleft between the two. The central cleft is composed of positively charged aa residues that can promote the binding of negatively charged small RNAs51. The N domain of AGO is involved in the cleavage of target RNA and the dissociation of sRNA double strands9,52. Further, L2 can connect the PAZ domain with MID, N domain with DUF1785 domains to help stabilize the entire protein structure51. The MID domain has a binding region named nucleotide-specific loop, which can recognize and bind the 5′ nucleotide of sRNA, making the binding of AGO and sRNA highly specific. For example, AtAGO1 containing the MID region preferentially recognizes the sRNA with a 5′ U, whereas AtAGO4 and AtAGO5 preferentially recognize sRNAs with 5′ A and 5′ C, respectively36. Furthermore, the QF-V motif of PIWI domain in AtAGO1 and AtAGO2 helps to recognize the #15 base pair in the sRNA duplex, and is essential for the effective sorting of miRNA into AtAGO1 and AtAGO253. In this study, all CqAGO members in the AtAGO1/5/10 clade and CqAGO4c, 6a and 6b in the AGO4/6/8/9 clade contained the MID domain, therefore, it can be inferred that they may recognize sRNAs through the QF-V motif or the MID domain. However, all CqAGOs grouped in the AtAGO2/3/7 clade did not contain an identifiable MID domain, suggesting that they may recognize sRNA through the QF-V motif only.
Furthermore, all CqAGOs contain PAZ and PIWI domains. The PAZ and PIWI domains are important domains that form RISC; the PAZ domain can recognize the 2-nt 3′ end of sRNA54, while the PIWI domain can bind the 5′ end of siRNA to the target RNA, cleaving the target RNA complementary to the sRNA sequence55,56. The DEDD/H in the PIWI domain is required for RNase H-like endonuclease activity57. Most CqAGOs also exhibit the DEDD/H catalytic tetrad except for CqAGO2/3, in which the first D was replaced by N, and CqAGO5e, which is a short protein that lacks the last catalytic residue D/H. Incomplete catalytic residues may fail CqAGO2/3 and 5e to perform slice activity, may induce gene silencing by other means, or help in performing novel functions. However, studies have shown that even if the AGO protein has a conserved catalytic tetrad, it may not necessarily have endonuclease activity. It has been determined that AtAGO1, AtAGO2, AtAGO4, AtAGO7, and AtAGO10 have endonuclease activity57; however, other AGOs do not display endonuclease activity and may regulate PTGS by inhibiting the translation of target RNA58. In addition, the H798 residue in the PIWI domain is important for cleavage function, and its lack thereof leads to cleavage deficiency in AtAGO159. The P residue replaces the H residue in barley, adding HvAGO5b to act as a chromatin modifier38. In the AGO4/6/8/9 clade, CqAGO4b, 4c, 4d, 4e, 4f, 6a, 6b, and 8/9 contain P residues (Fig. 4A), which suggests that these CqAGOs lack cleavage function and may act as chromatin modifiers. The N residues in AtAGO1 and OsAGO1 in the PIWI domain may preferentially bind to 5′ U 21-nt sRNA36,60, whereas in the CqAGO family, 11 of 21 CqAGOs have retained the N residues, including 1a, 1b, 4a, 4b, 4c, 4d, 4e, 4f, 8/9, 10a, and 10b (Fig. 4B). This indicates that these AGOs may have similar preferences. AGOs in the same clade of quinoa and Arabidopsis were conserved in terms of aa sequence, secondary, and 3D structures, suggesting greater functional similarity among them. Nonetheless, these results warrant further investigation.
As a member of the ribonuclease III enzyme family, DCL can regulate gene expression and participate in antiviral defence via the RNAi pathway37. Arabidopsis encodes four DCL proteins that produce different sRNAs. AtDCL1 is related to the production of miRNAs, which can regulate gene expression in fundamental biological processes, such as development and metabolism11,13. In contrast, AtDCL2, AtDCL3, and AtDCL4 process long dsRNA into 22-, 24- and 21-nt-long siRNA, respectively61. Furthermore, AtDCL2 and AtDCL4 are also involved in antiviral defence response62, and AtDCL3 mainly guides chromatin modification and maintains genome stability63. DCL proteins mainly include six domains: DEXDc, HELICc, Dicer-dimer, PAZ, RIBOc, and DSRM. PAZ and RIBOc are essential for the removal of siRNA from the end of the dsRNA molecule64,65. In the AtDCL4 model, the spatial arrangement of PAZ and RIBOc helps ‘measure’ cleaved dsRNA37. Studies have shown that catalytic residues N and K of RNase III A and RNase III B in RIBOc are highly conserved66. In the AtDCL4–dsRNA complex, N and K residues can interact with dsRNA37, while in the yeast RNase III—RNA complex, the N and K residues can interact with the 5′-phosphate group of the cleavage bond67. The N and K of RNase III A and RNase III B in CqDCL are highly conserved (Fig. 5B,C), indicating that N and K in CqDCL may also be involved in the cleavage of the phosphodiester bond. Consistent with the prediction of AtDCL2 in the same clade68, only one DSRM domain was predicted for CqDCL2a and CqDCL2b. In terms of structure, they did not contain a second DSRM domain. DSRM may be involved in protein–protein interactions, such as the specific binding of AtDCL to the HYPONASTIC LEAVES (HYL) protein family69. As the DSRM domain also mediates the transfer of sRNA to the appropriate AGO protein70, the partial deletion of the DSRM domain may affect the binding of DCL and downstream genes of the RNAi pathway.
Single-stranded RNA molecules are used by RDR as templates to synthesize dsRNA, which is then cleaved by DCL into secondary siRNA to enhance and maintain the silent state of the target RNA71. Studies have shown that RDR can regulate reproductive development in Arabidopsis, including female gametophyte development, maternal-to-zygotic transition, self-fertilization, and double fertilization72,73,74,75. Furthermore, RDR is involved in the antiviral response. Thus, AtRDR1, AtRDR2, and AtRDR6 have lost or altered functions, thereby increasing susceptibility to a variety of plant viruses and viral RNA accumulation76. Under various stress conditions, the AtRDR6 gene is the most sensitive, it is induced in response to high temperatures and repressed during long exposure to salt or cold stress, while AtRDR1 and AtRDR5 expression decrease during prolonged exposure to high salinity or low temperatures77.
The RDR protein has only one conserved catalytic domain: RdRP. Of the three main types of RDRs (RDRα, RDRβ, and RDRγ), plants only contain RDRα and RDRγ78. In Arabidopsis, AtRDR1, 2, and 6 belong to RDRα and contain the typical C-terminal catalytic motif, DLDGD. RDR1, RDR2, or RDR6 can mediate the production of a variety of viral siRNAs and play an important role in defence against viruses in plants79,80,81,82,83. In quinoa, CqRDR1, 2a, 2b, 6a, and 6b, belonging to the AtRDR1/2/6 clade, share the DLDGD motif84. This similarity in structure implies that CqRDR1, 2a, 2b, 6a, and 6b play a role in plant defence responses against pathogens. Owing to their high sequence similarity, AtRDR3, 4, and 5, also named RDR3a, 3b, and 3c, belong to RDRγ78 and share an uncharacteristic catalytic DFDGD motif84. The CqRDR motif belonging to the AtRDR3 clade is DL/YDGD, in which F is replaced by L/Y, with an unknown function. Each CqRDR has an extension, except for AtRDR1, which may indicate the diversification of the quinoa RDR family.
Subcellular localization is important to understand the molecular functions of AGO, DCL, and RDR. Furthermore, miRNAs (such as miR-29b) that bind to AGO, contain a nuclear localization signal (NLS)85. AGO participates in the formation of heterochromatin by recruiting methyltransferase and acetyltransferase onto chromatin to perform TGS in various organisms and participates in transcriptional silencing in the nucleus86. Similarly, most CqAGOs are localized to the nucleus (Table S9). Studies have shown that DCL1-GFP and DCL4-GFP fusion proteins are localized in the nucleus69. Non-classical NLS have detected in the dsRNA C-terminal binding domains of DCL1 and DCL486, and these NLSs can likely guide DCL1 and DCL4 to the nucleus. This study predicted that all CqDCL members are localized in the nucleus, further implying that CqAGOs and CqDCLs may participate in the RNAi pathway.
In this study, the tissue-specific and abiotic stress expression patterns of CqAGO, CqDCL and CqRDR genes were investigated. RNA-seq results showed that most of these genes were expressed in five tissues including dry seeds, seedlings, internode stems, inflorescences and leaves, but the expression of the same gene varied in different tissues, indicating that these genes may be involved in different developmental processes. Studies have shown that AtAGO1 regulates leaf development, and AtAGO1 together with AtAGO10 regulates floral stem cell termination through miR172 and miR165/16687. CqAGO1a and CqAGO1b (the homologous genes of AtAGO1) were highly expressed in five tissues, thus, we speculated that they may be involved in the development or maintenance of leaves and flowers. OsDCLs and AtDCLs were expressed in different tissues, AtDCL1, AtDCL3, and AtDCL4 were expressed at higher levels in flowers88. Similarly, CqDCL1a, CqDCL1b, and CqDCL4a were also expressed in all five tested tissues, and showed the highest expression in inflorescences. OsRDR6 is required for floral organ development89, and CqRDR6a and CqRDR6b are also highly expressed in inflorescences in the RNA-seq results, suggesting that CqDCLs and CqRDRs are involved in floral organ development in quinoa. Plants resist the effects of stress in a variety of ways. In response to stresses such as drought, salt, and heat, the expression of many plant genes changes. For example, the expression of OsDCL was slightly inhibited under drought, cold and salt stress88. Under salt stress, the expression of AtDCL1 in roots and shoots showed a downward trend, and the expression of AtDCL4 decreased with the prolongation of salt treatment time88. Similar to these results, based on the result of the RNA-seq, the expression levels of most CqDCLs in both tissues were decreased in all stresses, which suggested that most CqDCLs are involved in abiotic stress responses.
Conclusion
In this study, 21 CqAGO, eight CqDCL and 11 CqRDR genes were identified in C. quinoa. Based on bioinformatics analyses, we aimed to improve the understanding of these gene families, including their genomic location, phylogenetic relationship, domain components, 3D structure, related functional annotations, subcellular localization and expression patterns. We show that these gene families have the potential to regulate gene transcription and translation, which may indicate a role in the typical RNAi pathway in quinoa. This is the first report that provides insight into important gene families involved in the biogenesis of sRNA in quinoa, which paves the way for further functional characterization of these genes. This information can be used to improve stress resistance and yield quality in quinoa. However, in addition to our bioinformatics analyses, further investigation is needed to confirm the functions of these proteins and pinpoint their roles in the involvement of the RNAi pathway in growth and development and disease resistance in quinoa.
Methods
Sequence acquisition and database search
Sequence information of the AGO, DCL, and RDR genes in Arabidopsis was obtained from TAIR (http://www.arabidopsis.org) (Table S1). The coding sequences (CDS), protein sequences, CDS length, and peptide length in quinoa corresponding to the primary transcripts of Arabidopsis homologous genes were downloaded from the Plant Comparative Genomics portal Phyzome 13 Chenopodium quinoa v1.0 database (https://phytozome-next.jgi.doe.gov/)90. The description and chromosomal location of CqAGO-, CqDCL-, and CqRDR-related sequences were determined using Expasy (https://www.expasy.org/), the chromosome location of these genes were represented using online tool Mapgene2chrom (http://mg2c.iask.in/mg2c_v2.1/)91.
Phylogenetic and structural analyses, gene ontology annotation, and subcellular localization
Phylogeny analysis was performed using the Phylogeny.fr web server (http://phylogeny.lirmm.fr/phylo_cgi/index.cgi)92, the CqAGO, CqDCL, and CqRDR genes predicted in the quinoa genome were named according to their phylogenetic relationship with the members of the same protein family in Arabidopsis. Multiple sequence alignment (MSA) was performed using Clustal Omega (https://www.ebi.ac.uk/Tools/msa/clustalo/)93, and multiple alignment viewer (Mview) (https://www.ebi.ac.uk/Tools/msa/mview/)93 was used to visualize the conserved residues and domains of related proteins. To identify the similarity of two sequences, we used the pairwise sequence alignment tool EMBOSS Needle (https://www.ebi.ac.uk/Tools/psa/)93. The simple modular architecture research tool (SMART) (http://smart.embl-heidelberg.de/)94 in normal SMART mode was used for the analysis of protein domains, and illustrator for biological sequences (IBS) (http://ibs.biocuckoo.org/)95 were used for domain visualization. The gene intron structures were predicted using the online software gene structure display server (GSDS) v.2.0 (http://gsds.gao-lab.org/)96. GO annotation was performed using Expasy (https://www.expasy.org/), and protein localization was predicted using the plant subcellular localization integrative predictor (PSI) (http://bis.zju.edu.cn/psi/).
3D structure modeling and verification
SWISS-MODEL, a homology-based modeling software (https://beta.Swissmodel.Expasy.Org/)97, was used to predict the 3D structure of proteins, and the template was checked using SAVES v 6.0 (https://saves.mbi.ucla.edu/). The Python molecular graphics system (PyMOL) (https://pymol.org/2) was used to visualize protein structures.
Expression profile analysis of CqAGOs, CqDCLs and CqRDRs
Transcriptome data of quinoa-related tissues were downloaded from NCBI’s SRA database, including different tissues (SRP116149), and different stresses of drought, heat, salt, and low phosphorus in the root and shoot (SRS1538629). The R package pheatmap was used to cluster and visualize the data with the following parameter settings: distance measure, Euclidean; clustering method, Median. Kallisto was used to calculate the expression level. Tissue expression heatmaps were drawn using TBtools98.
Plant materials, RNA extraction, PCR amplification and electrophoresis
Quinoa (QQ74) was provided by the Agricultural College, Shanxi Agricultural University, grown in growth chambers at 24 °C day/22 °C night, 16 day length. Dry seeds, one-week-old seedlings, stems, leaves, and inflorescences from six-week-old plants were collected and frozen in liquid nitrogen. Total RNA from different tissues was extracted according to the instructions of Trizol, and cDNA was obtained by reverse transcription. The genes that the reads of RNA-sequencing (RNA-seq) were greater than 100 in all five tissues were selected, designed primers, and amplified. After PCR amplification, PCR products were analysed via electrophoresis on 1.5% agarose gels, and the amplified target fragment was observed with Quantity One software. Primers were listed in Table S10. The complete electrophoretic diagrams were shown in Fig. S3. Experimental research on the plant(s)/plant material complied with the relevant institutional, national, and international guidelines and legislation.
Data availability
All data generated or analysed during this study are included in this published article and its supplementary information files.
References
Lisitskaya, L., Aravin, A. A. & Kulbachinskiy, A. DNA interference and beyond: Structure and functions of prokaryotic Argonaute proteins. Nat. Commun. 9, 5165 (2018).
Mello, C. C. & Conte, D. Jr. Revealing the world of RNA interference. Nature 431, 338–342 (2004).
Sarkies, P. & Miska, E. A. Small RNAs break out: The molecular cell biology of mobile small RNAs. Nat. Rev. Mol. Cell Biol. 15, 525–535 (2014).
Siomi, H. & Siomi, M. C. On the road to reading the RNA-interference code. Nature 457, 396–404 (2009).
Guo, Z., Li, Y. & Ding, S. W. Small RNA-based antimicrobial immunity. Nat. Rev. Immunol. 19, 31–44 (2019).
Mosharaf, M. P. et al. In silico identification and characterization of AGO, DCL and RDR gene families and their associated regulatory elements in sweet orange (Citrus sinensis L.). PLoS ONE 15, e0228233 (2020).
Fukudome, A. & Fukuhara, T. Plant dicer-like proteins: Double-stranded RNA-cleaving enzymes for small RNA biogenesis. J. Plant Res. 130, 33–44 (2017).
Iwasaki, S. et al. Hsc70/Hsp90 chaperone machinery mediates ATP-dependent RISC loading of small RNA duplexes. Mol. Cell 39, 292–299 (2010).
Kwak, P. B. & Tomari, Y. The N domain of Argonaute drives duplex unwinding during RISC assembly. Nat. Struct. Mol. Biol. 19, 145–151 (2012).
Niaz, S. The AGO proteins: An overview. Biol. Chem. 399, 525–547 (2018).
Voinnet, O. Origin, biogenesis, and activity of plant microRNAs. Cell 136, 669–687 (2009).
Moazed, D. Small RNAs in transcriptional gene silencing and genome defence. Nature 457, 413–420 (2009).
Bartel, D. P. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell 116, 281–297 (2004).
Bologna, N. G. & Voinnet, O. The diversity, biogenesis, and activities of endogenous silencing small RNAs in Arabidopsis. Annu. Rev. Plant Biol. 65, 473–503 (2014).
Kapoor, M. et al. Genome-wide identification, organization and phylogenetic analysis of Dicer-like, Argonaute and RNA-dependent RNA Polymerase gene families and their expression analysis during reproductive development and stress in rice. BMC Genomics 9, 451 (2008).
Qian, Y. et al. Identification and characterization of Dicer-like, Argonaute and RNA-dependent RNA polymerase gene families in maize. Plant Cell Rep. 30, 1347–1363 (2011).
Yadav, C. B., Muthamilarasan, M., Pandey, G. & Prasad, M. Identification, characterization and expression profiling of Dicer-like, Argonaute and RNA-dependent RNA polymerase gene families in foxtail millet. Plant Mol. Biol. Report. 33, 43–55 (2015).
Zhao, H. et al. Comprehensive analysis of Dicer-like, Argonaute, and RNA-dependent RNA polymerase gene families in grapevine (Vitis Vinifera). J. Plant Growth Regul. 34, 108–121 (2015).
Bai, M. et al. Genome-wide identification of Dicer-like, Argonaute and RNA-dependent RNA polymerase gene families and their expression analyses in response to viral infection and abiotic stresses in Solanum lycopersicum. Gene 501, 52–62 (2012).
Akond, Z. et al. Comprehensive in silico analysis of RNA silencing-related genes and their regulatory elements in wheat (Triticum aestivum L.). Biomed. Res. Int. 2022, 4955209 (2022).
Liu, X., Lu, T., Dou, Y., Yu, B. & Zhang, C. Identification of RNA silencing components in soybean and sorghum. BMC Bioinform. 15, 4 (2014).
Qin, L., Mo, N., Muhammad, T. & Liang, Y. Genome-wide analysis of DCL, AGO, and RDR gene families in pepper (Capsicum annuum L.). Int. J. Mol. Sci. 19, 1038 (2018).
Gan, D. et al. Genome-wide identification of the Dicer-like, Argonaute, and RNA-dependent RNA polymerase gene families in cucumber (Cucumis sativus L.). J. Plant Growth Regul. 35, 135–150 (2016).
Hamar, E. et al. Genome-wide identification of RNA silencing-related genes and their expressional analysis in response to heat stress in barley (Hordeum vulgare L.). Biomolecules 10, 929 (2020).
Cui, D. L. et al. Genome-wide identification and characterization of DCL, AGO and RDR gene families in Saccharum spontaneum. Sci. Rep. 10, 13202 (2020).
Krishnatreya, D. B. et al. Genome-wide identification, evolutionary relationship and expression analysis of AGO, DCL and RDR family genes in tea. Sci. Rep. 11, 8679 (2021).
Miranda, M. et al. Antimicrobial potential and phytochemical content of six diverse sources of quinoa seeds (Chenopodium quinoa Willd.). Agric. Sci. 5, 1015–1024 (2014).
Vega-Galvez, A. et al. Nutrition facts and functional potential of quinoa (Chenopodium quinoa willd.), an ancient Andean grain: A review. J. Sci. Food Agric. 90, 2541–2547 (2010).
Lin, M. et al. Quinoa secondary metabolites and their biological activities or functions. Molecules 24, 2512 (2019).
Graf, B. L. et al. Quinoa seeds leach phytoecdysteroids and other compounds with anti-diabetic properties. Food Chem. 163, 178–185 (2014).
Yao, Y., Yang, X., Shi, Z. & Ren, G. Anti-inflammatory activity of saponins from quinoa (Chenopodium quinoa Willd.) seeds in lipopolysaccharide-stimulated RAW 264.7 macrophages cells. J. Food Sci. 79, H1018-1023 (2014).
Yao, Y., Shi, Z. & Ren, G. Antioxidant and immunoregulatory activity of polysaccharides from quinoa (Chenopodium quinoa Willd.). Int. J. Mol. Sci. 15, 19307–19318 (2014).
Angeli, V. et al. Quinoa (Chenopodium quinoa Willd.): An overview of the potentials of the “golden grain” and socio-economic and environmental aspects of its cultivation and marketization. Foods 9, 216 (2020).
Seo, J. K., Wu, J., Lii, Y., Li, Y. & Jin, H. Contribution of small RNA pathway components in plant immunity. Mol. Plant Microbe Interact. 26, 617–625 (2013).
Wheeler, B. S. Small RNAs, big impact: Small RNA pathways in transposon control and their effect on the host stress response. Chromosome Res. 21, 587–600 (2013).
Mi, S. et al. Sorting of small RNAs into Arabidopsis argonaute complexes is directed by the 5′ terminal nucleotide. Cell 133, 116–127 (2008).
Mickiewicz, A. et al. Modeling of the catalytic core of Arabidopsis thaliana Dicer-like 4 protein and its complex with double-stranded RNA. Comput. Biol. Chem. 66, 44–56 (2017).
Secic, E., Zanini, S. & Kogel, K. H. Further elucidation of the Argonaute and Dicer protein families in the model grass species Brachypodium distachyon. Front. Plant Sci. 10, 1332 (2019).
Mallory, A. & Vaucheret, H. Form, function, and regulation of ARGONAUTE proteins. Plant Cell 22, 3879–3889 (2010).
Havecker, E. R. et al. The Arabidopsis RNA-directed DNA methylation argonautes functionally diverge based on their expression and interaction with target loci. Plant Cell 22, 321–334 (2010).
Ronemus, M., Vaughn, M. W. & Martienssen, R. A. MicroRNA-targeted and small interfering RNA-mediated mRNA degradation is regulated by argonaute, dicer, and RNA-dependent RNA polymerase in Arabidopsis. Plant Cell 18, 1559–1574 (2006).
Azevedo, J. et al. Argonaute quenching and global changes in Dicer homeostasis caused by a pathogen-encoded GW repeat protein. Genes Dev. 24, 904–915 (2010).
Liu, C. et al. Arabidopsis ARGONAUTE 1 binds chromatin to promote gene transcription in response to hormones and stresses. Dev. Cell 44, 348-361.e347 (2018).
Dolata, J. et al. Salt stress reveals a new role for ARGONAUTE1 in miRNA biogenesis at the transcriptional and posttranscriptional levels. Plant Physiol. 172, 297–312 (2016).
Roussin-Leveillee, C., Silva-Martins, G. & Moffett, P. ARGONAUTE5 represses age-dependent induction of flowering through physical and functional interaction with miR156 in Arabidopsis. Plant Cell Physiol. 61, 957–966 (2020).
Zhu, H. et al. Arabidopsis Argonaute10 specifically sequesters miR166/165 to regulate shoot apical meristem development. Cell 145, 242–256 (2011).
Hunter, C., Sun, H. & Poethig, R. S. The Arabidopsis heterochronic gene ZIPPY is an ARGONAUTE family member. Curr. Biol. 13, 1734–1739 (2003).
Kwon, J. et al. RNA silencing-related genes contribute to tolerance of infection with potato virus X and Y in a susceptible tomato plant. Virol. J. 17, 149 (2020).
Agorio, A. & Vera, P. ARGONAUTE4 is required for resistance to Pseudomonas syringae in Arabidopsis. Plant Cell 19, 3778–3790 (2007).
Song, J. J., Smith, S. K., Hannon, G. J. & Joshua-Tor, L. Crystal structure of Argonaute and its implications for RISC slicer activity. Science 305, 1434–1437 (2004).
Poulsen, C., Vaucheret, H. & Brodersen, P. Lessons on RNA silencing mechanisms in plants from eukaryotic argonaute structures. Plant Cell 25, 22–37 (2013).
Hauptmann, J. et al. Turning catalytically inactive human Argonaute proteins into active slicer enzymes. Nat. Struct. Mol. Biol. 20, 814–817 (2013).
Zhang, X. et al. ARGONAUTE PIWI domain and microRNA duplex structure regulate small RNA sorting in Arabidopsis. Nat. Commun. 5, 5468 (2014).
Ma, J. B., Ye, K. & Patel, D. J. Structural basis for overhang-specific small interfering RNA recognition by the PAZ domain. Nature 429, 318–322 (2004).
Hock, J. & Meister, G. The Argonaute protein family. Genome Biol. 9, 210 (2008).
Rivas, F. V. et al. Purified Argonaute2 and an siRNA form recombinant human RISC. Nat. Struct. Mol. Biol. 12, 340–349 (2005).
Fang, X. & Qi, Y. RNAi in plants: An argonaute-centered view. Plant Cell 28, 272–285 (2016).
Carthew, R. W. & Sontheimer, E. J. Origins and Mechanisms of miRNAs and siRNAs. Cell 136, 642–655 (2009).
Baumberger, N. & Baulcombe, D. C. Arabidopsis ARGONAUTE1 is an RNA Slicer that selectively recruits microRNAs and short interfering RNAs. Proc. Natl. Acad. Sci. USA 102, 11928–11933 (2005).
Wu, L. et al. Rice MicroRNA effector complexes and targets. Plant Cell 21, 3421–3435 (2009).
Henderson, I. R. et al. Dissecting Arabidopsis thaliana DICER function in small RNA processing, gene silencing and DNA methylation patterning. Nat. Genet. 38, 721–725 (2006).
Deleris, A. et al. Hierarchical action and inhibition of plant Dicer-like proteins in antiviral defense. Science 313, 68–71 (2006).
Chan, S. W. et al. RNA silencing genes control de novo DNA methylation. Science 303, 1336 (2004).
Zhang, H., Kolb, F. A., Jaskiewicz, L., Westhof, E. & Filipowicz, W. Single processing center models for human Dicer and bacterial RNase III. Cell 118, 57–68 (2004).
Macrae, I. J. et al. Structural basis for double-stranded RNA processing by Dicer. Science 311, 195–198 (2006).
Du, Z., Lee, J. K., Tjhen, R., Stroud, R. M. & James, T. L. Structural and biochemical insights into the dicing mechanism of mouse Dicer: A conserved lysine is critical for dsRNA cleavage. Proc. Natl. Acad. Sci. USA 105, 2391–2396 (2008).
Liang, Y. H., Lavoie, M., Comeau, M. A., Abou Elela, S. & Ji, X. Structure of a eukaryotic RNase III postcleavage complex reveals a double-ruler mechanism for substrate selection. Mol. Cell 54, 431–444 (2014).
Margis, R. et al. The evolution and diversification of Dicers in plants. FEBS Lett. 580, 2442–2450 (2006).
Hiraguri, A. et al. Specific interactions between Dicer-like proteins and HYL1/DRB-family dsRNA-binding proteins in Arabidopsis thaliana. Plant Mol. Biol. 57, 173–188 (2005).
Parker, G. S., Maity, T. S. & Bass, B. L. dsRNA binding properties of RDE-4 and TRBP reflect their distinct roles in RNAi. J. Mol. Biol. 384, 967–979 (2008).
Devert, A. et al. Primer-dependent and primer-independent initiation of double stranded RNA synthesis by purified Arabidopsis RNA-dependent RNA polymerases RDR2 and RDR6. PLoS ONE 10, e0120100 (2015).
Tantikanjana, T., Rizvi, N., Nasrallah, M. E. & Nasrallah, J. B. A dual role for the S-locus receptor kinase in self-incompatibility and pistil development revealed by an Arabidopsis rdr6 mutation. Plant Cell 21, 2642–2654 (2009).
Olmedo-Monfil, V. et al. Control of female gamete formation by a small RNA pathway in Arabidopsis. Nature 464, 628–632 (2010).
Ron, M., Alandete Saez, M., Eshed Williams, L., Fletcher, J. C. & McCormick, S. Proper regulation of a sperm-specific cis-nat-siRNA is essential for double fertilization in Arabidopsis. Genes Dev. 24, 1010–1021 (2010).
Autran, D. et al. Maternal epigenetic pathways control parental contributions to Arabidopsis early embryogenesis. Cell 145, 707–719 (2011).
Pumplin, N. & Voinnet, O. RNA silencing suppression by plant pathogens: Defence, counter-defence and counter-counter-defence. Nat. Rev. Microbiol. 11, 745–760 (2013).
Willmann, M. R., Endres, M. W., Cook, R. T. & Gregory, B. D. The functions of RNA-dependent RNA Polymerases in Arabidopsis. Arabidopsis Book 9, e0146 (2011).
Zong, J., Yao, X., Yin, J., Zhang, D. & Ma, H. Evolution of the RNA-dependent RNA polymerase (RdRP) genes: Duplications and possible losses before and after the divergence of major eukaryotic groups. Gene 447, 29–39 (2009).
Qu, F., Ye, X. & Morris, T. J. Arabidopsis DRB4, AGO1, AGO7, and RDR6 participate in a DCL4-initiated antiviral RNA silencing pathway negatively regulated by DCL1. Proc. Natl. Acad. Sci. USA 105, 14732–14737 (2008).
Garcia-Ruiz, H. et al. Arabidopsis RNA-dependent RNA polymerases and dicer-like proteins in antiviral defense and small interfering RNA biogenesis during Turnip Mosaic Virus infection. Plant Cell 22, 481–496 (2010).
Wang, X. B. et al. RNAi-mediated viral immunity requires amplification of virus-derived siRNAs in Arabidopsis thaliana. Proc. Natl. Acad. Sci. USA 107, 484–489 (2010).
Cao, M. et al. Virus infection triggers widespread silencing of host genes by a distinct class of endogenous siRNAs in Arabidopsis. Proc. Natl. Acad. Sci. USA 111, 14613–14618 (2014).
Diaz-Pendon, J. A., Li, F., Li, W. X. & Ding, S. W. Suppression of antiviral silencing by cucumber mosaic virus 2b protein in Arabidopsis is associated with drastically reduced accumulation of three classes of viral small interfering RNAs. Plant Cell 19, 2053–2063 (2007).
Wassenegger, M. & Krczal, G. Nomenclature and functions of RNA-directed RNA polymerases. Trends Plant Sci. 11, 142–151 (2006).
Hwang, H. W., Wentzel, E. A. & Mendell, J. T. A hexanucleotide element directs microRNA nuclear import. Science 315, 97–100 (2007).
Pong, S. K. & Gullerova, M. Noncanonical functions of microRNA pathway enzymes—Drosha, DGCR8, Dicer and Ago proteins. FEBS Lett. 592, 2973–2986 (2018).
Ji, L. et al. ARGONAUTE10 and ARGONAUTE1 regulate the termination of floral stem cells through two microRNAs in Arabidopsis. PLoS Genet. 7, e1001358 (2011).
Liu, Q., Feng, Y. & Zhu, Z. Dicer-like (DCL) proteins in plants. Funct. Integr. Genomics 9, 277–286 (2009).
Song, X. et al. Rice RNA-dependent RNA polymerase 6 acts in small RNA biogenesis and spikelet development. Plant J. 71, 378–389 (2012).
Goodstein, D. M. et al. Phytozome: A comparative platform for green plant genomics. Nucleic Acids Res. 40, D1178-1186 (2012).
Chao, J. et al. MG2C: A user-friendly online tool for drawing genetic maps. Mol. Hortic. 1, 16 (2021).
Dereeper, A. et al. Phylogeny.fr: Robust phylogenetic analysis for the non-specialist. Nucleic Acids Res. 36, W465–W469 (2008).
Madeira, F. et al. The EMBL-EBI search and sequence analysis tools APIs in 2019. Nucleic Acids Res. 47, W636–W641 (2019).
Letunic, I., Khedkar, S. & Bork, P. SMART: Recent updates, new developments and status in 2020. Nucleic Acids Res. 49, D458–D460 (2021).
Liu, W. et al. IBS: An illustrator for the presentation and visualization of biological sequences. Bioinformatics 31, 3359–3361 (2015).
Hu, B. et al. GSDS 2.0: An upgraded gene feature visualization server. Bioinformatics 31, 1296–1297 (2015).
Waterhouse, A. et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 46, W296–W303 (2018).
Chen, C. et al. TBtools: An integrative toolkit developed for interactive analyses of big biological data. Mol. Plant. 13, 1194–1202 (2020).
Acknowledgements
We gratefully acknowledge Editage (http://www.editage.cn) for English language editing.
Funding
This work was supported by the Research Program Sponsored by the State Key Laboratory of Sustainable Dryland Agriculture (in preparation), Shanxi Agricultural University (NO. 202105D121008), and the earmarked fund for Modern Agro-industry Technology Research System (2022-05).
Author information
Authors and Affiliations
Contributions
X.Z. and S.Y. designed the figures. S.Y. performed data analysis and wrote the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Yun, S., Zhang, X. Genome-wide identification, characterization and expression analysis of AGO, DCL, and RDR families in Chenopodium quinoa. Sci Rep 13, 3647 (2023). https://doi.org/10.1038/s41598-023-30827-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-023-30827-1
This article is cited by
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
