
Abstract
Within macrophages and amoeba, the Legionella-containing vacuole (LCV) membrane is derived from the ER. The bona fide F-box AnkB effector protein of L. pneumophila strain AA100/130b is anchored to the cytosolic side of the LCV membrane through host-mediated farnesylation of its C-terminal eukaryotic “CaaX” motif. Here we show that the AnkB homologue of the Paris strain has a frame shift mutation that led to a loss of the CaaX motif and a concurrent generation of a unique C-terminal KNKYAP motif, which resembles the eukaryotic di-lysine ER-retention motif (KxKxx). Our phylogenetic analyses indicate that environmental isolates of L. pneumophila have a potential positive selection for the ER-retention KNKYAP motif. The AnkB-Paris effector is localized to the LCV membrane most likely through the ER-retention motif. Its ectopic expression in HEK293T cells localizes it to the perinuclear ER region and it trans-rescues the ankB mutant of strain AA100/130b in intra-vacuolar replication. The di-lysine ER retention motif of AnkB-Paris is indispensable for function; most likely as an ER retention motif that enables anchoring to the ER-derived LCV membrane. Our findings show divergent evolution of the ankB allele in exploiting either host farnesylation or the ER retention motif to be anchored into the LCV membrane.
Similar content being viewed by others


Introduction
Legionella pneumophila is an environmental organism that proliferates within various protists hosts in the aquatic environment1,2,3. Co-evolution and adaptation of L. pneumophila to the intracellular lifestyle within protists is believed to have played a major role in its ability to exploit evolutionarily conserved eukaryotic processes that enables its proliferation within human cells1,2,3. Upon aerosol transmission from the aquatic environment to humans as planktonic or in biofilms4, L. pneumophila replicates within alveolar macrophages, causing Legionnaires’ disease5.
Upon attachment to macrophages and amoeba through pili and other attachment factors6,7,8, the bacteria are internalized. Within both evolutionarily distant host cells, the L. pneumophila-containing vacuole (LCV)9 evades endocytic fusion and intercepts ER-to-Golgi vesicle traffic to be remodeled into an ER-derived vacuole10,11,12,13,14,15. The Dot/Icm type IV secretion system16, 17 injects into the host cell a cadre of ~300 effectors18, 19 to modulate a myriad of cellular processes involved in biogenesis of the LCV and to re-program the host cell into a proliferation niche11, 12, 20,21,22,23. Most of the effectors are dispensable for intracellular proliferation of L. pneumophila 24. The Ankyrin B (AnkB) effector of L. pneumophila is one of very few effectors required for the intracellular proliferation and exploits the evolutionarily-conserved ubiquitin-proteasome machinery within mammalian and protozoan cells25, 26. The crystal structure of the AnkB effector shows that it is composed of three ankyrin repeats and an F-box domain27.
Anchored to the LCV membrane, the bona fide F-box AnkB effector interacts with the host SCF1 ubiquitin ligase complex25, 26, 28,29,30. On the LCV membrane, AnkB functions as a platform for the docking of Lys48- linked polyubiquitinated proteins to the LCV membrane within human cells and amoeba as well as Drosophila-derived cells, and the process is initiated upon bacterial attachment to the plasma membrane25, 26, 29, 31, 32. High throughput proteomic analyses of the LCV polyubiquitinated proteome have identified several host proteins specifically polyubiquitinated in an AnkB-dependent manner33. The AnkB-assembled Lys48 -linked polyubiquitinated proteins are targeted to proteasomal degradation34, which is essential to raise the concentration of host cell amino acids above the threshold needed as major sources of carbon and energy for the robust intra-vacuolar proliferation of L. pneumophila within mammalian and protozoan cells5, 29, 35, 36. Thus, AnkB is designated as a nutritional virulence effector37,38,39,40,41. Interestingly, AnkB has been recently shown to be to bind the Trim 21 host ubiquitin ligase and to become ubiquitinated by K11- linked polyubiquitination32, but the role of this ubiquitination is AnkB function is not known.
Post-translational modification of hydrophilic eukaryotic proteins through farnesylation is mediated by the covalent addition of a 15-carbon farnesyl isoprenoid lipid moiety that enables anchoring of the farnesylated proteins to the lipid bi-layer of eukaryotic membranes36, 42, 43. Anchoring of the AnkB effector of strain AA100/130b of L. pneumophila into the LCV membrane is mediated through host-mediated farnesylation of the cysteine residue at the -4 position from the C-terminus within the eukaryotic-like “CaaX” motif3, 36, 44. Similar to genetic ablation of ankB 25, 26, 45, substitution of the cysteine residue within the CaaX motif results in a total loss of biological function of AnkB of strain AA100/130b, which leads to a defect in intracellular proliferation within amoeba and human cells and in attenuation in the mouse model of Legionnaires disease44. In addition to farnesylation, AnkB has been recently shown to be modified through asparagine hydroxylation by the host asaparaginyl hydroxylase, which is recruited to the LCV in a Dot/Icm-dependent manner46.
Similar to strain AA100/130b, the AnkB homologue of strain Paris (AnkB-Paris) mediates decoration of the LCV with polyubiquitinated proteins, and is also required for intracellular proliferation and for virulence in the mouse model, yet to a lesser extent than strain AA100/130b26, 45. Surprisingly, while the C-terminal CaaX motif of AnkB-AA100/130b is indispensable for anchoring the effector to the LCV membrane through host-mediated farnesylation, which is essential for function, the CaaX motif is absent from AnkB-Paris. Ectopically expressed AnkB-AA100/130b in HEK293 cells or amoeba becomes farnesylated and is uniformly localized throughout the cytosolic side of the plasma membrane where it recruits polyubiquitinated proteins that are degraded by the proteasomes25, 36, 44. Importantly, farnesylation-mediated anchoring of AnkB-AA100/130b into the plasma membrane is essential for trans-rescue of the ankB mutant in intracellular proliferation25. In contrast to AnkB-AA100/130b, upon ectopic expression within A549 cells, AnkB-Paris is enriched at the leading edge of lamellipodium formation and co-localizes with α-actinin26. However, sub-cellular location of AnkB-Paris during infection is not known. Compared to the ankB-AA100/130b allele, there is a single nucleotide deletion in the ankB-Paris leading to a frame shift, which results in a truncation of the protein for the last 18 amino acids residues, which include a portion of the third ankyrin repeat domain and the CaaX farnesylation motif. However, the respective frame shift in AnkB-Paris generated a unique C-terminus, which resembles a eukaryotic di-lysine ER-retention motif (KxKxx)47. The crystal structure of AnkB-AA100/130b indicates that the C-terminal truncation of AnkB-Paris eliminates a large portion of the third ankyrin repeat compared to AnkB-AA100/130b48. Importantly, the AnkB lysine residues modified by K11-linked polyubiquitination are conserved in the two AnkB effectors32.
Here we show that among 51 unique clinical and environmental isolates there is predominance and selection of the ankB-Paris allele in environmental isolates. The divergent evolution of ankB-Paris allele has led to acquisition of a C-terminal putative di-lysine ER retention motif, which is indispensable for biological function. The di-lysine ER retention motif likely enables anchoring of AnkB-Paris to the ER-derived LCV membrane, in contrast to the CaaX motif farnesylation-mediated anchoring of AnkB-AA100/130b. Despite truncation of the third ankyrin repeat domain in AnkB-Paris, it can functionally substitute for AnkB-AA100/130b strain in decoration of the LCV with polyubiquitinated proteins for rescue of intra-vacuolar proliferation.
Results
Episodic Positive Selection in ankB Evolution
Compared to strain AA100/130b, the ankB gene of the Paris strain (ankB-Paris) has a deletion of an adenine at position 450 (ΔA450), which resulted in a frame shift mutation (Fig. 1). This has led to a truncation of the last 18 amino acids that included the CaaX farnesylation motif, which is essential for anchoring AnkB-AA100/130b into the LCV membrane, which is indispensable for its biologic function in decorating the LCV with polyubiquitinated proteins36, 44. Despite this frame shift mutation and deletion of the C-terminal CaaX farnesylation motif, AnkB-Paris is required for decoration of the LCV with polyubiquitinated proteins26. Concurrently, a unique NKYAP sequence motif is generated at amino acids 150–154 in AnkB-Paris. To determine whether this frame shift mutation was unique to the Paris strain or more widespread among other Legionella isolates, we examined the abundance of the ΔA450 mutation in fifty-one isolates of clinical (N = 25) and environmental (N = 26) origin. Analysis of full-length ankB sequences revealed 15 distinct ankB alleles (Fig. 1b; Fig. S1). Interestingly, among the 15 ankB alleles, ankB1 (Paris strain) was the only ankB allele to harbor the ΔA450 mutation (Fig. 1b). The data showed that 19 of 51 isolates (37.25%), of which 17 were environmental and 2 were of clinical origin, contained ankB1 allele (ankB1/ankB-Paris). We conclude that the ankB1/ankB-Paris allele is wide-spread and is predominant among environmental isolates.

Molecular evolution of ankB. (a) Representative sequence chromatograms indicating the frame shift mutation at nucleotide position 450 (ΔA) in ankB1 compared to other ankB alleles. This mutation in ankB1 alters the reading frame and predicts a prematurely terminated AnkB protein at residue 154 and generation of a unique NKYAP sequence motif. Arrows mark the location of the Dot-Icm translocation signal. (b) ML analysis of ankB alleles variously from clinical (in black text) and environmental (in red text) isolates. All alleles are indicated. Diamonds indicate ankB sequence from strains whose complete genomes has been determined. Bootstrap values are shown above the branches. The tree is drawn to scale, with branch lengths measured in the number of substitutions per site.
To better understand the forces that shaped ankB evolution, in particular the maintenance and spread of the ankB1 allele in environmental Legionella isolates, we next analyzed the selective pressures acting on ankB codons and also on the ankB1 branch using CODEML and a variety of other methods (materials and methods). We found preponderance of sites that were constrained by either negative selection or evolved neutrally. Comparisons of models M7 and M8 also suggested that at least 4 ankB codons had been subject to positive selection (Table S1). However, we also found evidence of recombination in the ankB alignment (Fig. S2, Table S2). Although the M7 vs M8 comparison should be robust to model violations introduced due to recombination, we sought additional evidence to verify positive selection using REL, FEL, IFEL and MEME methods (materials and methods). Each of these methods found significant statistical support for site-specific positive selection in ankB codons (Tables S3–S7). Branch site tests implemented in CODEML, and the GA branch test both provided statistical support for the hypotheses that the internal branch leading up to ankB1/ankB-Paris and its branch both had experienced positive selection. Moreover, the NKYAP C-terminal motif itself was identified as the target of positive selection (H S1; Fig. S3 and Table S7).
Decoration of the LCV with Polyubiquitinated Proteins Independent of the ankB Genotype
We next asked whether the altered C-terminus of AnkB variant encoded by ankB1 (Fig. 1a) either modified or significantly reduced the ability of AnkB1 strains to recruit polyubiquitinated proteins to the LCV. Overall, we found extensive variation in the ability of 23 distinct Legionella isolates to recruit polyubiquitinated proteins to the LCV (Fig. 2a and b). Specifically, among AnkB1 strains, polyubiquitination varied from 30% to 54%. In contrast, six isolates with full-length ankB alleles were either similar to the ΔankB mutant or the ΔdotA translocation-defective mutant in their ability to recruit polyubiquitinated proteins to the LCV (Fig. 2a and b). However, a comparison of environmental and clinical isolates revealed a modest, but statistically significant difference (Student t-test, P < 0.05), in their ability to recruit polyubiquitinated proteins (Fig. S4). Thus, while the environmental isolates seem less capable than clinical isolates to recruit polyubiquitinated proteins to the LCV, the ability to recruit polyubiquitinated proteins itself seems independent of the ankB genotype.

AnkB-genotype does not predict the ability to recruit polyubiquitinated proteins to the LCV. (a) Representative confocal microscopy images of polyubiquitinated protein recruitment to the LCV among 23 different isolates of L. pneumophila expressing various ankB alleles. Percentages indicate the number of LCVs positive for ubiquitin. Images in the right panel were taken using the AA100 strain. (b) Distribution of percent polyubiquitin recruitment among 23 different isolates of L. pneumophila. Environmental isolates are shown in red and clinical isolates are shown in black. ‡, indicates strains carrying the ankB1 allele.
Localization of AnkB-Paris to the LCV membrane
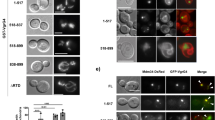
Ectopic expression of AnkB-AA100/130b within amoeba and HEK293T cells results in farnesylation-mediated anchoring to the plasma membrane of both evolutionarily distinct host cells36, 44, 49, 50. In contrast to AnkB-AA100/130b, AnkB-Paris ectopically expressed within A549 cells is enriched at the leading edge of lamellipodium formation and co-localizes with α-actinin26. Our data showed that in contrast to AnkB-AA100/130b (Fig. 3a), when AnkB-Paris is ectopically expressed in HEK293T cells, it did not localize to the plasma membrane; but it exhibited a punctate appearance and a perinuclear distribution, which is characteristic of sub-cellular localization of the ER (Fig. 3c). This perinuclear distribution is distinct from the diffuse cytosolic pattern characteristic of AnkB-C169A, which lacks the farnesylation motif (Fig. 3b). Interestingly, mutation of both lysines within the di-lysine motif of AnkB-Paris to arginine (AnkB-Paris K149,151R) did not grossly alter the distribution upon ectopic expression (Fig. 3d). In addition, labeling cells for co-localization with calnexin failed to show that either AnkB-Paris or AnkB-ParisK149,151R localized to the ER (data not shown). Therefore, ectopically expressed AnkB-Paris was localized to the perinuclear ER region while AnkB-ParisK149,151R was distributed throughout the cytoplasm, suggesting the loss of ER localization mediated by the ER retention motif (Fig. 3). Our data clearly show a distinct sub-cellular localization of AnkB-Paris and AnkB-AA100/130b.

AnkB-Paris localizes to the LCV during infection. (a) Macrophages infected with WT L. pneumophila AA100/130b, ankB mutant, or the ankB mutant complemented with either ankB from AA100/130b or ankB from Paris strain were fixed at 2 hours post-infection and stained with antibodies to AnkB and Legionella (Lpn). The percentage of bacteria staining positive for AnkB (mean ± 1 SD) was determined by analysis of 100 infected cells in triplicate. Data are representative of 2 independent experiments. (b) Representative confocal images of LCVs isolated from macrophages infected with the indicated strains. To differentiate between intact LCVs and extracellular bacteria, the LCVs were labeled prior to permeabilization with mouse anti-Lpn antisera and rabbit anti-AnkB antisera for 1 h. LCVs were then permeabilized with −20 °C methanol and counter-labeled with goat anti-Lpn antisera to detect intact LCVs. Abbreviations: ankB- (ankB null mutant in AA100/130b strain), dotA (dotA null mutant in AA100/130b strain), WT (wild type AA100/130b strain). Plasmids indicate the ankB allele used to complement the indicated strain.

Ectopically expressed AnkB-Paris localizes to the cytoplasm with a perinuclear distribution. (a–d) Localization of AnkB-AA100, AnkB-Paris, AnkB-Paris K149,151R, and AnkB-C169A in HEK293T cells transiently transfected with 3X Flag-tagged versions of each and stained with anti-flag antibodies and DAPI. Representative confocal images are shown.
While AnkB-AA100/130b is localized to the LCV membrane during infection by host-mediated farnesylation, sub-cellular location of AnkB-Paris during infection is not known. Since the farnesylation motif is missing from AnkB-Paris, we set out to determine sub-cellular localization of AnkB-Paris during infection of hMDMs. We created an identical ankB-Paris allele and introduced it into the isogenic ankB null mutant of strain AA100/130b to determine its potential anchoring to the LCV membrane despite the lack of the farnesylation motif. At 2 hours post-infection, the LCVs were semi-purified from infected hMDMs. Prior to their permeabilization, the LCVs were labeled with anti-AnkB antibodies to detect AnkB on the cytosolic side of the LCV membrane, as we described previously44. Analyses by confocal microscopy showed that AnkB-AA100/130b was anchored to the cytosolic side of the LCV membrane of 80% of WT strain-containing LCVs (Fig. 4a and b). As expected, complementation of the ankB mutant of strain AA100/130b with ankB-AA100/130b restored localization of AnkB to the LCV membrane similar to the wild type strain44. Interestingly, despite the lack of the farnesylation motif, complementation of the AA100/130b-derived ankB mutant with the ankB-Paris allele resulted in anchoring AnkB-Paris to the LCV membrane, similar to AnkB-AA100/130b. Therefore, despite lacking the farnesylation motif, which is indispensable for anchoring AnkB-AA00/130b to the ER-derived LCV membrane, AnkB-Paris is also anchored to the LCV membrane, and this is likely to be mediated the di-lysine ER retention motif.
Functional substitution of AnkB-AA100/130b by AnkB-Paris
Compared to AnkB-AA100/130b, the AnkB-Paris has a truncation of the last 18 amino acids. The crystal structure of AnkB indicates that the C-terminal truncation of AnkB-Paris eliminates a large portion of the third ankyrin repeat compared to AnkB-AA100/130b (Fig. 5)48. To determine if AnkB-Paris can functionally substitute for AnkB-AA100/130b strain, we complemented the ankB null mutant of the AA100/130b strain with the ankB-Paris allele and assessed intracellular replication and decoration of the LCV with polyubiquitinated proteins within human monocytes-derived macrophages (hMDMs)36, 44.

The crystal structure of AnkB. (a) The structure of AnkB from the Philadelphia strain spanning from Lys2 to Ala16848. The F-box domain near the N-terminus is indicated. Three ankyrin repeats are present rather than the predicted two repeats based on the sequence (yellow, cyan, and magenta). (b) Predicted structure of AnkB-Paris, which maintains all three ankyrin repeats by keeping the last half of the last repeat (magenta).
We assessed polyubiquitination of the vacuole at 2 hours post-infection by confocal microscopy. The data showed that 20% of ankB mutant-containing LCVs were decorated with polyubiquitinated proteins (Fig. 6a). In contrast, approximately 75% of strain AA100/130b-containing LCVs were decorated with polyubiquitinated proteins. Despite truncation of the third ankyrin domain, the ankB-Paris allele fully complemented the AA100/130b isogenic ankB null mutant for accumulation of polyubiquitinated proteins, similar to the wild type strain.

AnkB-Paris complements the ankB mutant of strain AA100/130b. (a) Co-localization of polyubiquitinated proteins with the LCV at 2 hours post-infection of hMDMs. Macrophages were infected with either wild type L. pneumophila strain AA100/130b, its isogenic ankB or dotA mutant, or the ankB mutant complemented with ankB-Paris or ankB-AA100. Numbers indicate the percentage of LCVs ( ± 1 SD) that co-localize with polyubiquitinated proteins. The data are based on analysis of 100 infected cells performed in triplicate and are representative of three independent experiments. (b) At 10 hours post-infection hMDMs were fixed, stained with anti-Lpn, and analyzed by confocal microscopy. The number of bacteria per cell was determined and the data are based on analysis of 100 infected cells (mean ± 1 SD) performed in triplicate and are representative of three independent experiments. *p < 0.05, **p < 0.01, ***p < 0.001 compared to corresponding value for ankB null mutant.
We assessed the ability of AnkB-Paris to restore intracellular replication of the ankB mutant of strain AA100/130b by determination of the frequency of formation of replicative vacuoles at 10 hours post-infection, by confocal microscopy (Fig. 6b). The majority of cells infected with the ankB mutant contained a single bacterium. In contrast, the majority of cells infected with WT bacteria contained 2–4 bacteria per cell and >20% of the LCVs harbored more than 5 bacteria per cell. Similarly, the ankB mutant complemented with the ankB-Paris allele formed replicative vacuoles at a frequency similar to the WT strain (Fig. 6b). Taken together, these results indicate that AnkB-Paris is functionally equivalent to AnkB-AA100/130b in its ability to decorate the LCV with polyubiquitinated proteins and to power intracellular proliferation of L. pneumophila.
An indispensable role for the C-terminal Di-lysine Motif of AnkB-Paris in biological function
The C-terminal 5 residues of AnkB strain Paris (K149NK151YAP) resemble a eukaryotic di-lysine motif (KxKxx) responsible for ER-to-golgi retrograde protein trafficking and retention in the ER51,52,53,54. The AnkB-AA100/130b is anchored to the LCV membrane by host-mediated farnesylation, which is essential for biological function. Therefore, we tested the hypotheses that the generated di-lysine ER-retention motif is also required for biological function of AnkB-Paris. We constructed single and double substitutions of lysine149 and lysine151 in the ankB-Paris allele with arginine. Since L. pneumophila effectors often have Dot/Icm translocation signals encoded in their C-terminus, we tested the ER retention motif AnkB-ParisK149R and AnkB-ParisK151R substitution mutants45 for Dot/Icm-mediated translocation using an adenylate cyclase reporter assay, as we described previously25, 44, 45. The cya reporter fusions of ankB-Paris, ankB-Paris K149R, or ankB-Paris K151R were transformed into either the WT strain AA100/130b or its isogenic translocation-deficient dotA mutant. After 2 hours of infection, cells were lysed and cAMP levels were determined via ELISA (Fig. 7a and b). Cells infected with WT bacteria expressing the Cya-AnkB-Paris reporter fusion showed robust cAMP production compared to cells infected with dotA mutant bacteria expressing the same reporter fusion or cells infected with the WT strain expressing the catalytic domain of Cya alone. This indicates that AnkB-Paris is translocated by the AA100/130b strain. In contrast, substitution of either K149 or K151 completely abolished translocation of AnkB-Paris. These results indicate that the two lysine residues in the putative di-lysine ER-retention motif are essential for translocation of AnkB-Paris during infection.

The putative di-lysine motif in the C-terminus of AnkB-Paris is essential for translocation by the Dot/Icm system. (a) Translocation of AnkB-Paris into U937 cells at 2 hours post-infection by WT or dotA mutant bacteria expressing either Cya (negative control) or the indicated Cya::AnkB-Paris fusions. Data represent the mean cAMP concentration of 3 wells (±1 SD). *p < 0.005 compared to dotA harboring Cya::AnkB-Paris. (b) Proteins derived from 1 × 108 bacteria were loaded onto an SDS-PAGE gel and expression of fusion constructs was detected by Western blot using an antibody to the M45 epitope present in all Cya fusions. Blots were re-probed with anti-CAT antibodies. Lanes 1: WT Cya, 2: dotA Cya-AnkB-Paris K151R, 3: WT Cya-AnkB-Paris K151R, 4: dotA Cya-AnkB-Paris K151R, 5: dotA Cya-AnkB-Paris K149R, 6: WT Cya-AnkB-Paris K149R, 7: dotA Cya-AnkB-Paris, 8: WT Cya-AnkB-Paris.
In eukaryotic cells, the di-lysine motif is recognized by the coatomer complex (COPI). Coatomer is a multiprotein complex composed of two subcomplexes that include a trimer of α-COP, β’-COP, and ε-COP and a tetramer composed of β-COP, γ-COP, δ-COP, and ζ-COP55. The α-COP and β’-COP subunits of coatomer are responsible for binding di-lysine motifs. We tested for a physical interaction between AnkB-Paris and α-COP or β’-COP in vivo by Co-IP but were unable to detect any interaction. It is possible that overexpression of multiple members of the COPI complex is required to detect a physical interaction with AnkB-Paris.
The Putative Di-lysine Motif of AnkB-Paris is Required for in-trans Rescue of the ankB Mutant
Since AnkB-ParisK149R and AnkB-ParisK151R are not translocated by the Dot/Icm system, we could not test the potential effect of these mutations on intracellular growth or decoration of the LCV with polyubiquitinated proteins. We have previously shown that the ankB null mutant of strain AA100/130b is rescued for intra-vacuolar growth within HEK293T ectopically expressing AnkB but not by the farnesylation-defective AnkB variant that has a substitution of the cysteine within the C-terminal CaaX farnesylation motif44. This is due to the ability of ectopically expressed AnkB to be farnesylated and anchored to the cytosolic side of the plasma membrane where polyubiquitinated proteins are assembled, while the farnesylation defective variant of AnkB is defective. Since this approach bypasses the need for translocation, we transfected 3X Flag-tagged versions of AnkB-Paris, AnkB-ParisK149R and AnkB-ParisK151R or 3X Flag vector control into HEK293T cells and then infected with the ankB mutants. At 10 hours post-infection, cells were fixed and examined for formation of replicative vacuoles using confocal microscopy. Our data showed that replication of the ankB mutant was efficiently trans-rescued by ectopically-expressed AnkB-Paris compared to the vector control (Fig. 8a and b). In contrast, replication of the ankB mutant was not rescued in cells ectopically expressing AnkB-ParisK149R, AnkB-ParisK151R, or AnkB-ParisK149,151R substitution mutants. Ectopically expressed AnkB-Paris was localized to the perinuclear ER region while the AnkB-ParisK149,151R was distributed throughout the cytoplasm (Fig. 3). These data indicate that the putative di-lysine ER-retention motif is indispensable for function of AnkB-Paris; likely through membrane anchoring to the ER-derived LCV membrane.

Requirement of the putative di-lysine motif in the C-terminus of ectopically expressed AnkB-Paris for trans-rescue of the ankB mutant. (a) HEK293T cells were first transfected with plasmids encoding 3X-Flag empty vector, 3X-Flag AnkB-Paris, 3X-Flag AnkB-Paris K149,151R, 3X-Flag AnkB-Paris K149R, or 3X-Flag AnkB-Paris K151R and then infected with the ankB mutant. Intracellular replication was analyzed at 10 hours post infection by confocal microscopy. The results are based on examination of 50 infected/transfected cells using three biological replicates. The mean number of bacteria per transfected HEK293T cell is shown. Error bars represent 1 standard deviation. *p < 0.01 compared to cells transfected with empty vector. (b) Representative confocal microscopy images of cells infected with the ankB mutant and expressing the indicated 3X Flag-ankB fusion. Anti-flag staining is shown in green and anti-Lpn is shown in red.
Discussion
Among the more than 300 confirmed and predicted effectors of L. pneumophila, very few of them are required for intracellular proliferation and AnkB is one the effectors indispensable for intracellular proliferation56. These have been an emerging common theme of variations in the number of effectors and their paralogues among various strains and phenotypic differences between various strains associated with these differences56, 57. Loss of the AnkB AA100/130b effector results in a more severe intracellular defect in macrophage and amoeba and in vivo 25, 45 compared to AnkB-Paris26, despite the observations that both function similarly in decorating the LCV with polyubiquitinated proteins. Although host proteasomal degradation is essential for intracellular replication of the Philadelphia-derived Lp02 strain31, its AnkB homologue does not contribute to decoration of the LCV with polyubiquitinated proteins or intracellular replication28. This suggests that other F-box proteins or ubiquitin ligases, such as SidE and LubX, are involved in decorating the LCV with polyubiquitinated proteins58,59,60,61. It is also becoming clear that L. pneumophila translocate deubiquitinases that remove ubiquitin from the modified protein, and variation in translocated deubiquitinases between various L. pneumophila isolates is likely to be a contributing factor for differences between them in polyubiquitination of the vacuoles and the effectors and metaeffectors (effectors of effectors) involved60. Whether metaeffectors of AnkB varies between various isolates is not known. In addition, modification of AnkB by K11-linked polyubiquitination and by asparagine hydroxylation has been shown for AnkB-AA100 but it is not known how that differs between isolates32, 46. Considering the phenotypic differences between isolates as a consequence of the loss of AnkB and the structural differences in AnkB between the two characterized strains Paris and AA100/130b, it is important to decipher the biological bases of these differences for one of the very few effectors required for intracellular proliferation of L. pneumophila.
Despite the frame shift mutation and deletion of the C-terminal CaaX farnesylation motif, AnkB-Paris (AnkB1) is required for decoration of the LCV with polyubiquitinated proteins26. Concurrently, a unique NKYAP sequence ER retention motif is generated at amino acids 150–154 of AnkB-Paris. The crystal structure of AnkB indicates that the third ankyrin repeat is truncated in AnkB-Paris48. Each ankyrin repeat domain is composed of two α-helices connected by a β-loop where the substrate binding domain is located62. Our phylogenetic data show that the ankB1 allele is predominant among environmental isolates. Statistical support for positive selection in ankB codons and lineages, and variable effects of ankB genotype on recruitment of polyubiquitinated proteins suggest that AnkB may be functionally pleiotropic and may engage diverse cellular pathways triggered by various strains to ensure survival during intracellular residency. Other possibilities include differential regulation of ankB in different isolates; read through of the stop codon (encoding a modified aa in place of stop codon) resulting in a full-length functional AnkB similar to AnkB-AA100/130b. We conclude that positive selection acts on few ankB codons; that the ankB1 allele itself is maintained in natural populations by positive selection specifically on the NKYAP ER retention motif; and that the relatively high frequency of the ankB1 allele in environmental isolates likely reflects a functionally advantageous trait conferred by the ankB1 allele. The selective advantage to harbor the ankB1 allele among environmental isolates of L. pneumophila could be due to a more efficient anchoring to the LCV membrane through the di-lysine ER retention motif compared to farnesylation in some unicellular hosts and/or the third ankyrin domain that is truncated in AnkB1 does not interact with host targets in environmental host but interacts with a specific human target. It is also possible that other effectors expressed by various strains may compensate for the loss of the third ankyrin domain in the ankB-Paris allele. Identification of the AnkB-interacting targets and their interacting domains in AnkB should facilitate deciphering these possibilities.
Despite the lack of the farnesylation motif, AnkB-Paris is anchored to the cytosolic side of the LCV membrane. However, substitutions of the di-lysine ER retention motif results in failure to translocate the effector. This indicates an overlap in the signal for membrane anchoring and Dot/Icm-mediated translocation of the AnkB-Paris effector. Unfortunately, it is not possible to determine whether the di-lysine ER retention motif of AnkB-Paris was responsible for localization to the ER-derived LCV membrane, since the di-lysine ER-retention motif substitution in AnkB-Paris resulted in loss of translocation by the Dot/Icm system. In addition, the perinuclear ER-like distribution of ectopically-expressed AnkB-Paris is lost upon alternation of the di-lysine ER retention motif. Importantly, the trans-rescue of the ankB mutant within cells ectopically expressing AnkB-Paris and the failure of the ER retention di-lysine mutant in trans-rescue clearly shows that the ER retention di-lysine motif is essential for the function of AnkB-Paris. This may not be surprising, since substitution of the cysteine within the CaaX farnesylation motif AnkB-AA100 results in a total loss of function of the effector in trans-rescue of the ankB mutant for intra-vacuolar proliferation44. We conclude that anchoring of AnkB variants to host membranes is essential for function, regardless of the mechanism of membrane anchoring by farnensylation or by the di-lysine ER-retention motif.
Materials and Methods
Bacterial strains, cell cultures, and infections
L. pneumophila strain AA100/130b (ATCC BAA-74), its isogenic dotA and ankB mutants, and complemented mutants were grown on BCYE agar plates for 3–4 days at 37 °C prior to infection as previously described. When required, antibiotics were used at a concentration of 50 µg/mL for kanamycin and 5 µg/mL for chloramphenicol. The E. coli strain DH5α was used for cloning. E. coli was grown in Luria-Bertani (LB) and antibiotics were used at a concentration of 100 µg/mL for ampicillin and 40 µg/mL for chloramphenicol. HEK293T cell line was maintained in DMEM (Gibco, Grand Island, NY) supplemented with 10% FBS.
Purification and preparation of human monocyte-derived macrophages (hMDMs) was performed as previously described. Monocytes were isolated from whole blood of healthy donors and then allowed to adhere to 6 well low adherence cell culture plates for 3 days at 37 °C and 5% CO2 in RPMI 1640 supplemented with 20% FBS. Monocytes were then counted and re-suspended RPMI 1640 supplemented with 10% FBS and plated on coverslips at a density of 2 × 105 cells per well of a 24 well cell culture plate and incubated for a further 2 days. The cell culture media was then replaced with RPMI 1640 supplemented with 5% FBS for one day, and then with RPMI 1640 supplemented with 1% FBS for one day. The resulting hMDMs were then used for infection.
All methods were carried out in accordance with relevant guidelines and regulations. We confirm that all experimental protocols were approved by the Institutional Review Board (IRB) committee. We confirm that informed consent was obtained from all subjects, as required per our approved IRB protocol.
Infection of hMDMs was performed as previously described. Bacteria were resuspended in RPMI 1640 with 10% FBS and macrophages were infected in triplicate for 1 hour at a multiplicity of infection (MOI) of 10. Plates were centrifuged at 200 g for 5 minutes to synchronize the infection. Infected cells were treated with 50 µg/mL gentamicin for 1 hour to kill extracellular bacteria. Following gentamicin treatment, cells were washed three times with Hank’s buffered saline solution (HBSS) and then RPMI containing 10% FBS was added. At 10 hours post infection, cells were fixed and processed for confocal microscopy. Infection of HEK-293 cells was performed at an MOI of 50 for 1 hour followed by treatment with gentamicin 50 µg/mL for 1 hour. At 10 hours post infection, cells were fixed and processed for confocal microscopy.
HEK293T cell transfection and infection
To create ankB-Paris and ankB-ParisK149,151R, ankB from strain AA100/130b cloned into the mammalian vector p3XFlag-CMV-10 (Sigma) was used as a template for site directed mutagenesis by PCR. HEK293T cells (85% confluent) were re-plated onto poly-L lysine treated coverslips in 24 well plates at a density of 5 × 104 cells/well. After overnight incubation, cells were transfected with 0.625 µg plasmid DNA per well using 1.5 µg polyethylenimine (PEI) per well. After 24 hours, cells were infected with bacteria suspended in DMEM at an MOI of 100 for 1 hour at 37 °C and 5% CO2. Plates were centrifuged at 200 g for 10 minutes to synchronize the infection. Extracellular bacteria were eliminated by treatment with gentamicin 50 µg/mL for 1 hour. At 10 hours post infection, cells were fixed and processed for confocal microscopy.
LCV AnkB localization
To the determine localization of AnkB on the LCV surface during infection, post-nuclear supernatants of infected hMDMs were prepared and then differentially labeled as described previously44. Briefly, a total of 1 × 106 hMDMs were infected with L. pneumophila at an MOI of 10 for 2 h. Post nuclear supernatants were prepared as described previously44, and LCVs were allowed to adhere to poly-L-lysine coated glass coverslips and fixed using 4% paraformaldehyde. To differentiate between intact LCVs and extracellular bacteria, the LCVs were labeled prior to permeabilization with mouse anti-Legionella antisera (1/1000 dilution) and rabbit anti-AnkB antisera (1/200 dilution) for 1 h. LCVs were then permeabilized with −20 °C methanol and counter-labeled with goat anti-Legionella antisera (1/1000 dilution) for 1 h to detect intact LCVs. The LCVs were then labeled with Alexa-Fluor conjugated secondary antibodies (anti-mouse 488, anti-rabbit 555 and anti-goat 647) following the manufacturers recommendations (Invitrogen).
Confocal microscopy
Processing of infected cells for confocal microscopy was performed as we described previously44. Purification of the LCVs and their labeling prior to permeabilization to localize AnkB on the cytosolic side of the LCV membrane was performed as we described previously44. For antibody labeling, goat polyclonal anti-L. pneumophila was used at a dilution of 1:500 and detected by Alexa-Fluor 488-conjugated donkey anti-goat IgG (Invitrogen, Carlsbad, CA). Poly-ubiquitinated proteins were detected using mouse anti-polyubiquitin FK1 antibody at a dilution of 1:50 (BIOMOL International/Affiniti, Exeter, United Kingdom), followed by Alexa-Fluor 647-conjugated goat anti-mouse IgM (Invitrogen, Carlsbad, CA). For detection of 3X-Flag tagged proteins during transfection experiments, mouse monoclonal anti-Flag (Sigma) antibodies were used followed by detection with Alexa-Fluor 488-conjugated donkey anti-mouse (Invitrogen, Carlsbad, CA). An Olympus FV1000 laser scanning confocal microscope was used to examine cells as we described previously. On average, 8–15 0.2 µm serial Z sections of each image were captured and stored for further analyses, using Adobe Photoshop CS3.
Adenylate cyclase and Western blot analysis
L. pneumophila strain AA100/130b (ATCC BAA-74) or its isogenic dotA mutant harboring pCya-AnkB-Paris, pCya empty vector, or pCya-AnkB-Paris with K149R or K151R were grown on BCYE agar plates for 3–4 days at 37 °C prior to infection as previously described45. U937 macrophages differentiated with PMA were infected at MOI 50 in triplicate and plates were centrifuged to synchronize the infection. After 2 hours at 37 °C and 5% CO2, cells were washed three times with PBS and lysed by adding 250 µl of 0.1 N HCl containing 0.5% Triton X-100 and incubating at room temperature for 20 minutes. Lysates were assayed for cAMP using the Direct Cyclic AMP Enzyme Immunoassay kit (Enzo Life Sciences, Inc.). Aliquots of bacteria used for infection (1 × 108 bacteria) were lysed by adding SDS-PAGE loading buffer and boiling for 5 minutes. Fusion protein expression was assessed by Western blot using anti-M45 (1:50 dilution) according to standard procedures. Blots were re-probed with anti-CAT (1:2000).
PCR and Sequencing of ankB alleles
The ankB gene was amplified with the following primers: ankB1F: 5′-GGATCCCAAGAGATTTTTAG-3′ and ankB1R: 5′-CATTTAACAAACAAGGCACT-3′ using standard PCR conditions. PCR primers were located in genes flanking ankB. Briefly, 25 ng of genomic DNA was used as template in a 25 µL PCR reaction containing 1U of Taq polymerase (Midsci, St. Louis MO), 150 µM dNTPs, 20 pm/ml of each primer with the following cycling parameters: 94 °C-5′ – 1 cycle followed by 30 cycles of 94 °C-1′, 55 °C-1, 72 °C 1′ and a final 5 min extension at 72 °C. DNA sequencing was performed on both strands at the University of Washington Sequencing Core, and the sequence data was assembled and edited using the DNASTAR suite (DNASTAR Inc., Madison, WI).
Phylogenetic Analysis
Maximum likelihood (ML) tree was constructed using MEGA version 663 assuming the TN93 + G substitution model. The percentage of trees in which the associated taxa clustered together was determined by a bootstrap analysis of 1000 trees. Initial tree for the heuristic search was obtained automatically by applying Neighbor-Joining and BioNJ algorithms to a matrix of pairwise distances estimated using the Maximum Composite Likelihood (MCL) approach, and then selecting the topology with superior log likelihood value. A discrete gamma distribution was used to model evolutionary rate differences among sites (5 categories (+G, parameter = 0.2388)). Nucleotide sequence data has been submitted to GenBank® and assigned the following accession numbers: KM276667-KM276681.
Analysis of Selection Pressures
-
(a)
Site Models: To identify the different selective forces, i.e., negative, neutral or positive selection, that acted upon ankB codons during its evolutionary history we tested the fit of the sequence data to several codon-based models implemented in CODEML package of PAML ver 4.764 accessed via its GUI interface PAMLX1.265 essentially as described before66. In brief, we used site models to determine selective pressures on each ankB codon by comparing the differences in the likelihood score of each model’s fit to the sequence data via a series of likelihood ratio tests [LRTs)64. To verify or supplement CODEML outcomes, we conducted several other alternate tests including GARD (genetic algorithms for recombination detection) to detect recombination among ankB sequences, and SLAC (single-likelihood ancestor counting), FEL (fixed effects likelihood), IFEL (internal fixed effects likelihood), REL (random effects likelihood), and MEME (mixed effects models of evolution), which can each detect positive and negatively selected codon in protein coding genes and can explicitly account and correct for recombination within sequences. All these methods were accessed and their outcomes analyzed via the www.datamonkey.org server67.
-
(b)
Branch Site Models. To determine whether the ankB1 allele branch experienced positive selection in its evolutionary history we used two versions of the branch-site models A (M2N2) implemented in CODEML (Table S1): (1) M2N2A1, which specifically tested for evidence of positive selection in the clade leading up to ankB1 and ankB8; and (2) M2N2A2, which specifically sought evidence for positive selection in the ankB1 branch itself. The fit of each model to the data was tested via LRTs with 1 degree of freedom and that measured the difference in the likelihood score of each model (e.g., M2N2A1) with a constrained version whereby ω for the branch suspected to be under positive selection was fixed at 1 (e.g. M2N2A1ωf). It has been suggested that selection of branches of interest to test for selection, or testing one branch at a time can sometimes lead to statistical instability or acceptance of poorly supported models67. Thus to confirm the outcomes of our CODEML branch site results, we performed supplemental analysis for detecting all branches that may have significantly experienced positive selection in their evolutionary history with the GA (genetic algorithm) branch method implemented at www.datamonkey.org.
References
Franco, I. S., Shuman, H. A. & Charpentier, X. The perplexing functions and surprising origins of Legionella pneumophila type IV secretion effectors. Cellular microbiology 11, 1435–1443, doi:10.1111/j.1462-5822.2009.01351.x (2009).
Richards, A. M., Von Dwingelo, J. E., Price, C. T. & Abu Kwaik, Y. Cellular microbiology and molecular ecology of Legionella-amoeba interaction. Virulence 4, 307–314, doi:10.4161/viru.24290 (2013).
Al-Quadan, T., Price, C. T. & Abu Kwaik, Y. Exploitation of evolutionarily conserved amoeba and mammalian processes by Legionella. Trends in microbiology 20, 299–306, doi:10.1016/j.tim.2012.03.005 (2012).
Abu Khweek, A. et al. Biofilm-derived Legionella pneumophila evades the innate immune response in macrophages. Frontiers in cellular and infection microbiology 3, doi:10.3389/fcimb.2013.00018 (2013).
Price, C. T., Richards, A. M., Von Dwingelo, J. E., Samara, H. A. & Abu Kwaik, Y. Amoeba host-Legionella synchronization of amino acid auxotrophy and its role in bacterial adaptation and pathogenic evolution. Environ Microbiol 16, 350–358, doi:10.1111/1462-2920.12290 (2014).
Hoppe, J. et al. PilY1 Promotes Legionella pneumophila Infection of Human Lung Tissue Explants and Contributes to Bacterial Adhesion, Host Cell Invasion, and Twitching Motility. Frontiers in cellular and infection microbiology 7, doi:10.3389/fcimb.2017.00063 (2017).
Stone, B. J. & Abu Kwaik, Y. Expression of multiple pili by Legionella pneumophila: identification and characterization of a type IV pilin gene and its role in adherence to mammalian and protozoan cells. Infection and immunity 66, 1768–1775 (1998).
Stone, B. J. & Kwaik, Y. A. Natural competence for DNA transformation by Legionella pneumophila and its association with expression of type IV pili. Journal of bacteriology 181, 1395–1402 (1999).
Herweg, J.-A. et al. Purification and proteomics of pathogen-modified vacuoles and membranes. Frontiers in cellular and infection microbiology 5, doi:10.3389/fcimb.2015.00048 (2015).
Kagan, J. C. & Roy, C. R. Legionella phagosomes intercept vesicular traffic from endoplasmic reticulum exit sites. Nature cell biology 4, 945–954 (2002).
Isberg, R. R., O’Connor, T. J. & Heidtman, M. The Legionella pneumophila replication vacuole: making a cosy niche inside host cells. Nat Rev Microbiol 7, 13–24, doi:10.1038/nrmicro1967 (2009).
Shin, S. & Roy, C. R. Host cell processes that influence the intracellular survival of Legionella pneumophila. Cellular microbiology 10, 1209–1220, doi:10.1111/j.1462-5822.2008.01145.x (2008).
Luo, Z. Q. Legionella secreted effectors and innate immune responses. Cellular microbiology, doi:10.1111/j.1462-5822.2011.01713.x (2011).
Abu Kwaik, Y. The phagosome containing Legionella pneumophila within the protozoan Hartmanella vermiformis is surrounded by the rough endoplasmic reticulum. Appl. Environ. Microbiol. 62, 2022–2028 (1996).
Li, L. & Faucher, S. P. The Membrane Protein LasM Promotes the Culturability of Legionella pneumophila in Water. Frontiers in cellular and infection microbiology 6, doi:10.3389/fcimb.2016.00113 (2016).
Segal, G., Purcell, M. & Shuman, H. A. Host cell killing and bacterial conjugation require overlapping sets of genes within a 22-kb region of the Legionella pneumophila chromosome. Proc. Natl. Acad. Sci. USA 95, 1669–1674 (1998).
Vogel, J. P., Andrews, H. L., Wong, S. K. & Isberg, R. R. Conjugative transfer by the virulence system of Legionella pneumophila. Science (New York, NY) 279, 873–876 (1998).
Zhu, W. et al. Comprehensive identification of protein substrates of the Dot/Icm type IV transporter of Legionella pneumophila. PLoS One 6, e17638, doi:10.1371/journal.pone.0017638 (2011).
Luo, Z. Q. Targeting One of its Own: Expanding Roles of Substrates of the Legionella Pneumophila Dot/Icm Type IV Secretion System. Frontiers in microbiology 2, 31, doi:10.3389/fmicb.2011.00031 (2011).
de Felipe, K. S. et al. Legionella eukaryotic-like type IV substrates interfere with organelle trafficking. PLoS pathogens 4, e1000117, doi:10.1371/journal.ppat.1000117 (2008).
Isaac, D. T. & Isberg, R. Master manipulators: an update on Legionella pneumophila Icm/Dot translocated substrates and their host targets. Future Microbiology 9, 343–359, doi:10.2217/fmb.13.162 (2014).
Casson, C. & Shin, S. Inflammasome-mediated cell death in response to bacterial pathogens that access the host cell cytosol: lessons from legionella pneumophila. Frontiers in cellular and infection microbiology 3, doi:10.3389/fcimb.2013.00111 (2013).
Speir, M. et al. Legionella pneumophila Strain 130b Evades Macrophage Cell Death Independent of the Effector SidF in the Absence of Flagellin. Frontiers in cellular and infection microbiology 7, doi:10.3389/fcimb.2017.00035 (2017).
O’Connor, T. J., Adepoju, Y., Boyd, D. & Isberg, R. R. Minimization of the Legionella pneumophila genome reveals chromosomal regions involved in host range expansion. Proceedings of the National Academy of Sciences of the United States of America 108, 14733–14740, doi:10.1073/pnas.1111678108 (2011).
Price, C. T. et al. Molecular mimicry by an F-box effector of Legionella pneumophila hijacks a conserved polyubiquitination machinery within macrophages and protozoa. PLoS pathogens 5, e1000704, doi:10.1371/journal.ppat.1000704 (2009).
Lomma, M. et al. The Legionella pneumophila F-box protein Lpp2082 (AnkB) modulates ubiquitination of the host protein parvin B and promotes intracellular replication. Cellular microbiology 12, 1272–1291, doi:10.1111/j.1462-5822.2010.01467.x (2010).
Wong, K. et al. Structural Mimicry by a Bacterial F Box Effector Hijacks the Host Ubiquitin-Proteasome System. Structure 25, 376–383, doi:10.1016/j.str.2016.12.015 (2017).
Ensminger, A. W. & Isberg, R. R. E3 ubiquitin ligase activity and targeting of BAT3 by multiple Legionella pneumophila translocated substrates. Infection and immunity 78, 3905–3919, doi:10.1128/IAI.00344-10 (2010).
Bruckert, W. M., Price, C. T. & Abu Kwaik, Y. Rapid nutritional remodeling of the host cell upon attachment of Legionella pneumophila. Infection and immunity 82, 72–82, doi:10.1128/IAI.01079-13 (2014).
Price, C. T. & Abu Kwaik, Y. Exploitation of Host Polyubiquitination Machinery through Molecular Mimicry by Eukaryotic-Like Bacterial F-Box Effectors. Frontiers in microbiology 1, 122, doi:10.3389/fmicb.2010.00122 (2010).
Dorer, M. S., Kirton, D., Bader, J. S. & Isberg, R. R. RNA interference analysis of Legionella in Drosophila cells: exploitation of early secretory apparatus dynamics. PLoS pathogens 2, e34 (2006).
Bruckert, W. M. & Abu Kwaik, Y. Lysine11-linked polyubiquitination of the AnkB F-Box effector of Legionella pneumophila. Infection and immunity, doi:10.1128/iai.01165-15 (2015).
Bruckert, W. M. & Abu Kwaik, Y. Complete and ubiquitinated proteome of the Legionella-containing vacuole within human macrophages. Journal of proteome research 14, 236–248, doi:10.1021/pr500765x (2015).
Price, C. T., Al-Quadan, T., Santic, M., Rosenshine, I. & Abu Kwaik, Y. Host proteasomal degradation generates amino acids essential for intracellular bacterial growth. Science (New York, NY) 334, 1553–1557, doi:10.1126/science.1212868 (2011).
Price, C. T. D., Richards, A. M., Von Dwingelo, J. E. & Samara, H. A. Legionella pneumophila synchronization of amino acid auxotrophy and its role in adaptation and pathogenic evolution to the amoeba host. Environ Microbiol 14, 759–764 (2013).
Al-Quadan, T. & Kwaik, Y. A. Molecular Characterization of Exploitation of the Polyubiquitination and Farnesylation Machineries of Dictyostelium Discoideum by the AnkB F-Box Effector of Legionella Pneumophila. Frontiers in microbiology 2, 23, doi:10.3389/fmicb.2011.00023 (2011).
Eisenreich, W., Heesemann, J., Rudel, T. & Goebel, W. Metabolic host responses to infection by intracellular bacterial pathogens. Frontiers in cellular and infection microbiology 3, 24, doi:10.3389/fcimb.2013.00024 (2013).
Fonseca, M. V. & Swanson, M. S. Nutrient salvaging and metabolism by the intracellular pathogen Legionella pneumophila. Frontiers in cellular and infection microbiology 4, 12, doi:10.3389/fcimb.2014.00012 (2014).
Fonseca, M. & Swanson, M. Nutrient salvaging and metabolism by the intracellular pathogen Legionella pneumophila. Frontiers in cellular and infection microbiology 4, doi:10.3389/fcimb.2014.00012 (2014).
Manske, C. & Hilbi, H. Metabolism of the vacuolar pathogen Legionella and implications for virulence. Frontiers in cellular and infection microbiology 4, doi:10.3389/fcimb.2014.00125 (2014).
Price, C. T. D., Richards, A. M. & Abu Kwaik, Y. Nutrient generation and retrieval from the host cell cytosol by intra-vacuolar Legionella pneumophila. Frontiers in cellular and infection microbiology 4, doi:10.3389/fcimb.2014.00111 (2014).
Wright, L. P. & Philips, M. R. Thematic review series: lipid posttranslational modifications. CAAX modification and membrane targeting of Ras. J Lipid Res 47, 883–891, doi:10.1194/jlr.R600004-JLR200 (2006).
Al-Quadan, T., Price, C. & Abu Kwaik, Y. Exploitation of evolutionarily conserved amoeba and mammalian processes by Legionella. Trends in microbiology 20, 299–306, doi:10.1016/j.tim.2012.1003.1005 (2012).
Price, C. T., Al-Quadan, T., Santic, M., Jones, S. C. & Abu Kwaik, Y. Exploitation of conserved eukaryotic host cell farnesylation machinery by an F-box effector of Legionella pneumophila. The Journal of experimental medicine 207, 1713–1726, doi:10.1084/jem.20100771 (2010).
Al-Khodor, S., Price, C. T., Habyarimana, F., Kalia, A. & Abu Kwaik, Y. A Dot/Icm-translocated ankyrin protein of Legionella pneumophila is required for intracellular proliferation within human macrophages and protozoa. Molecular microbiology 70, 908–923, doi:10.1111/j.1365-2958.2008.06453.x (2008).
Price, C. et al. Host FIH-Mediated Asparaginyl Hydroxylation of Translocated Legionella pneumophila Effectors. Frontiers in cellular and infection microbiology 7, 54, doi:10.3389/fcimb.2017.00054 (2017).
Gao, C. et al. Retention mechanisms for ER and Golgi membrane proteins. Trends in plant science 19, 508–515, doi:10.1016/j.tplants.2014.04.004 (2014).
Wang, K. et al. Structural Mimicry by a Bacterial F Box Effector Hijacks the Host Ubiquitin-Proteasome System. Structure doi:10.1016/j.str.2016.12.015 (2017).
Al-Quadan, T., Price, C. T., London, N., Schueler-Furman, O. & AbuKwaik, Y. Anchoring of bacterial effectors to host membranes through host-mediated lipidation by prenylation: a common paradigm. Trends in microbiology 19, 573–579, doi:10.1016/j.tim.2011.08.003 (2011).
Price, C. T., Jones, S. C., Amundson, K. E. & Kwaik, Y. A. Host-mediated post-translational prenylation of novel dot/icm-translocated effectors of legionella pneumophila. Frontiers in microbiology 1, 131, doi:10.3389/fmicb.2010.00131 (2010).
Trujillo, C., Taylor-Parker, J., Harrison, R. & Murphy, J. R. Essential lysine residues within transmembrane helix 1 of diphtheria toxin facilitate COPI binding and catalytic domain entry. Molecular Microbiology 76, 1010–1019, doi:10.1111/j.1365-2958.2010.07159.x (2010).
Custer, S. K., Todd, A. G., Singh, N. N. & Androphy, E. J. Dilysine motifs in exon 2b of SMN protein mediate binding to the COPI vesicle protein α-COP and neurite outgrowth in a cell culture model of spinal muscular atrophy. Human Molecular Genetics 22, 4043–4052, doi:10.1093/hmg/ddt254 (2013).
Gao, C. et al. Retention mechanisms for ER and Golgi membrane proteins. Trends in Plant Science 19, 508–515, doi:10.1016/j.tplants.2014.04.004 (2014).
Ma, W. & Goldberg, J. Rules for the recognition of dilysine retrieval motifs by coatomer Vol. 32 (2013).
Popoff, V., Adolf, F., Brugger, B. & Wieland, F. COPI budding within the Golgi stack. Cold Spring Harbor perspectives in biology 3, a005231, doi:10.1101/cshperspect.a005231 (2011).
Burstein, D. et al. Genomic analysis of 38 Legionella species identifies large and diverse effector repertoires. Nature genetics 48, 167–175, doi:10.1038/ng.3481 (2016).
Shi, X., Halder, P., Yavuz, H., Jahn, R. & Shuman, H. A. Direct targeting of membrane fusion by SNARE mimicry: Convergent evolution of Legionella effectors. Proceedings of the National Academy of Sciences of the United States of America 113, 8807–8812, doi:10.1073/pnas.1608755113 (2016).
Qiu, J. et al. Ubiquitination independent of E1 and E2 enzymes by bacterial effectors. Nature 533, 120–124, doi:10.1038/nature17657 (2016).
Quaile, A. T. et al. Molecular Characterization of LubX: Functional Divergence of the U-Box Fold by Legionella pneumophila. Structure 23, 1459–1469, doi:10.1016/j.str.2015.05.020 (2015).
Urbanus, M. L. et al. Diverse mechanisms of metaeffector activity in an intracellular bacterial pathogen, Legionella pneumophila. Molecular systems biology 12, 893, doi:10.15252/msb.20167381 (2016).
Speir, M. et al. Legionella pneumophila strain 130b evades macrophage cell death independent of the effector SidF in the absence of flagellin. Front. Cell. Infect. Microbiol. doi:10.3389/fcimb.2017.00035 (2017).
Al-Khodor, S., Price, C. T., Kalia, A. & Abu Kwaik, Y. Functional diversity of ankyrin repeats in microbial proteins. Trends in microbiology 18, 132–139, doi:10.1016/j.tim.2009.11.004 (2010).
Tamura, K., Stecher, G., Peterson, D., Filipski, A. & Kumar, S. MEGA6: Molecular Evolutionary Genetics Analysis Version 6.0. Molecular Biology and Evolution 30, 2725–2729, doi:10.1093/molbev/mst197 (2013).
Yang, Z. PAML 4: phylogenetic analysis by maximum likelihood. Mol Biol Evol 24, 1586–1591, doi:10.1093/molbev/msm088 (2007).
Xu, B. & Yang, Z. PAMLX: a graphical user interface for PAML. Mol Biol Evol 30, 2723–2724, doi:10.1093/molbev/mst179 (2013).
Putty, K. et al. Robustness of Helicobacter pylori infection conferred by context-variable redundancy among cysteine-rich paralogs. PLoS One 8, e59560, doi:10.1371/journal.pone.0059560 (2013).
Delport, W., Poon, A. F. & Frost, S. D. & Kosakovsky Pond, S. L. Datamonkey 2010: a suite of phylogenetic analysis tools for evolutionary biology. Bioinformatics 26, 2455–2457, doi:10.1093/bioinformatics/btq429 (2010).
Author information
Authors and Affiliations
Contributions
J.P., A.K., C.P., and S.J. did the experimental work and contributed to experimental design. K.W. and K.G. contributed to experimental design and prepared Figure 5. Y.A., J.P., and C.P. wrote the manuscript. All authors reviewed the manuscript.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Perpich, J.D., Kalia, A., Price, C.T.D. et al. Divergent evolution of Di-lysine ER retention vs. farnesylation motif-mediated anchoring of the AnkB virulence effector to the Legionella-containing vacuolar membrane. Sci Rep 7, 5123 (2017). https://doi.org/10.1038/s41598-017-05211-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-05211-5
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
