
Abstract
Transition metal-catalysed C–H hydroxylation is one of the most notable advances in synthetic chemistry during the past few decades and it has been widely employed in the preparation of alcohols and phenols. The site-selective hydroxylation of aromatic C–H bonds under mild conditions, especially in the context of substituted (hetero)arenes with diverse functional groups, remains a challenge. Here, we report a general and mild chelation-assisted C–H hydroxylation of (hetero)arenes mediated by boron species without the use of any transition metals. Diverse (hetero)arenes bearing amide directing groups can be utilized for ortho C–H hydroxylation under mild reaction conditions and with broad functional group compatibility. Additionally, this transition metal-free strategy can be extended to synthesize C7 and C4-hydroxylated indoles. By utilizing the present method, the formal synthesis of several phenol intermediates to bioactive molecules is demonstrated.
Similar content being viewed by others
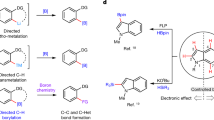
Introduction
Phenols are structural constituents of pharmaceuticals, agrochemicals, polymers, and naturally occurring compounds and serve as versatile synthetic intermediates1,2,3,4. Bioactive molecules of particular interest are (hetero)arenes such as amides, indolines, and indoles-containing hydroxyl groups (Fig. 1a)5,6,7,8,9,10,11. The site-selective introduction of a hydroxyl group to a (hetero)arene is an important task in both chemical industry and organic synthesis. Traditional methods used for phenol preparation include nucleophilic aromatic substitution of activated aryl halides12 and Sandmeyer-type hydroxylation13, as well as the transition-metal-catalysed hydroxylation of (hetero)aryl halides with hydroxide salts (e.g., KOH and NaOH)14,15,16,17,18, all of which require the presence of a (pseudo)halide in the (hetero)arenes. During the past decade, C–H functionalization has gained considerable momentum, holding great promise for avoiding the preinstalled functional groups19,20,21,22,23,24,25,26,27,28. Among these C–H functionalization techniques, hydroxylation is one of the most important C–H functionalization reactions29,30. As early as 1990, Fujiwara et al. explored the hydroxylation of benzene using O2 as the oxidant enabled by Pd catalysis31. However, this pioneering work had several limitations, such as a low efficiency, poor selectivity, and harsh reaction conditions. Substrates bearing a chelating functional group can coordinate with the metal catalyst and undergo further C–H functionalization32,33,34. In this context, several groups have explored transition-metal-catalysed directed aromatic C–H hydroxylation using organic oxidants, hydrogen peroxide or molecular oxygen (Fig. 1b)35,36,37,38,39,40,41,42,43,44,45,46. While synthetically very attractive, most of these protocols still suffer from the use of expensive noble metals, such as Pd, Rh, Ru, and Ir, as catalysts. This requirement may be a significant limitation, especially for applications needing large-scale synthesis methods and for the removal of toxic trace metals from pharmaceutical products. From a synthetic perspective, the ability to prepare synthetically relevant scaffolds via regio-controlled C–H hydroxylation under mild conditions by using cheap oxidants and avoiding the use of transition metals would be of great importance.

a Phenol-based bioactive molecules. b Transition-metal-catalysed directed aromatic C–H hydroxylation. c Our approach for directed aromatic C–H hydroxylation under transition-metal-free conditions.
The transition-metal-catalysed C–H borylation reaction has emerged as an effective method for the construction of arylboronic acids and their derivatives47,48,49. Recently, our group50 and the Ingleson group51 reported a general strategy for the mild directed C–H borylation of (hetero)arenes using BBr3 as both the reagent and catalyst under metal-free conditions52,53,54,55. BBr3 is an attractive borylation agent because it is a commercially available in multigram to kilogram quantities and is cheaper than most common boron reagents. To further extend the utility of this strategy, here, we developed a one-pot directed C–H borylation/oxidation protocol to access numerous structurally diverse phenols, whose regioselectivity is not easily accessed by traditional strategies (Fig. 1c). Replacing the transition-metal-catalysed C–H hydroxylation process by a boron-mediated strategy offers an alternative pathway for synthesizing phenols and has exciting possibilities because of the superior practicality, low cost, and environmental friendliness of this alternate method.
Results
Reaction design
We initiated our study by investigating the reaction of N-pivaloyl amide 1a with BBr3 (Table 1). As a result, we discovered that the use of 1.0 equivalent of 1a with 1.1 equivalents of BBr3 in DCM at room temperature for 1 h led to the full conversion of the precursors and formation of boron complex 1b. Then, the treatment of 3.0 equivalents of NaBO3 in a THF and H2O (1:1) co-solvent led to the in situ formation of the hydroxylated product, and phenol 1c was isolated with a 92% yield (Table 1, entry 1). Other boron halides, such as BF3, were not efficient for this reaction (Table 1, entry 2), and BCl3 only afforded a trace amount of the product (Table 1, entry 3). When the reaction was carried out using ClBcat or 9-BBN, we did not observe any C–H borylation or hydroxylation products (Table 1, entries 4–5). The substrate 1a′ bearing an N-Me group failed to achieve this transformation, confirming the importance of the N-pivaloyl moiety for achieving both a high reactivity and selectivity (Table 1, entry 6). To our delight, other common oxidants, such as oxone and H2O2, were also effective for this hydroxylation process, generating the desired product 1c in slightly lower yields (Table 1, entries 7–8).
Scope of the methodology
We first examined the scope of the ortho-selective C–H hydroxylation of amides (Fig. 2). When the simple N-pivaloyl amide 2a was employed as a substrate, hydroxylation proceeded at the ortho C–H bond, affording 2c with a 85% yield. Amides bearing methyl (3–5a), tBu (6a), phenyl (7a), and halogen-containing motifs (8–13a) at the ortho, meta, and para positions underwent facile hydroxylation and afforded the corresponding products 3–13c in good to excellent yields. The amides bearing electron-withdrawing groups such as CF3 (14–15a), COOMe (16a), and CN (17a) are particular noteworthy; these substrates produced ortho-hydroxylated products 14–17c with 66–80% yields. Electron-donating groups such as OTBS (18c) and SMe (19c) at the para position of the amides are tolerated. Substrate 20a bearing a methoxy group can undergo ortho C–H hydroxylation and O-demethylation to generate the corresponding product 20c with a 81% yield. Notably, the phenyldiazenyl substituent in substrate 21a, which is also susceptible to C–H borylation, remained intact during the reaction. Other N-pivaloyl amides, including N-methylaniline (22c), tetrahydroquinoline (23c), and indoline (24c), are also tolerated for C–H hydroxylation. This protocol is compatible with heterocyclic motifs such as thiophene 25c. Polyaromatic substrates 26–28c were also shown to be highly reactive. As a prominent structural motif, N-arylpyrrolidinones have been used in Ru(II)-catalysed C–H hydroxylation56,57. We found that the boron-mediated directed C–H hydroxylation of N-phenylpyrrolidinone (29a) in the presence of BBr3 could provide the desired product 29c with a 79% yield. The system was compatible with the different para- and meta-substitution patterns in the phenyl ring of the N-arylpyrrolidinone backbone (30–36c). In addition, this C–H hydroxylation method is not limited to N-arylpyrrolidinones. Lactams such as 37–38a, oxazolidin-2-one 39a, and thiophene 40a could also undergo C–H hydroxylation at the ortho position, affording good yields of products 37–40c. Subjecting N-pivaloyl amides 41–45a, which are substrates bearing two N-pivaloyl directing bonds, to our system resulted in the selective formation of the difunctionalization products 41–45c in 60–89% yields. These bisphenols could be utilized as precursors for construction of polymers58.

Reaction conditions: substrates 1a–28a (0.20 mmol), BBr3 (0.22 mmol) in 0.5 mL DCM at room temperature, 1 h, under Ar; NaBO3·4H2O (0.60 mmol) in 0.5 mL THF and 0.5 mL H2O, at room temperature, 1 h. 29a–40a (0.20 mmol), BBr3 (0.60 mmol) in 0.5 mL DCM at 60 °C, 24 h; NaBO3·4H2O (0.60 mmol) in 0.5 mL THF and 0.5 mL K2CO3 (aq), at room temperature, 1 h. 41a–45a (0.20 mmol), BBr3 (0.40 mmol) in 0.5 mL DCM at room temperature, 1 h, under Ar; NaBO3·4H2O (1.50 mmol) in 0.5 mL THF and 0.5 mL H2O, at room temperature, 1 h. aUsing BBr3 (2.0 mmol) in 0.1 mL of DCM. bN-(4-methoxyphenyl)pivalamide (0.20 mmol), BBr3 (0.5 mmol).
We next investigated the scope of the C7 selective C–H hydroxylation of indoles (Fig. 3a). We found that indole 46a could generate 7-hydroxyindole 46c with a 88% yield by a cascade C–H borylation/oxidation/DG removal protocol, in which the N-Piv group can be removed automatically during work-up with K2CO3. Indoles bearing methyl (47–49a) substituents at the 4–6 positions underwent facile hydroxylation and afforded the corresponding products in 74-85% yields. Again, halogen-containing motifs (F, Cl, and Br, 50–56a) work very well in the C7 selective borylation process. In addition, substrate 57a contains a phenyl substituent also delivering coupled product 57c with a 70% yield. We further examined the scope of using C3-pivaloyl indoles as coupling partners with BBr3; these compounds reacted with a high regioselectivity to produce C4-hydroxylated indoles (Fig. 3b)59. We first evaluated the influence of the N–H protection groups on the indoles. Notably, the free indole 58a could provide the desired product 58c with a 59% yield. The treatment of the indoles 59–60a bearing N-Me and N-Bn groups in the system provided a 71% and 54% isolated yields of the corresponding C4-hydroxylation products 59–60c. Indole 61a bearing an N-Ts protection group can promote the reactivity of this transformation, affording the product 61c with a 85% yield. Regarding the scope of the indole framework, diverse substituents, including methyl (62c), F (63a), Cl (64–65c), Br (66–67c), I (68c), and phenyl (69c) are tolerated.

a Directed C–H hydroxylation of indoles at the C7 position. b Directed C–H hydroxylation of indoles at the C4 position. Reaction conditions: substrates 46–57a (0.20 mmol), BBr3 (0.22 mmol) in 0.5 mL DCM at room temperature, 1 h, under Ar; NaBO3·4H2O (0.60 mmol) in 0.5 mL THF and 0.5 mL K2CO3 (aq), at room temperature, 1 h; 58–60a (0.20 mmol), BBr3 (0.60 mmol) in 0.5 mL DCM at 60 °C, 10 h, under Ar; NaBO3·4H2O (1.50 mmol) in 0.5 mL THF and 0.5 mL H2O, at 60 °C, 6 h; 61–69a (0.20 mmol), BBr3 (0.22 mmol) in 0.5 mL DCM at room temperature, 9 h, under Ar; NaBO3·4H2O (1.0 mmol) in 0.5 mL THF and 0.5 mL H2O, at room temperature, 2 h.
Synthetic applications
To further demonstrate the potential synthetic applications of this method, we showed three examples to compare existing strategies with our developed C–H hydroxylation method. Previously, using N-acetylindoline 70 as a model substrate for the total synthesis of the potent caspase-8 inhibitor (+)-haplocidine and its N1-amide congener (+)-haplocine, the precursor acetoxy-indoline 71′ was generated with a 84% yield by the palladium-catalysed C7 hydroxylation of indoline60. Based on the boron-mediated strategy, we prepared product 71 from substrate 70 with a 71% yield, in which N-acetyl can be used as a directing group (Fig. 4a). Trauner and co-workers61 reported the evolution of the total synthesis of exiguamines, where nitrovinylindole 74 was a key intermediate. To simplify this synthesis process, we provided an alternative route to 74 using the developed C–H hydroxylation protocol. The indole substrate 72 was regio-selectively hydroxylated at the C7 position and further deprotected and then protected as a benzyl ether to yield 7-hydroxy-6-bromoindole derivative 73 with a 63% yield. Then, indole 73 was formylated and condensed with nitromethane to yield nitrovinylindole 74 with a 89% yield (Fig. 4b). The Renata group62 recently identify a concise synthetic route to access tambromycin. During the study, they were drawn to a thallium-mediated C–H hydroxylation of indoles at the C4 position, suffering from highly variable yields and a lack of scalability. Inspired by this result, we finally focused our attention on the boron-mediated strategy to synthesize indole 78. Using N-methyl indole 75 as a substrate, C4-hydroxylation was identified as a viable approach to access the desired indole fragment 76 after etherification with MeI. To our delight, the removal of a pivaloyl group from 76 was readily accomplished by a reverse Friedel-Crafts reaction in the presence of TsOH and glycol, providing a good yield of 77. Further C3 formylation and oxidation could provide a good yield of the key building block 78, which was facile to convert to tambromycin (Fig. 4c).

a Using N-acylindoline 70 as a model substrate for the synthesis of (+)-haplocidine and (+)-haplocine. b Synthesis of the key intermediate 73 for the synthesis of the exiguamines. c Synthesis of the key intermediate 78 for the synthesis of tambromycin. Reagents and conditions: (a) 70 (0.2 mmol) and BBr3 (0.6 mmol) in 0.5 mL DCM at 110 °C, 24 h; NaBO3·4H2O (1.0 mmol) in 0.5 mL of THF and 0.5 mL of sat. K2CO3, at 60 °C, 6 h. (b) 72 (0.2 mmol) and BBr3 (0.22 mmol) in 0.5 mL of DCM at 25 °C, 1 h; NaBO3·4H2O (0.6 mmol) in 0.5 mL of THF and 0.5 mL of K2CO3 (aq) at 25 °C, 1 h; K2CO3 (0.6 mmol) and BnBr (0.24 mmol) in 2.0 mL of acetone at 25 °C, 24 h; (c) 73 (0.2 mmol) and POCl3 (0.25 mmol) in 2.0 mL of dry DMF, reflux at 160 °C; NH4OAc (0.22 mmol) in 1.0 mL MeNO2, reflux at 115 °C; (d) 75 (0.2 mmol) and BBr3 (0.6 mmol) in 0.5 mL of DCM, at 60 °C, 6 h; NaBO3·4H2O (2.0 mmol) in 0.5 mL of THF and 0.5 mL of K2CO3 (aq) at 25 °C, 1 h; NaH (0.24 mmol) in 1.0 mL of THF and MeI (0.24 mmol) at 25 °C, 1 h; (e) 76 (0.2 mmol), TsOH (0.3 mmol), and ethylene glycol (1,6 mmol) in 2.0 mL of toluene at 120 °C, 22 h; (f) 77 (0.2 mmol) and POCl3 (0.25 mmol) in 2.0 mL of dry DMF, reflux at 160 °C; 2-methylbut-2-ene (2.6 mmol) in 3 mL of tBuOH, NaClO2 (0.74 mmol), NaH2PO4 (1.0 mmol) at 25 °C, 24 h.
Discussion
In summary, we have developed an efficient boron-mediated system that is capable of mimicking the chelation-assisted metallic system to achieve directed C–H hydroxylation. The use of this method for the preparation of substituted phenols and downstream-functionalized products showcases the strategic opportunity to use this strategy for the synthesis of biologically active compounds. The reaction provides a simple new bond disconnection protocol for constructing these motifs with different regioselectivities and broader functional group compatibilities than existing methods.
Methods
General procedure for the synthesis of phenol 1c
A flame-dried 25 mL Schlenk tube was flushed with argon, and N-pivaloyl amide 1a (0.2 mmol, 1.0 equiv) and dry DCM (0.5 mL, 0.4 M) were introduced. A solution of BBr3 (1.0 M in DCM, 0.22 mL, 1.1 equiv) was added slowly under an argon atmosphere. The mixture was stirred at room temperature for 1 h. After stirring, the solvent was removed under vacuum directly. NaBO3·4H2O (92.3 mg, 0.6 mmol, 3.0 equiv), 0.5 mL of THF, and 0.5 mL of H2O were sequentially added to the reaction mixture and stirred at room temperature for another 1 h (monitored by TLC). After that, the excess water was removed by filtration with MgSO4 and then washed with EtOAc (10.0 mL × 3). The filtrate was collected, and the crude mixture was directly subjected to column chromatography on a silica gel, using petrol ether/EtOAc (10/1) as the eluent to give the desired product 1c as a white solid (41.5 mg, 92%).
Data availability
The authors declare that the data supporting the findings of this study are available within the article and Supplementary Information file or from the corresponding author upon reasonable request. The X-ray crystallographic coordinates for the structures reported in this study have been deposited at the Cambridge Crystallographic Data Centre (CCDC) under deposition numbers CCDC 1910134. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif.
References
Tyman, J. H. P. Synthetic and Natural Phenols (Elsevier, New York, 1996).
Rappoport, Z. The Chemistry of Phenols (Wiley-VCH, Weinheim, 2003).
Alonso, D. A., Nájera, C., Pastor, I. M. & Yus, M. Transition-metal-catalysed synthesis of hydroxylated arenes. Chem. Eur. J. 16, 5274–5284 (2010).
Roughley, S. D. & Jordan, A. M. The medicinal chemist’s toolbox: an analysis of reactions used in the pursuit of drug candidates. J. Med. Chem. 54, 3451–3479 (2011).
Warabi, K., Matsunaga, S., van Soest, R. W. M. & Fusetani, N. Dictyodendrins A−E, the first telomerase-inhibitory marine natural products from the sponge Dictyodendrilla verongiformis. J. Org. Chem. 68, 2765–2770 (2003).
Tichenor, M. S. et al. Asymmetric total synthesis of (+)- and ent-(-)-yatakemycin and duocarmycin SA: evaluation of yatakemycin key partial structures and its unnatural enantiomer. J. Am. Chem. Soc. 128, 15683–15696 (2006).
Nicolaou, K. C., Li, A., Edmonds, D. J., Tria, G. S. & Ellery, S. P. Total synthesis of platensimycin and related natural products. J. Am. Chem. Soc. 131, 16905–16918 (2009).
Pandey, S. K., Guttormsen, Y., Haug, B. E., Hedberg, C. & Bayer, A. A concise rotal synthesis of Breitfussin A and B. Org. Lett. 17, 122–125 (2015).
Yamaguchi, A. D., Chepiga, K. M., Yamaguchi, J., Itami, K. & Davies, H. M. L. Concise syntheses of dictyodendrins A and F by a sequential C–H functionalization strategy. J. Am. Chem. Soc. 137, 644–647 (2015).
Matsuoka, J., Matsuda, Y., Kawada, Y., Oishi, S. & Ohno, H. Total synthesis of dictyodendrins by the gold-catalysed cascade cyclization of conjugated diynes with pyrroles. Angew. Chem. Int. Ed. 56, 7444–7448 (2017).
Miley, G. P., Rote, J. C., Silverman, R. B., Kelleher, N. L. & Thomson, R. J. Total synthesis of tambromycin enabled by indole C−H functionalization. Org. Lett. 20, 2369–2373 (2018).
Bunnett, J. F. & Zahler, R. E. Aromatic nucleophilic substitution reactions. Chem. Rev. 49, 273–412 (1951).
Satyamurthy, N., Barrio, J. R., Bida, G. T. & Phelps, M. E. Efficient conversion of 1-aryl-3,3-dialkyltriazenes to phenols and oxygen-18 labeled phenols. Tetrahedron Lett. 31, 4409–4412 (1990).
Anderson, K. W., Ikawa, T., Tundel, R. E. & Buchwald, S. L. The selective reaction of aryl halides with KOH: synthesis of phenols, aromatic ethers, and benzofurans. J. Am. Chem. Soc. 128, 10694–10695 (2006).
Schulz, T. et al. Practical imidazole-based phosphine ligands for selective palladium-catalysed hydroxylation of aryl halides. Angew. Chem. Int. Ed. 48, 918–921 (2009).
Tlili, A., Xia, N., Monnier, F. & Taillefer, M. A very simple copper-catalysed synthesis of phenols employing hydroxide salts. Angew. Chem. Int. Ed. 48, 8725–8728 (2009).
Zhao, D. et al. Synthesis of phenol, aromatic ether, and benzofuran derivatives by copper-catalysed hydroxylation of aryl halides. Angew. Chem. Int. Ed. 48, 8729–8732 (2009).
Xia, S., Gan, L., Wang, K., Li, Z. & Ma, D. Copper-catalysed hydroxylation of (hetero)aryl halides under mild conditions. J. Am. Chem. Soc. 138, 13493–13496 (2016).
Engle, K. M., Mei, T.-S., Wasa, M. & Yu, J.-Q. Weak coordination as a powerful means for developing broadly useful C−H functionalization reaction. Acc. Chem. Res. 45, 788–802 (2012).
Yamaguchi, J., Yamaguchi, A. D. & Itami, K. C−H Bond Functionalization: Emerging Synthetic Tools for Natural Products and Pharmaceuticals. Angew. Chem. Int. Ed. 51, 8960–9009 (2012).
Colby, D. A., Tsai, A. S., Bergman, R. G. & Ellman, J. A. Rhodium catalysed chelation-assisted C−H bond functionalization reactions. Acc. Chem. Res. 45, 814–825 (2012).
Arockiam, P. B., Bruneau, C. & Dixneuf, P. H. Ruthenium(II)-catalysed C–H bond activation and functionalization. Chem. Rev. 112, 5879 (2012).
Gao, K. & Yoshikai, N. Low-valent cobalt catalysis: new opportunities for C–H functionalization. Acc. Chem. Res. 47, 1208–1219 (2014).
Gensch, T., Hopkinson, M. N., Glorius, F. & Wencel-Delord, J. Mild metal-catalysed C–H activation: examples and concepts. Chem. Soc. Rev. 45, 2900–2936 (2016).
He, J. et al. Palladium-catalysed alkyl C–H bond activation. Chem. Rev. 117, 8754 (2017).
Yang, Y., Lan, J. & You, J. Oxidative C–H/C–H coupling reactions between two (hetero)arenes. Chem. Rev. 117, 8787–8863 (2017).
Gandeepan, P. et al. 3d transition metals for C–H activation. Chem. Rev. 119, 2192–2452 (2019).
Loup, J., Dhawa, U., Pesciaioli, F., Wencel-Delord, J. & Ackermann, L. Enantioselective C−H activation with earth-abundant 3d transition metals. Angew. Chem. Int. Ed. 58, 12803–12818 (2019).
Yuan, C. et al. Metal-free oxidation of aromatic carbon-hydrogen bonds through a reverse-rebound mechanism. Nature 499, 192–196 (2013).
Zhang, Y.-W. et al. Photocatalytic hydrogen-evolution cross-couplings: benzene C−H amination and hydroxylation. J. Am. Chem. Soc. 138, 10080–10083 (2016).
Jintoku, T., Nishimura, K., Takaki, K. & Fujiwara, Y. Palladium-catalysed transformation of benzene to phenol with molecular oxygen. Chem. Lett. 1687–1688 (1990).
Lyons, T. W. & Sanford, M. S. Palladium-catalysed ligand-directed C−H functionalization reactions. Chem. Rev. 110, 1147–1169 (2010).
Zhang, F. & Spring, D. R. Arene C–H functionalisation using a removable/modifiable or a traceless directing group strategy. Chem. Soc. Rev. 43, 6906–6919 (2014).
Sambiagio, C. et al. A comprehensive overview of directing groups applied in metal-catalysed C–H functionalization chemistry. Chem. Soc. Rev. 47, 6603–6743 (2018).
Chen, X., Hao, X.-S., Goodhue, C. E. & Yu, J.-Q. Cu(II)-catalysed functionalizations of aryl C−H bonds using O2 as an oxidant. J. Am. Chem. Soc. 128, 6790–6791 (2006).
Zhang, Y.-H. & Yu, J.-Q. Pd(II)-catalysed hydroxylation of arenes with 1 atm of O2 or Air. J. Am. Chem. Soc. 131, 14654–14655 (2009).
Zhang, H.-Y., Yi, H.-M., Wang, G.-W., Yang, B. & Yang, S.-D. Pd(II)-catalysed C(sp2)–H hydroxylation with R2(O)P-coordinating group. Org. Lett. 15, 6186–6189 (2013).
Yan, Y. P. et al. PdCl2 and N-hydroxyphthalimide cocatalysed Csp2-H hydroxylation by dioxygen activation. Angew. Chem. Int. Ed. 52, 5827–5831 (2013).
Gallardo-Donaire, J. & Martin, R. Cu-catalysed mild C(sp2)−H functionalization assisted by carboxylic acids en route to hydroxylated arenes. J. Am. Chem. Soc. 135, 9350–9353 (2013).
Li, X. et al. Copper-mediated hydroxylation of arenes and heteroarenes directed by a removable bidentate auxiliary. Org. Lett. 16, 3904–3907 (2014).
Yang, F. Z., Rauch, K., Kettelhoit, K. & Ackermann, L. Aldehyde-assisted ruthenium(II)-catalysed C–H oxygenations. Angew. Chem. Int. Ed. 53, 11285–11288 (2014).
Liang, Y.-F. et al. Ligand-promoted Pd-catalysed oxime ether directed C−H hydroxylation of arenes. ACS Catal. 5, 6148–6152 (2015).
Sun, Y. H., Sun, T. Y., Wu, Y. D., Zhang, X. H. & Rao, Y. A diversity-oriented synthesis of bioactive benzanilides via a regioselective C(sp2)-H hydroxylation strategy. Chem. Sci. 7, 2229–2238 (2016).
Das, P., Saha, D., Saha, D. & Guin, J. Aerobic direct C(sp2)-H hydroxylation of 2-arylpyridines by palladium catalysis induced with aldehyde auto-oxidation. ACS Catal. 6, 6050–6054 (2016).
Chen, X.-Y., Ozturk, S. & Sorensen, E. J. Pd-catalysed ortho C−H hydroxylation of benzaldehydes using a transient directing group. Org. Lett. 19, 6280–6283 (2017).
Shang, M. et al. Identification of monodentate oxazoline as a ligand for copper-promoted ortho-C–H hydroxylation and amination. Chem. Sci. 8, 1469–1473 (2017).
Mkhalid, I. A. I. et al. C–H activation for the construction of C–B bonds. Chem. Rev. 110, 890–931 (2010).
Ros, A., Fernández, R. & Lassaletta, J. M. Functional group directed C–H borylation. Chem. Soc. Rev. 43, 3229–3243 (2014).
Jiang, Z.-T., Wang, B.-Q. & Shi, Z.-J. Transition metal catalysed direct oxidative borylation of C–H bonds. Chin. J. Chem. 36, 950–954 (2018).
Lv, J. et al. Metal-free directed sp2-C–H borylation. Nature 575, 336–341 (2019).
Iqbal, S. A., Cid, J., Procter, R., Uzelac, M., Yuan, K. & Ingleson, M. J. Acyl directed ortho-borylation of anilines and C7 borylation of indoles using just BBr3. Angew. Chem. Int. Ed. 58, 15381–15385 (2019).
Ishida, N., Moriya, T., Goya, T. & Murakami, M. Synthesis of pyridine-borane complexes via electrophilic aromatic borylation. J. Org. Chem. 75, 8709–8712 (2010).
Niu, L., Yang, H., Wang, R. & Fu, H. Metal-free ortho C–H borylation of 2-phenoxypyridines under mild conditions. Org. Lett. 14, 2618–2621 (2012).
Ingleson, M. J. Metal-free acyl-directed electrophilic C–H borylation using just BBr3. Sci. China Chem. 62, 1547–1548 (2019).
Li, Y. & Wu, X.-F. Direct C−H bond borylation of (hetero)arenes: evolution from noble metal to metal free. Angew. Chem. Int. Ed. 59, https://doi.org/10.1002/anie.201914914 (2020).
Yang, X., Shan, G. & Rao, Y. Synthesis of 2-aminophenols and heterocycles by Ru-catalyzed C–H mono- and dihydroxylation. Org. Lett. 15, 2334–2337 (2013).
Yuan, Y.-C., Bruneau, C., Dorcet, V., Roisnel, T. & Gramage-Doria, R. Ru-xatalyzed selective C–H bond hydroxylation of cyclic imides. J. Org. Chem. 84, 1898–1907 (2019).
Wang, Y. et al. Functionalized boron nitride nanosheets: a thermally rearranged polymer nanocomposite membrane for hydrogen separation. Angew. Chem. Int. Ed. 57, 16056–16061 (2018).
Yang, Y., Gao, P., Zhao, Y. & Shi, Z. Regiocontrolled direct C–H arylation of Indoles at the C4 and C5 positions. Angew. Chem. Int. Ed. 56, 3966–3971 (2017).
White, kL. & Movassaghi, M. Concise total syntheses of (+)-haplocidine and (+)-haplocine via late-stage oxidation of (+)-fendleridine derivatives. J. Am. Chem. Soc. 138, 11383–11389 (2016).
Sofiyev, V., Lumb, J.-P., Volgraf, M. & Trauner, D. Total synthesis of exiguamines A and B inspired by catecholamine. Chem. Chem. Eur. J. 18, 4999–5005 (2012).
Zhang, X., King-Smith, E. & Renata, H. Total synthesis of tambromycin by combining chemocatalytic and biocatalytic C–H functionalization. Angew. Chem. Int. Ed. 57, 5037–5041 (2018).
Acknowledgements
This study was supported by National Natural Science Foundation of China (Grant 21972064 and 21672097), the Excellent Youth Foundation of Jiangsu Scientific Committee (Grant BK20180007), and the “Innovation & Entrepreneurship Talents Plan” of Jiangsu Province.
Author information
Authors and Affiliations
Contributions
Z.S. conceived the concept, directed the project, and wrote the paper. J.L. and B.Z. performed the experiments. Y.Y. and Y.H. discussed the results.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information Nature Communications thanks the anonymous reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Lv, J., Zhao, B., Yuan, Y. et al. Boron-mediated directed aromatic C–H hydroxylation. Nat Commun 11, 1316 (2020). https://doi.org/10.1038/s41467-020-15207-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-020-15207-x
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
