
Key Points
-
The operon that is responsible for the production, processing and export of streptolysin S (SLS), the post-translationally modified cytolytic toxin that causes the β-haemolytic phenotype of group A Streptococcus (GAS; also known as Streptococcus pyogenes), has been identified and characterized.
-
Initial steps have been made towards the elucidation of the structure of SLS.
-
SLS contributes to virulence through soft-tissue damage, its impact on host phagocytes and its role in the translocation of GAS across the epithelial barrier. It also functions as a signalling molecule, and there is speculation that it has a role in iron acquisition.
-
It has become evident that SLS-like toxins are more widespread among streptococci than was previously appreciated; SLS-like toxins are produced by invasive human isolates of β-haemolytic group C and G streptococci, by the zoonotic fish pathogen Streptococcus iniae and by the horse pathogen Streptococcus equi.
-
The SLS-like peptide family has been further extended to beyond the genus Streptococcus following the identification, initially using in silico approaches, of related gene clusters in a number of notorious Gram-positive pathogens, including Listeria monocytogenes, Clostridium botulinum and Staphylococcus aureus.
-
SLS-like peptides belong to a recently defined large class of bioactive natural products known as thiazole/oxazole-modified microcins (TOMMs). TOMMs are characterized by a biosynthetic gene cluster that encodes a small precursor peptide and three adjacent synthetase proteins which serve to introduce thiazole, oxazole and methyloxazole heterocycles onto a ribosomally produced protoxin scaffold.
Abstract
Streptolysin S (SLS) is a potent cytolytic toxin and virulence factor that is produced by nearly all Streptococcus pyogenes strains. Despite a 100-year history of research on this toxin, it has only recently been established that SLS is just one of an extended family of post-translationally modified virulence factors (the SLS-like peptides) that are produced by some streptococci and other Gram-positive pathogens, such as Listeria monocytogenes and Clostridium botulinum. In this Review, we describe the identification, genetics, biochemistry and various functions of SLS. We also discuss the shared features of the virulence-associated SLS-like peptides, as well as their place within the rapidly expanding family of thiazole/oxazole-modified microcins (TOMMs).
Similar content being viewed by others

Main
Humans are the natural host and sole reservoir of group A Streptococcus (GAS; also known as Streptococcus pyogenes)1,2. The bacterium can survive and replicate in a variety of body locations, including the skin, throat and blood3, and is a common cause of illness, especially in children. The more typical infections include self-limiting skin disorders such as impetigo, and respiratory-tract infections such as pharyngitis. In rare cases, complications can lead to destructive soft-tissue infections, including necrotizing fasciitis and the multisystem disorder streptococcal toxic shock syndrome3. In recent decades, a dramatic increase in severe invasive GAS infections has been documented worldwide4,5. These infections carry a considerable risk of mortality6,7, with an estimated 500,000 deaths per year worldwide, most of which are attributed to invasive infection or acute rheumatic fever and subsequent rheumatic heart disease8,9.
Although the ability of certain strains of streptococci to haemolyse red blood cells (β-haemolysis)10,11,12 was first observed as early as 1895 (Refs 13, 14), it was not until 1938 that streptolysin S (SLS) was identified as being one of the two distinct toxins that are responsible for the ability of GAS to lyse mammalian erythrocytes15; the other toxin is the unrelated large cholesterol-dependent, oxygen-sensitive streptolysin O (SLO)16,17. Although SLS has been subject to rigorous investigation (Timeline), isolation of the mature SLS toxin and elucidation of its molecular structure have proved difficult18.

SLS is a 2.7-kDa peptide that is extensively post-translationally modified before export, resulting in the formation of a distinctive heterocyclic compound19,20. In addition to having an unusual structure, SLS is cytolytic only when associated with the bacterial cell surface or in the presence of certain carrier molecules21,22,23. The cytolytic spectrum of the toxin is broad and includes erythrocytes, leukocytes, platelets and subcellular organelles24,25,26,27, but excludes bacteria with intact cell walls28. SLS is not immunogenic in the course of natural infection29, a fact that may reflect its small size and its highly modified nature (which removes proteolytic sites that are critical for antigen digestion and display) as well as, perhaps most importantly, its potent cytotoxicity against cells that are involved in both innate and adaptive immunity30,31. Although the exact mechanism of SLS toxicity is not yet fully understood, it has been suggested that its accumulation in cell membranes results in the formation of transmembrane pores and irreversible osmotic lysis32.
It has become apparent that SLS represents the founding member of a class of post-translationally modified virulence peptides, as homologous toxins have now been identified in other Streptococcus species. The haemolytic activity of this distinct group of streptococcal toxins is abolished by the addition of trypan blue33. Furthermore, gene clusters that are similar to the SLS-associated cluster have been identified in other disease-causing pathogens such as Listeria monocytogenes, Clostridium botulinum and Staphylococcus aureus. In this Review, we describe the identification of SLS, and the GAS chromosomal locus (the SLS-associated gene (sag) operon) that encodes both the toxin and the proteins involved in its production, as well as the initial steps towards the characterization of its structure. We provide a brief overview of the role of SLS in virulence and other putative functions. We then discuss the identification of sag-like loci in non-GAS streptococci and advances in the identification of similar loci in other genera. Finally, we highlight the recent discovery that similar biosynthetic clusters are found in numerous microbial phyla and the subsequent definition of a new family of peptides called the thiazole/oxazole-modified microcins (TOMMs).
The GAS sag cluster
The chromosomal locus that is responsible for SLS production was first identified following the characterization of GAS transposon insertion mutants that did not produce SLS34,35. The transposons disrupted the promoter for the gene (designated sagA) that encodes the 53 amino acid SLS precursor, SagA. Subsequent chromosome-walking studies and genome sequence data identified the contiguous nine-gene sag operon (sagABCDEFGHI)36,37 (Fig. 1). Further analysis confirmed that the sag operon encodes all the accessory proteins that are required for proper processing and export of SLS36.

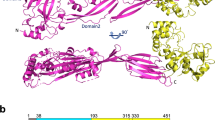
The carboxy-terminal core peptides of streptolysin (SLS)-associated gene A protein (SagA) (part a), which is produced by group A Streptococcus (GAS; also known as Streptococcus pyogenes), and of microcin B17 protein A (McbA) (part b), which is produced by Escherichia coli, are post-translationally modified by the SagBCD and McbBCD complexes to form biologically active SLS (a cytotoxin) and microcin B17 (MccB17; a DNA gyrase inhibitor), respectively. The amino-terminal leader sequence (black) is cleaved from the mature core peptide following modification, resulting in a mature peptide product. The 'leaky' Rho-independent terminator sequence between sagA and sagB, and mcbA and mcbB, acts as a regulatory mechanism, giving rise to an excess of sagA and mcbA transcripts compared with the amount of transcripts for the genes encoding the modification and transport machinery.
Following an in silico analysis of the sag operon, it became apparent that SLS is related to the bacteriocin family of antimicrobial peptides — or, more specifically, to a number of class I bacteriocins36,38. Like SLS, class I bacteriocins are encoded by an operon that contains a structural gene for a precursor peptide with an amino-terminal 'leader' region and a carboxy-terminal 'core' peptide39. The operon also includes genes encoding the machinery that is required for post-translational modification of the core peptide, as well as for cleavage of the leader and export of the mature form of the bacteriocin. In addition to the antimicrobial activity of bacteriocins, in rare cases they also exhibit broader haemolytic and cytolytic properties40, as is the case for the cytolysin that is produced by Enterococcus faecalis41.
SagA possesses features that are reminiscent of bacteriocin precursor peptides, such as a potential Gly-Gly leader cleavage site that would yield a 23 amino acid leader peptide and a 30 amino acid structural peptide42,43. This structural peptide contains an abundance of residues that are frequently the target of post-translational modification in bacteriocins, including Ser, Thr, Cys and Gly43. Site-directed mutagenesis of conserved residues in SagA supports its designation as a bacteriocin-like toxin19, as does the fact that SagA exhibits features that are reminiscent of McbA36, the precursor of the bacteriocin microcin B17 (MccB17) of Escherichia coli44. Furthermore, in common with many bacteriocin operons, a 'leaky' Rho-independent terminator sequence after sagA acts as a regulator of transcript abundance36 (Fig. 1a).
The sagA gene is followed by sagBCD, genes that exhibit low sequence identity to the mcbBCD genes that are located in the E. coli mcb cluster20 (Fig. 1b). McbBCD form an enzyme complex — containing a dehydrogenase (McbC), a cyclodehydratase (McbB) and a 'docking' protein (McbD) — that is required for the post-translational conversion of four Ser residues and four Cys residues within McbA into oxazole and thiazole heterocycles, respectively45,46,47 (Fig. 2). These modifications are essential for the activity of mature MccB17 (Ref. 48). SagB is a 36-kDa species that has some similarity to McbC (22% identity), SagC is a 40.3-kDa protein that is 13% identical to McbB, and SagD is a 51.6-kDa docking protein that is 18% identical to McbD20,36. Owing to these similarities, it was hypothesized that SagBCD functions in a similar manner to the McbBCD synthetase complex. It was subsequently established that SagBCD can also catalyse heterocycle formation, as recombinant SagBCD can successfully substitute for McbBCD to process McbA in vitro20.

Streptolysin S (SLS) heterocycles are formed via two distinct steps, catalysed by the combined activity of a zinc tetrathiolate-containing cyclodehydratase (SLS-associated gene C protein (SagC); green) and a dehydrogenase (SagB; yellow) from within a three-protein complex that also contains the docking protein (SagD; blue). SagC removes water from Cys, Ser and Thr residues in the peptide backbone to generate thiazoline, oxazoline and methyloxazoline rings. Subsequently, a flavin mononucleotide (FMN)-dependent dehydrogenation reaction catalysed by SagB removes hydrogen to generate the aromatic thiazole, oxazole and methyloxazole heterocycles. SagD is proposed to have a role in the formation of the SagBCD complex and in the regulation of its enzymatic activity. An example of each heterocyclizable residue is shown for illustrative purposes, with Gly included as a typical residue that is found on the amino-terminal side of the cyclized residues to facilitate the orbital alignment that is required for cyclodehydration45,50,159,160.
Further studies with SagBCD established that the complex is indeed responsible for the conversion of SagA into SLS20. Separate sites within the N-terminal leader peptide provide SagC with a high-affinity substrate-binding site, leading to efficient modification of the structural peptide by the SagBCD complex49. The modifications involve the conversion, via two distinct steps, of Cys, Ser and Thr residues to thiazole, oxazole and methyloxazole heterocycles, respectively20 (Fig. 2). SagC removes water from the peptide backbone to produce thiazoline, oxazoline and methyloxazoline rings, and then a dehydrogenation reaction that is catalysed by SagB formally removes hydrogen to generate aromatic thiazole, oxazole and methyloxazole heterocycles, respectively20. SagD is proposed to have a role in the formation of the SagBCD complex and in the regulation of its enzymatic activity20. The incorporation of these heterocycles into the unmodified precursor peptide restricts backbone conformational flexibility and provides the mature SLS with a more rigid structure, which is essential for bioactivity, as unstructured peptides would pay too large an entropic penalty to bind to their molecular targets efficiently50.
Recent research has yielded insights into the structure–activity relationships of SLS, but the precise chemical structure of mature SLS has yet to be fully elucidated. However, site-directed mutagenesis with Ala (to reduce backbone rigidity) and/or Pro (to retain rigidity) suggests that oxazoles are formed at residues Ser34, Ser39, Ser46 and Ser48; a thiazole is formed at Cys32 (Ref. 49); and Cys24 and Cys27 are also important residues with respect to haemolytic activity19,49. The incorporation of thiazoles, oxazoles or methyloxazoles results in a net loss of 20 Da in peptide mass, and this has been exploited for liquid chromatography–tandem mass spectrometry (LC–MS/MS) with in vitro-modified SLS, which has shown that heterocycle formation is catalysed by SagBCD and has confirmed the incorporation of oxazole moieties at Ser46 and Ser48 (Ref. 51).
The role of the remaining Sag proteins is less clear. SagF (26.2 kDa) is predicted to be membrane associated, and SagE (25.4 kDa) is expected to be a membrane-spanning peptidase20,52. Although it is reasonable to suggest that SagE is the enzyme responsible for leader cleavage, it has also been noted that intact sagE is required for viability and that the encoded protein has weak similarity with a candidate bacteriocin immunity (or self-protection) protein, PlnP, of Lactobacillus plantarum19. SagG (41.7 kDa), SagH (42.2 kDa) and SagI (41.7 kDa) are thought to be membrane proteins that form an ABC-type transporter19,20 similar to those frequently involved in bacteriocin export36,53, with SagG displaying the signature ATP-binding-pocket motifs19.
sag -like loci in other streptococci
The production of SLS-like cytolysins is not exclusively a GAS-associated trait (Table 1), as invasive human isolates of the β-haemolytic group C Streptococcus (GCS) and group G Streptococcus (GGS), which belong to Streptococcus dysgalactiae subsp. equisimilis, produce SLS-like peptides. Related peptides are also produced by the animal pathogens Streptococcus iniae and Streptococcus equi.
GCS is a common cause of acute pharyngitis among young adults54,55. Acute pharyngitis can also be caused by the normally commensal GGS56, and GGS strains have been associated with necrotising soft-tissue infections in patients with underlying medical conditions57. Both GCS and GGS display a hallmark β-haemolytic phenotype on blood agar57,58. The GCS and GGS sagA genes are identical, and the corresponding peptides have 89% identity with the GAS SagA57. Other conserved features include the sag operon promoter, a Rho-independent attenuator located downstream of sagA, and eight genes resembling the remaining SLS-associated genes57. Targeted mutagenesis of GGS sagA has been found to abolish β-haemolytic activity, a phenotype that is partially restored on transformation of the mutant with the GAS sagA homologue57.
S. iniae is a β-haemolytic pathogen of commercial fish species and can also be a rare human pathogen59,60,61. Characterization of the genomic region responsible for the haemolytic phenotype of S. iniae identified a nine-gene locus that has 73% sequence similarity to the GAS sag operon62. Furthermore, heterologous expression of sagA from S. iniae restored haemolytic activity to sagA-mutant GAS63. Finally, S. equi is the causative agent of 'strangles', a prevalent and highly contagious disease of horses64. Although an associated sag cluster has not yet been identified in this species, the haemolytic activity of S. equi has been characterized and found to be caused by an SLS-like toxin65.
Functions of SLS
A recent screen of clinical GAS samples has shown that 99% of all isolates are haemolytic, whereas the remaining 1% are not predicted to produce SLS and are assumed to be non-haemolytic (Lowry-type) strains66. Although such atypical strains have occasionally been associated with human infection66,67,68, there is little doubt that SLS has an important role in the pathogenicity of streptococci. Unsurprisingly, the specific mechanisms by which SLS contributes to virulence have been the subject of much investigation (Fig. 3) and include soft-tissue damage, an impact on host phagocytes and a contribution to GAS translocation across the epithelial barrier. SLS also functions as a quorum sensing molecule, and sagA mRNA has been implicated in the regulation of other virulence genes. In addition, it has been speculated that SLS contributes to iron acquisition from the host by providing a means through which GAS can access intracellular iron (through the lysis of host red blood cells)69,70.

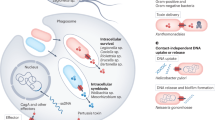
The mechanisms by which streptolysin S (SLS) is known to contribute to the virulence of group A Streptococcus (GAS; also known as Streptococcus pyogenes) include soft-tissue damage, an impact on host phagocytes and a contribution to the paracellular translocation of GAS. In addition, SLS-associated gene A (sagA) mRNA and the associated 'pleiotropic effects locus' (pel) mRNA affect virulence through their impact on the expression of other virulence genes. SLS also functions as a signalling molecule, and it has been proposed to contribute to iron acquisition from the host.
Role of SLS in tissue injury. In vivo studies have demonstrated that SLS is an important virulence factor in skin and soft-tissue infection, in which it contributes to tissue injury19. The virulence of an SLS-negative GAS mutant is substantially reduced in a mouse soft-tissue infection model35. Further studies confirming the importance of SLS with respect to GAS and GGS pathogenesis57,71,72,73 have revealed that even a single point mutation that is predicted to interfere with heterocycle formation can render a strain of GAS avirulent in a mouse model of skin infection49. The direct toxicity of SLS towards cells of the deep soft tissues and feeding vessels, leading to cell death and provoking neutrophil influx, is thought to promote the development of necrotising fasciitis57. It has been suggested that other virulence factors — such as SLO19 and the antiphagocytic surface protein M19,74,75 — as well as host neutrophil-derived oxidants and proteases23,76,77,78, interact synergistically with SLS to accelerate necrosis. It has also been established that expression of SLS is required for S. iniae-induced local tissue necrosis62,63.
Role of SLS in phagocytic clearance. The role of SLS in the resistance of GAS to phagocytic clearance was first uncovered when it was revealed that a sagA mutant did not survive as well as wild-type GAS in killing assays with human whole blood and purified neutrophils19. It was subsequently revealed that cell-associated SLS actively destroys neutrophils that are recruited to the site of infection79. The elimination of neutrophils (the phagocytic cells that are primarily responsible for the ingestion and killing of GAS) may be a specific virulence mechanism that effectively allows the bacterium to evade the innate immune system79. In support of this hypothesis, a paucity or lack of neutrophils in the affected tissues is regarded as an unfavourable prognostic sign in patients suffering from necrotizing fasciitis80, and a failure of neutrophil influx is also observed in primates that do not survive GAS-associated necrotizing fasciitis and myositis78. Macrophages constitute another crucial component of the host's phagocytic defence against GAS infection. GAS can kill macrophages through the SLS- and SLO-mediated activation of an inflammatory programmed cell death pathway81. It is important to note, however, that the relative contribution of SLS to phagocyte resistance is subject to both species and strain variation62,63,72,73, and other factors may also have a prominent role.
GAS is known to produce virulence factors that diminish the host's immune response to infection82,83,84. SLS is thought to contribute to this by affecting the ability of the host cell to produce signals that are chemotactic for neutrophils85. Using a zebrafish model, it was shown that an SLS mutant was significantly less virulent than wild-type GAS and was associated with a more robust recruitment of neutrophils85.
Finally, little is known about the entry and subsequent multiplication of S. equi following the exposure of a susceptible equine host86, but data indicate that virulence is the result of a potent antiphagocytic effect and the failure of innate immune defences87, suggesting that the SLS-like toxin of this bacterium may play an important part.
Role of SLS in GAS translocation. Systemic dissemination of GAS involves bacterial colonization of the pharynx or damaged skin, followed by penetration of the epithelial barrier. The mechanisms underlying the adherence of GAS to epithelial cells and its subsequent internalization have been extensively studied88,89,90,91. Most internalized GAS cells are eliminated by intracellular killing. However, it has been speculated that the programmed cell death induced in epithelial cells by massive adherence or internalization of GAS reduces the barrier function of the epithelium, providing GAS with access to deeper tissues through intracellular invasion92,93,94. Paracellular invasion by GAS has also been noted95 and, notably, recent research has identified SLS as a critical factor in this process96. SLS recruits the host Cys protease calpain to the plasma membrane by an as-yet-uncharacterized mechanism and then uses the proteolytic activity of this enzyme to degrade intercellular junctions and thus allow invasion through a paracellular route96.
SLS as a signalling molecule. Many species of bacteria use a cell density-dependent response system, or quorum sensing, to regulate gene expression97, and several GAS virulence genes are modulated by quorum sensing98,99. luxS (which encodes S-ribosylhomocysteine lyase) is essential for the production of autoinducer 2 (AI2) of the AI2 quorum sensing pathway in a diverse range of bacterial species100, and GAS strains with mutations in the luxS homologue display a number of altered phenotypes101. Notably, the SLS activity of a luxS mutant was found to be enhanced as a result of increased sagA transcription101.
Indeed, SLS expression increases with cell density35, and it was initially suspected that SLS itself might act as a quorum sensing molecule102. Several bacteriocins are known to be regulated by quorum sensing38, and in some instances (for example, the lantibiotic nisin) the structural peptides also function as signalling molecules and induce their own expression on activation of the density-dependent autoinduction loop103. Importantly, it has since been established that expression of sagA is upregulated by exposure to SLS102. It should also be noted that the expression of sagA is under transcriptional control of the GAS global regulators that are encoded by covR (also known as csrR) and covS (also known as csrS), mga, rofA, the fas operon and nra104,105,106,107,108,109,110.
Finally, global regulatory functions have been attributed to an untranslated mRNA, designated pel for 'pleiotropic effects locus', that matches the sagA gene111,112. It has been suggested that sagA mRNA is involved in the pre- and post-translational control of other GAS virulence factors, including M proteins, the capsule protein streptopain (SpeB) and streptokinase111,112,113. In contrast to these results, it has been found by others that elimination of sagA does not produce significant pleiotrophic effects19, suggesting that the role of sagA and/or pel mRNA in the regulation of other virulence factors may differ in a strain-specific manner.
sag -like loci in other bacterial genera
Until recently, post-translationally modified virulence peptides had rarely been reported and SLS-like cytolysins had been exclusively associated with the genus Streptococcus. As noted above, however, many similarities exist between the GAS sag operon and the MccB17 biosynthesis operon that is present in some E. coli. This observation prompted a search of public genomic databases, in the belief that other bacteria would use related machinery to introduce Ser-, Thr- and Cys-derived heterocycles into a wider variety of ribosomally produced peptides20,114. From this approach, it became evident that a number of sag-like gene clusters are present in some of the most notorious Gram-positive pathogens, including L. monocytogenes, C. botulinum and S. aureus20,114 (Fig. 4; Table 1). In addition, more distantly related clusters were identified in an even more diverse collection of bacteria20. This led to the definition of a new class of compounds that are characterized by a biosynthetic gene cluster encoding a small precursor peptide and three adjacent synthetase proteins that serve to introduce thiazole, oxazole and methyloxazole heterocycles onto the ribosomally produced protoxin scaffold. These bioactive natural products and their biosynthetic gene clusters are now referred to as TOMMs20,50. Although the biological purposes of the majority of newly identified TOMMs have not been uncovered, it is likely, based on their similarity to McbA and SagA, that some will act as either DNA gyrase inhibitors or membrane-damaging agents115.

a | The amino acid sequence of the unmodified stapholysin S precursor (StsA), listeriolysin S precursor (LlsA), streptolysin S precursor (streptolysin S-associated gene A protein (SagA)) and clostridolysin precursor (ClosA) from Staphylococcus aureus, Listeria monocytogenes, group A Streptococcus (GAS; also known as Streptococcus pyogenes) and Clostridium botulinum, respectively. The predicted leader regions are to the left and terminate in putative leader cleavage sites (purple). Residues that are potentially involved in the modification of the precursor peptides are indicated in orange (Cys), yellow (Ser), green (Thr) and blue (Gly). Amino acids that cannot be modified are shown in grey. Based on the sequence of sagA and assuming cleavage after the Gly-Gly site, the molecular weight of modified SagA is estimated to be approximately 2.7 kDa (although this does not preclude the possibility that mature streptolysin S (SLS) is an assemblage of modified SagA peptides). b | sag-like gene clusters in S. aureus, L. monocytogenes, S. pyogenes and C. botulinum strains. Related genes are indicated by colour. In each case, the respective gene designations are: A, structural gene; B, dehydrogenase; C and Y, cyclodehydratase; D, docking protein; E and P, CAAX protease; G, H and I, ABC-type transporter components; Z, X and F, proteins of unknown function. In the case of S. aureus str. RF122, the genomic map appears to be fragmented, giving multiple ORFs for several biosynthetic proteins.
Within TOMM-associated gene clusters, the genes encoding the SagB-like dehydrogenase, SagC-like cyclodehydratase, and SagD-like docking protein are often found as adjacent ORFs. It is probable that this will aid in the identification of additional orthologous clusters as more genome sequences become available. However, the associated TOMM structural genes are frequently overlooked during annotation because of their small and hypervariable nature114,116. Furthermore, although short ORFs encoding proteins of 50–70 residues that are rich in Cys, Ser and Thr are usually found in organisms with sagBCD-like genes as adjacent ORFs20,114, TOMM peptides are sometimes situated far from the genes encoding the thiazole-, oxazole- and methyloxazole-forming machinery, and in such cases it can be difficult to assign the substrate for a particular TOMM pathway117. The situation is further complicated by the observation that numerous substitutions in the C-terminal core sequence of the propeptide can often be tolerated49. This can result in the production of many similar peptide products50,118,119,120 by 'natural combinatorial biosynthesis'. In addition, new families of more unusual modifying enzymes are often not sufficiently related to characterized relatives to be identified by BLAST (basic local alignment search tool) searching20,121,122.
Several factors can increase confidence in the designation of an ORF as encoding a TOMM precursor, including the predicted protein having sequence similarity to previously identified TOMM precursors, having a suitable leader peptide cleavage motif and having a C-terminal core region that is rich in heterocyclizable residues in the predicted protein20,49 (Fig. 4). The detection of genes encoding enzymes involved in thiazole and oxazole synthesis, dehydroalanine production123 and peptide macrocyclization116,118,122,124 provides further support when annotating gene clusters that are responsible for the biosynthesis of post-translationally modified peptides. The identification of a TOMM biosynthesis cluster is also facilitated by the tendency of the modification enzymes to cluster with other genes that are associated with the cleavage and export of the final product20,36,46.
It is clear that the extent of structural variation in and the biological impact of TOMMs is gaining greater appreciation. Thus, it is likely that novel bioinformatics-based approaches such as those reported by Haft et al., whereby multiple highly sensitive profile-based search models are used to analyse large numbers of sequenced genomes50,117, will be applied to uncover additional members of this family of modified peptides, as well as to help identify cognate modification and export genes.
Listeriolysin S of L. monocytogenes. L. monocytogenes is an intracellular pathogen that is usually transmitted to humans through contaminated food products125. It has a high mortality rate in pregnant women, neonates and individuals with a compromised immune system126. Strains of L. monocytogenes are divided into three evolutionary lineages127, with lineage I (which consists of strains of serotype 1/2b and the notorious 'epidemic' serotype 4b) contributing to the majority of sporadic cases and epidemics that are associated with this often fatal pathogen128,129.
Since it was first established that listeriolysin O (LLO)-negative mutants of L. monocytogenes did not lyse blood cells, it was believed that this cholesterol-dependent virulence factor was the only cytotoxin to be produced by L. monocytogenes130,131. Recent in silico analysis identified a gene cluster in a number of L. monocytogenes strains that resembles the sag operon and was designated the listeriolysin S (lls) gene cluster114. The LLS structural gene within this cluster, llsA, encodes a peptide consisting of an N-terminal leader region, and a C-terminal core region with an extreme predominance of Cys, Ser and Thr residues, as well as a putative Ala-Gly leader cleavage motif114. As the predicted llsA promoter was found to be induced only under oxidative stress conditions, constitutive expression of the operon was used to establish that these genes did indeed encode an SLS-like cytolysin114. Like SLS, LLS was found to be active in a cell-associated form but inactive in cell-free situations in the absence of a stabilizer114. It is now apparent that previous detection of LLS-mediated haemolysis was hindered by the absence of the LLS cluster from the majority of the most frequently used laboratory strains, coupled with the inducible nature of the llsA promoter and the masking effect of LLO activity.
With respect to the rest of the LLS operon, the products of llsB (42% similarity to SagB), llsY (37% similarity to SagC) and llsD (46% similarity to SagD) are predicted to form a synthetase complex that is necessary for the production of mature, active LLS114,132. LLS is believed to be post-translationally modified in a similar manner to SLS, a theory that is supported by the fact that a SagA–LlsA chimaera (the SagA leader sequence fused to the LlsA core peptide) is converted into a cytolytic entity by SagBCD in vitro49. Other proteins encoded by the operon include LlsG and LlsH, which are two components of an ABC transporter (represented by three ORFs, sagGHI, in the sag operon), and LlsP, which has been annotated as a CAAX protease with 36% similarity to SagE114,132,133. The gene llsX is not homologous to any known gene and thus can be used to identify LLS-positive strains of L. monocytogenes by real-time PCR132. Deletion mutagenesis has established that six of the seven genes (llsGHXBYDP) that are located downstream of llsA in the lls operon are essential for LLS activity, with only llsP being non-essential114,132.
LLS contributes to pathogenesis, as evidenced by the significantly reduced levels of an LLS-negative mutant in the liver and spleen of mice following intraperitoneal inoculation, compared with levels of the corresponding wild-type bacteria114. Furthermore, wild-type L. monocytogenes survives significantly better than the LLS-negative mutant in purified human polymorphonuclear neutrophils. Notably, polymorphonuclear neutrophils are crucial for the resolution of L. monocytogenes infections134. Given the oxidative stress-inducible nature of the lls promoter and the contribution of LLS to neutrophil endurance, it has been suggested that LLS contributes to virulence by enhancing the survival of bacteria that are still retained in the phagosome upon phagosome–lysosome fusion114. The LLS-encoding TOMM cluster is also known as Listeria pathogenicity island 3 (LIPI-3)114. Given that LIPI-3 is consistently absent from all lineage II and III L. monocytogenes strains, and that few of the lineage I strains that are associated with outbreaks lack LIPI-3, it has been postulated that this gene cluster is the long-sought-after explanation for the enhanced virulence of a proportion of lineage I L. monocytogenes strains114.
Stapholysin S of Staphylococcus aureus str. RF122. A sag-like cluster is also present in the genome of S. aureus str. RF122 (Refs 20, 114), a pathogenic strain that is responsible for bovine mastitis135. Of the known sag-like clusters, this S. aureus cluster is most closely related to the lls cluster20,49,114. The putative structural gene again encodes a peptide consisting of a proposed N-terminal leader region, a C-terminal precursor peptide with an abundance of Cys, Ser and Thr residues, and an Ala-Gly suspected leader cleavage site20,114. Thus, it was predicted that mature 'stapholysin S' would act as a cytolysin20. This hypothesis was experimentally validated when a chimeric substrate comprising the leader peptide of SagA fused to the C terminus of stapholysin S was converted into a cytolytic entity by SagBCD in vitro49.
Clostridiolysin S of clostridia. Using comparative genomic analysis, nine-gene sag-like clusters were identified in the genomes of clostridia, including the biological warfare-associated pathogen C. botulinum and the food pathogen Clostridium sporogenes51. C. botulinum is an anaerobic, spore-forming, rod-shaped bacterium that causes the potentially fatal neuroparalytic diseases food-borne, infant, wound and inhalation botulism, as well as other invasive infections, through the elaboration of potent neurotoxins136. C. sporogenes is the C. botulinum counterpart that does not produce botulinum toxin137.
Before the functionality of the C. botulinum sag-like cluster had been confirmed, in vitro production of the bioactive natural product via SagBCD modification supported its designation as a TOMM, as did the haemolytic nature of the biotoxin that was generated20. This is not unexpected in light of the gene organization in the C. botulinum cluster (closABCDEFGHI) and the amino acid sequences of the encoded proteins, which correspond closely to the organization and encoded proteins of the sagABCDEFGHI cluster. In fact, C. botulinum harbours the most closely related sag-like genetic cluster known outside of the streptococci49. The cytolytic gene product of closA has been named clostridiolysin S, or CLS51. The functional equivalence of the clostridial sag-like gene clusters to the SLS biosynthesis pathway genes was confirmed by the complementation of targeted GAS sag operon knockouts with the corresponding clostridial genes51. The availability of the complete genome sequence of C. sporogenes138 allowed this safer organism to be used for mutagenesis studies, which established that the clos cluster is indeed responsible for a haemolytic phenotype51.
A more in-depth investigation of the clos genes further highlighted their TOMM-associated features. The first gene in the operon, closA, encodes the inactive precursor peptide that is post-translationally modified by the synthetase complex ClosBCD to form the mature biotoxin51. ClosG, ClosH and ClosI are ABC transporters and therefore probably export the mature haemolytic product. ClosE has been annotated as an immunity protein but, like SagE, is similar to the CAAX protease superfamily133 and so might be responsible for leader cleavage from the post-translationally modified propeptide51. ClosF is a protein of unknown function.
The in vitro reconstitution of CLS activity allowed a direct confirmation by mass spectrometry that the non-toxic precursor, ClosA, is post-translationally modified by the synthetase enzymes to contain heterocyclic moieties51. A heterocycle was identified at position Thr46 within ClosA, and the dramatically negative impact of a Thr46Ala substitution on haemolytic activity confirmed the importance of heterocyclic conversion at this site51. These investigations represent the first step towards the elucidation of the structure of mature CLS.
Other related clusters. The research of Lee et al. first highlighted the fact that, in addition to the Gram-positive pathogens that are discussed above, similar TOMM-associated clusters are widely disseminated in the genomes of a diverse group of microorganisms spanning six phyla20. Indeed, even archaea such as the thermophile Pyrococcus furiosus, can contain heterocycle-forming synthetases20. In addition to the SLS-like toxins described above, which have established or suspected roles in virulence, there are several other TOMM biosynthesis clusters of note (Table 1). As a consequence of the recent TOMM family expansion, this family now encompasses such diverse bacterial products as the microcins139, thiazolyl peptides140,141 and cyanobactins142. Emerging TOMM subfamilies include putative thiazole-containing heterocyclic bacteriocins associated with the genus Bacillus117, and a group of nitrile hydratase and Nif11-related precursor peptides50.
A gene cluster that has an identical arrangement to the mcb cluster has been found in the plant symbiont Pseudomonas putida str. KT2440 (Ref. 139), and it is speculated that, like MccB17, the product of this gene cluster functions as an inhibitor of DNA gyrase20. Many other bacteria harbour a sag-like gene cluster that is not associated with the production of a cytolysin, such as the goadsporin-producing microorganism, Streptomyces sp. TP-A0584 (Ref. 143). The molecular targets of this secondary metabolite have yet to be elucidated, but it is known that goadsporin promotes secondary metabolism and sporulation in actinomycetes, as well as exhibiting antibiotic activity115,123,144. Other diverse functions encoded by TOMM peptides include the inhibition of ribosome-mediated protein synthesis145 by the thiazolyl peptides, a family of >50 bactericidal antibiotics146,147, and the reversal of multidrug resistance in a human leukaemia cell line by the cyanobactin patellamide D148,149.
Last, the genome of Bacillus amyloliquefaciens str. FZB42, a Gram-positive saprophyte that promotes plant growth and can suppress the growth of bacterial and fungal pathogens of plants150, was recently shown to contain a novel TOMM cluster20. Subsequent investigations have revealed that this unique TOMM, plantazolicin, functions as a narrow-spectrum antibacterial compound151. It has been suggested that the biological role of this natural product is to suppress the growth of closely related competitors within the plant rhizosphere151.
It is apparent that these ribosomally produced natural peptides represent a largely hidden arsenal of active small molecules that, although grouped as a consequence of their possessing the same general modifications, can perform a wide variety of functions other than contributing to pathogenicity.
Conclusion
With the ongoing discovery and analysis of the biosynthetic genes that are associated with the production of SLS-like cytolytic peptides, evidence is rapidly mounting that these pathways are both more common and more diverse than previously suspected152 and can contribute substantially to the virulence potential of pathogenic bacteria. Of the myriad putative TOMM-encoding clusters that have been identified, those associated with L. monocytogenes, C. botulinum, C. sporogenes and S. aureus are particularly noteworthy as a consequence of their similarity to the sag cluster of GAS and the notoriety of the associated pathogens.
The number and variety of potential producers of SLS-like toxins (and, by extension, TOMMs), coupled with the fact that secondary metabolites produced by similar biosynthetic clusters have been an abundant source of pharmaceuticals in the past, suggest that further research into this group of peptides will lead to the identification of novel targets for antibiotic and vaccine development. Vaccine or chemotherapeutic strategies designed to neutralize SLS activity could be beneficial as adjuncts to surgical and antibiotic management of human streptococcal infection. The observation that synthetic peptides corresponding to the SLS precursor retain sufficient features of the mature toxin to raise neutralizing antibodies32,153 suggests that subunits or inactivated toxoids of each haemolysin could have substantial immunogenicity. Insights gained through the in vitro application of the SagBCD machinery and the SagA peptide49 could also be used to guide the design of artificial toxins with a view to structure-based vaccines. Furthermore, because the targeting of virulence factors is becoming an attractive anti-infective strategy, selective inhibition of the family of TOMM synthetase enzymes may be of value.
In summary, the structural diversity and biological impact of both the SLS-like peptide group and the wider TOMM family of natural products is just beginning to be fully appreciated50. The proven success of in silico analyses suggests that further studies of this type will help uncover the biosynthetic systems for novel TOMM (and other natural product) secondary metabolites and will aid in the definition of the evolutionary routes and interrelationships of the rapidly expanding TOMM ribosomal-peptide group. Such studies inform structural investigations into natural products and pave the way for combinatorial-biosynthesis studies and rational engineering.
References
Lancefield, R. C. A serological differentiation of human and other groups of hemolytic streptococci. J. Exp. Med. 57, 571–595 (1933). The original report of the now-standard classification technique based on specific carbohydrate 'group' antigens that are associated with the β-haemolytic streptococci.
Facklam, R. What happened to the streptococci: overview of taxonomic and nomenclature changes. Clin. Microbiol. Rev. 15, 613–630 (2002). A review covering changes in the nomenclature and taxonomy of the genus Streptococcus , arising from the additional use of genetic comparisons in the classification of the different species.
Cunningham, M. W. Pathogenesis of group A streptococcal infections. Clin. Microbiol. Rev. 13, 470–511 (2000).
Efstratiou, A. Group A streptococci in the 1990s. J. Antimicrob. Chemother. 45 (Suppl. 1), 3–12 (2000).
Stevens, D. L. The flesh-eating bacterium: what's next? J. Infect. Dis. 179, S366–S374 (1999).
O'Loughlin, R. E. et al. The epidemiology of invasive group A streptococcal infection and potential vaccine implications: United States, 2000–2004. Clin. Infect. Dis. 45, 853–862 (2007).
Ben-Abraham, R. et al. Invasive group A streptococcal infections in a large tertiary center: epidemiology, characteristics and outcome. Infection 30, 81–85 (2002).
O'Brien, K. L. et al. Epidemiology of invasive group A Streptococcus disease in the United States, 1995–1999. Clin. Infect. Dis. 35, 268–276 (2002).
WHO. The current evidence for the burden of group A streptococcal diseases. WHO [online], (2005).
Centor, R. M., Meier, F. A. & Dalton, H. P. Throat cultures and rapid tests for diagnosis of group A streptococcal pharyngitis. Ann. Intern. Med. 105, 892–899 (1986).
Brown, J. H. The Use of Blood Agar for the Study of Streptococci. (The Rockefeller Institute for Medical Research, New York, 1919).
Taranta, A., Moody, M. D. Diagnosis of streptococcal pharyngitis and rheumatic fever. Pediatr. Clin. North Am. 18, 125–143 (1971).
Marmorek, A. Le streptocoque et le sérum antistreptococcique. Ann. Inst. Pasteur 9, 593–620 (1895) (in French).
Marmorek, A. L'Unité des streptocoques pathogènes pour l'homme. Ann. Inst. Pasteur 16, 172 (1902) (in French).
Todd, E. W. The differentiation of two distinct serologic varieties of streptolysin, streptolysin O and streptolysin S. J. Pathol. Bacteriol. 47, 423–445 (1938). The paper that established that GAS produces two distinct haemolysins, one designated SLO to indicate its sensitivity to oxygen and another, an oxygen-stable haemolysin, designated SLS because of its high solubility in serum.
McCormick, J. K., Peterson, M. L. & Schlievert, P. M. in Gram-Positive Pathogens 2nd edn (eds Fischetti, V. A. et al.) 47–58 (American Society for Microbiology Press, Washington DC, 2006).
Halbert, S. P. Streptolysin O. Microb. Toxins 3, 69–98 (1970).
De Azavedo, J. C., Salim, K. Y. & Bast, D. J. in The Comprehensive Sourcebook of Bacterial Protein Toxins (eds Alouf, J. E. & Popoff, M. R.) 728–736 (Academic Press, San Diego, 2006).
Datta, V. et al. Mutational analysis of the group A streptococcal operon encoding streptolysin S and its virulence role in invasive infection. Mol. Microbiol. 56, 681–695 (2005). Work showing that the sagABCDEFG genes in the sag operon are each required for the expression of functional SLS, and providing support for the designation of SLS as a bacteriocin-like toxin.
Lee, S. W. et al. Discovery of a widely distributed toxin biosynthetic gene cluster. Proc. Natl Acad. Sci. USA 105, 5879–5884 (2008). A study demonstrating that, after modification by in vitro reconstitution, SagA is converted into a cytolysin; this article provides the first molecular-level description of the factor that is responsible for the β-haemolytic phenotype of GAS.
Alouf, J. E. Streptococcal toxins (streptolysin O, streptolysin S, erythrogenic toxin). Pharmacol. Ther. 11, 661–717 (1980).
Theodore, T. S. & Calandra, G. B. Streptolysin S activation by lipoteichoic acid. Infect. Immun. 33, 326–328 (1981).
Ginsburg, I. Is streptolysin S of group A streptococci a virulence factor? APMIS 107, 1051–1059 (1999).
Keiser, H., Weissmann, G. & Bernheimer, A. W. Studies on lysosomes. IV. Solubilization of enzymes during mitochondrial swelling and disruption of lysosomes by streptolysin S and other hemolytic agents. J. Cell Biol. 22, 101–113 (1964).
Hryniewicz, W. & Pryjma, J. Effect of streptolysin S on human and mouse T and B lymphocytes. Infect. Immun. 16, 730–733 (1977).
Bernheimer, A. W. & Schwartz, L. L. Lysosomal disruption by bacterial toxins. J. Bacteriol. 87, 1100–1104 (1964).
Bernheimer, A. W. & Schwartz, L. L. Effect of staphylococcal and other bacterial toxins on platelets in vitro. J. Pathol. Bacteriol. 89, 209–223 (1965).
Bernheimer, A. W. Disruption of wall-less bacteria by streptococcal and staphylococcal toxins. J. Bacteriol. 91, 1677–1680 (1966).
Roggiani, M., Assimacopoulos, A. P. & Schlievert, P. M. in Streptococcal Infections: Clinical Aspects, Microbiology, and Molecular Pathogenesis (eds Stevens, D. L. & Kaplan, E. L.) 57–75 (Oxford Univ. Press, New York, 2000).
Nizet, V. Streptococcal β-hemolysins: genetics and role in disease pathogenesis. Trends Microbiol. 10, 575–580 (2002).
Ofek, I., Bergner-Rabinowitz, S. & Ginsburg, I. Oxygen-stable hemolysins of group A streptococci. VIII. Leukotoxic and antiphagocytic effects of streptolysins S and O. Infect. Immun. 6, 459–464 (1972).
Carr, A., Sledjeski, D. D., Podbielski, A., Boyle, M. D. & Kreikemeyer, B. Similarities between complement-mediated and streptolysin S-mediated hemolysis. J. Biol. Chem. 276, 41790–41796 (2001).
Loridan, C. & Alouf, J. E. Purification of RNA-core induced streptolysin S, and isolation and haemolytic characteristics of the carrier-free toxin. J. Gen. Microbiol. 132, 307–315 (1986).
Borgia, S. M., Betschel, S., Low, D. E. & de Azavedo, J. C. Cloning of a chromosomal region responsible for streptolysin S production in Streptococcus pyogenes. Adv. Exp. Med. Biol. 418, 733–736 (1997).
Betschel, S. D., Borgia, S. M., Barg, N. L., Low, D. E. & De Azavedo, J. C. Reduced virulence of group A streptococcal Tn916 mutants that do not produce streptolysin S. Infect. Immun. 66, 1671–1679 (1998). After more than a century of research into the β-haemolytic phenotype of GAS, this study reports the discovery of a potential genetic determinant involved in SLS production: sagA , a gene that encodes a small peptide of 53 amino acids.
Nizet, V. et al. Genetic locus for streptolysin S production by group A Streptococcus. Infect. Immun. 68, 4245–4254 (2000). Research characterizing the nine-gene sag operon that is involved in SLS production.
Ferretti, J. J. et al. Complete genome sequence of an M1 strain of Streptococcus pyogenes. Proc. Natl Acad. Sci. USA, 98, 4658–4663 (2001).
Cotter, P. D., Hill, C. & Ross, R. P. Bacteriocins: developing innate immunity for food. Nature Rev. Microbiol. 3, 777–788 (2005).
Oman, T. J. & van der Donk, W. A. Follow the leader: the use of leader peptides to guide natural product biosynthesis. Nature Chem. Biol. 6, 9–18 (2010).
Nes, I. F. & Tagg, J. R. Novel lantibiotics and their pre-peptides. Antonie Van Leeuwenhoek 69, 89–97 (1996).
Cox, C. R., Coburn, P. S. & Gilmore, M. S. Enterococcal cytolysin: a novel two component peptide system that serves as a bacterial defense against eukaryotic and prokaryotic cells. Curr. Protein Pept. Sci. 6, 77–84 (2005).
McAuliffe, O., Ross, R. P. & Hill, C. Lantibiotics: structure, biosynthesis and mode of action. FEMS Microbiol. Rev. 25, 285–308 (2001).
Jack, R. W., Tagg, J. R. & Ray, B. Bacteriocins of gram-positive bacteria. Microbiol. Rev. 59, 171–200 (1995).
Yorgey, P., Davagnino, J. & Kolter, R. The maturation pathway of microcin B17, a peptide inhibitor of DNA gyrase. Mol. Microbiol. 9, 897–905 (1993).
Li, Y. M., Milne, J. C., Madison, L. L., Kolter, R. & Walsh, C. T. From peptide precursors to oxazole and thiazole-containing peptide antibiotics: microcin B17 synthase. Science 274, 1188–1193 (1996). This investigation determines that the MccB17-asscociated cyclodehydratase, dehydrogenase and docking protein form a complex and act collectively to install thiazoles and oxazoles onto the inactive precursor; the work also highlights the importance of the leader peptide.
Millan, J. L. S., Kolter, R. & Moreno, F. Plasmid genes required for microcin-B17 production. J. Bacteriol. 163, 1016–1020 (1985).
Yorgey, P. et al. Posttranslational modifications in microcin B17 define an additional class of DNA gyrase inhibitor. Proc. Natl Acad. Sci. USA 91, 4519–4523 (1994).
Milne, J. C. et al. Cofactor requirements and reconstitution of microcin B17 synthetase: a multienzyme complex that catalyzes the formation of oxazoles and thiazoles in the antibiotic microcin B17. Biochemistry 38, 4768–4781 (1999).
Mitchell, D. A. et al. Structural and functional dissection of the heterocyclic peptide cytotoxin streptolysin S. J. Biol. Chem. 284, 13004–13012 (2009). A paper providing the first steps towards the elucidation of the SLS structure, and describing the identification of the requisite features of SagBCD substrate recognition and SagA residues of functional importance.
Haft, D. H., Basu, M. K. & Mitchell, D. A. Expansion of ribosomally produced natural products: a nitrile hydratase- and Nif11-related precursor family. BMC Biol. 8, 70 (2010). As well as uniting numerous TOMM biosynthesis gene clusters with their cognate substrates, this study defines the TOMM family of natural products.
Gonzalez, D. J. et al. Clostridiolysin S: a post-translationally modified biotoxin from Clostridium botulinum. J. Biol. Chem. 285, 28220–28228 (2010).
Nakai, K. & Horton, P. PSORT: a program for detecting sorting signals in proteins and predicting their subcellular localization. Trends Biochem. Sci. 24, 34–36 (1999).
Sahl, H. G. & Bierbaum, G. Lantibiotics: biosynthesis and biological activities of uniquely modified peptides from gram-positive bacteria. Annu. Rev. Microbiol. 52, 41–79 (1998).
Meier, F. A., Centor, R. M., Graham, L. Jr & Dalton, H. P. Clinical and microbiological evidence for endemic pharyngitis among adults due to group C streptococci. Arch. Intern. Med. 150, 825–829 (1990).
Turner, J. C., Hayden, F. G., Lobo, M. C., Ramirez, C. E. & Murren, D. Epidemiologic evidence for Lancefield group C beta-hemolytic streptococci as a cause of exudative pharyngitis in college students. J. Clin. Microbiol. 35, 1–4 (1997).
Lindbaek, M., Hoiby, E. A., Lermark, G., Steinsholt, I. M. & Hjortdahl, P. Clinical symptoms and signs in sore throat patients with large colony variant β-haemolytic streptococci groups C or G versus group A. Br. J. Gen. Pract. 55, 615–619 (2005).
Humar, D. et al. Streptolysin S and necrotising infections produced by group G Streptococcus. Lancet 359, 124–129 (2002).
Hashikawa, S. et al. Characterization of group C and G streptococcal strains that cause streptococcal toxic shock syndrome. J. Clin. Microbiol. 42, 186–192 (2004).
McNulty, S. T., Klesius, P. H., Shoemaker, C. A. & Evans, J. J. Streptococcus iniae infection and tissue distribution in hybrid striped bass (Morone chrysops x Morone saxatilis) following inoculation of the gills. Aquaculture 220, 165–173 (2003).
Weinstein, M. R. et al. Invasive infections due to a fish pathogen, Streptococcus iniae. N. Engl. J. Med. 337, 589–594 (1997).
Facklam, R., Elliott, J., Shewmaker, L. & Reingold, A. Identification and characterization of sporadic isolates of Streptococcus iniae isolated from humans. J. Clin. Microbiol. 43, 933–937 (2005).
Fuller, J. D. et al. Identification of a streptolysin S-associated gene cluster and its role in the pathogenesis of Streptococcus iniae disease. Infect. Immun. 70, 5730–5739 (2002).
Locke, J. B. et al. Streptococcus iniae β-hemolysin streptolysin S is a virulence factor in fish infection. Dis. Aquat. Organ. 76, 17–26 (2007).
Timoney, J. F. The pathogenic equine streptococci. Vet. Res. 35, 397–409 (2004).
Flanagan, J. et al. Characterization of the haemolytic activity of Streptococcus equi. Microb. Pathog. 24, 211–221 (1998).
Yoshino, M. et al. Nonhemolytic Streptococcus pyogenes isolates that lack large regions of the sag operon mediating streptolysin S production. J. Clin. Microbiol. 48, 635–638 (2010).
James, L. & McFarland, R. B. An epidemic of pharyngitis due to a nonhemolytic group A Streptococcus at Lowry Air Force Base. N. Engl. J. Med. 284, 750–752 (1971).
Turner, D. P. & Gunn, S. L. Fatal case of sepsis caused by a non-haemolytic strain of Streptococcus pyogenes. J. Clin. Pathol. 60, 1057 (2007).
Bates, C. S., Montanez, G. E., Woods, C. R., Vincent, R. M. & Eichenbaum, Z. Identification and characterization of a Streptococcus pyogenes operon involved in binding of hemoproteins and acquisition of iron. Infect. Immun. 71, 1042–1055 (2003).
Eichenbaum, Z., Muller, E., Morse, S. A. & Scott, J. R. Acquisition of iron from host proteins by the group A Streptococcus. Infect. Immun. 64, 5428–5429 (1996).
Engleberg, N. C., Heath, A., Vardaman, K. & DiRita, V. J. Contribution of CsrR-regulated virulence factors to the progress and outcome of murine skin infections by Streptococcus pyogenes. Infect. Immun. 72, 623–628 (2004).
Fontaine, M. C., Lee, J. J. & Kehoe, M. A. Combined contributions of streptolysin O and streptolysin S to virulence of serotype M5 Streptococcus pyogenes strain Manfredo. Infect. Immun. 71, 3857–3865 (2003).
Sierig, G., Cywes, C., Wessels, M. R. & Ashbaugh, C. D. Cytotoxic effects of streptolysin O and streptolysin S enhance the virulence of poorly encapsulated group A streptococci. Infect. Immun. 71, 446–455 (2003).
Ofek, I., Zafriri, D., Goldhar, J. & Eisenstein, B. I. Inability of toxin inhibitors to neutralize enhanced toxicity caused by bacteria adherent to tissue culture cells. Infect. Immun. 58, 3737–3742 (1990).
Smeesters, P. R., McMillan, D. J. & Sriprakash, K. S. The streptococcal M protein: a highly versatile molecule. Trends Microbiol. 18, 275–282 (2010).
Ginsburg, I. Could synergistic interactions among reactive oxygen species, proteinases, membrane-perforating enzymes, hydrolases, microbial hemolysins and cytokines be the main cause of tissue damage in infectious and inflammatory conditions? Med. Hypotheses 51, 337–346 (1998).
Kwinn, L. A. & Nizet, V. How group A Streptococcus circumvents host phagocyte defenses. Future Microbiol. 2, 75–84 (2007).
Taylor, F. B. Jr et al. Staging of the baboon response to group A streptococci administered intramuscularly: a descriptive study of the clinical symptoms and clinical chemical response patterns. Clin. Infect. Dis. 29, 167–177 (1999).
Miyoshi-Akiyama, T. et al. Cytocidal effect of Streptococcus pyogenes on mouse neutrophils in vivo and the critical role of streptolysin S. J. Infect. Dis. 192, 107–116 (2005).
Bakleh, M. et al. Correlation of histopathologic findings with clinical outcome in necrotizing fasciitis. Clin. Infect. Dis. 40, 410–414 (2005).
Goldmann, O., Sastalla, I., Wos-Oxley, M., Rohde, M. & Medina, E. Streptococcus pyogenes induces oncosis in macrophages through the activation of an inflammatory programmed cell death pathway. Cell. Microbiol. 11, 138–155 (2009).
Wexler, D. E., Chenoweth, D. E. & Cleary, P. P. Mechanism of action of the group A streptococcal C5a inactivator. Proc. Natl Acad. Sci. USA 82, 8144–8148 (1985).
Sumby, P. et al. A Chemokine-degrading extracellular protease made by group A Streptococcus alters pathogenesis by enhancing evasion of the innate immune response. Infect. Immun. 76, 978–985 (2008).
Zinkernagel, A. S. et al. The IL-8 protease SpyCEP/ScpC of group A Streptococcus promotes resistance to neutrophil killing. Cell Host Microbe 4, 170–178 (2008).
Lin, A., Loughman, J. A., Zinselmeyer, B. H., Miller, M. J. & Caparon, M. G. Streptolysin S inhibits neutrophil recruitment during the early stages of Streptococcus pyogenes infection. Infect. Immun. 77, 5190–5201 (2009).
Harrington, D. J., Sutcliffe, I. C. & Chanter, N. The molecular basis of Streptococcus equi infection and disease. Microbes Infect. 4, 501–510 (2002).
Timoney, J. F. & Kumar, P. Early pathogenesis of equine Streptococcus equi infection (strangles). Equine Vet. J. 40, 637–642 (2008).
Bisno, A. L., Brito, M. O. & Collins, C. M. Molecular basis of group A streptococcal virulence. Lancet Infect. Dis. 3, 191–200 (2003).
Terao, Y., Kawabata, S., Nakata, M., Nakagawa, I. & Hamada, S. Molecular characterization of a novel fibronectin-binding protein of Streptococcus pyogenes strains isolated from toxic shock-like syndrome patients. J. Biol. Chem. 277, 47428–47435 (2002).
Joh, D., Wann, E. R., Kreikemeyer, B., Speziale, P. & Hook, M. Role of fibronectin-binding MSCRAMMs in bacterial adherence and entry into mammalian cells. Matrix Biol. 18, 211–223 (1999).
Kawabata, S. et al. Capsular hyaluronic acid of group A streptococci hampers their invasion into human pharyngeal epithelial cells. Microb. Pathog. 27, 71–80 (1999).
Nakagawa, I., Nakata, M., Kawabata, S. & Hamada, S. Cytochrome c-mediated caspase-9 activation triggers apoptosis in Streptococcus pyogenes-infected epithelial cells. Cell. Microbiol. 3, 395–405 (2001).
Cywes Bentley, C., Hakansson, A., Christianson, J. & Wessels, M. R. Extracellular group A Streptococcus induces keratinocyte apoptosis by dysregulating calcium signalling. Cell. Microbiol. 7, 945–955 (2005).
Klenk, M. et al. Streptococcus pyogenes serotype-dependent and independent changes in infected HEp-2 epithelial cells. ISME J. 1, 678–692 (2007).
Cywes, C. & Wessels, M. R. Group A Streptococcus tissue invasion by CD44-mediated cell signalling. Nature 414, 648–652 (2001).
Sumitomo, T. et al. Streptolysin S contributes to group A streptococcal translocation across an epithelial barrier. J. Biol. Chem. 286, 2750–2761 (2011).
Fuqua, W. C., Winans, S. C. & Greenberg, E. P. Quorum sensing in bacteria: the LuxR-LuxI family of cell density-responsive transcriptional regulators. J. Bacteriol. 176, 269–275 (1994).
Chaussee, M. S., Phillips, E. R. & Ferretti, J. J. Temporal production of streptococcal erythrogenic toxin B (streptococcal cysteine proteinase) in response to nutrient depletion. Infect. Immun. 65, 1956–1959 (1997).
Griffiths, B. B. & McClain, O. The role of iron in the growth and hemolysin (Streptolysin S) production in Streptococcus pyogenes. J. Basic Microbiol. 28, 427–436 (1988).
Surette, M. G., Miller, M. B. & Bassler, B. L. Quorum sensing in Escherichia coli, Salmonella typhimurium, and Vibrio harveyi: a new family of genes responsible for autoinducer production. Proc. Natl Acad. Sci. USA 96, 1639–1644 (1999).
Lyon, W. R., Madden, J. C., Levin, J. C., Stein, J. L. & Caparon, M. G. Mutation of luxS affects growth and virulence factor expression in Streptococcus pyogenes. Mol. Microbiol. 42, 145–157 (2001).
Salim, K. Y., de Azavedo, J. C., Bast, D. J. & Cvitkovitch, D. G. Role for sagA and siaA in quorum sensing and iron regulation in Streptococcus pyogenes. Infect. Immun. 75, 5011–5017 (2007).
Kleerebezem, M. Quorum sensing control of lantibiotic production; nisin and subtilin autoregulate their own biosynthesis. Peptides 25, 1405–1414 (2004).
Kihlberg, B. M., Cooney, J., Caparon, M. G., Olsen, A. & Bjorck, L. Biological properties of a Streptococcus pyogenes mutant generated by Tn916 insertion in mga. Microb. Pathog. 19, 299–315 (1995).
Federle, M. J., McIver, K. S. & Scott, J. R. A response regulator that represses transcription of several virulence operons in the group A Streptococcus. J. Bacteriol. 181, 3649–3657 (1999).
Heath, A., DiRita, V. J., Barg, N. L. & Engleberg, N. C. A two-component regulatory system, CsrR-CsrS, represses expression of three Streptococcus pyogenes virulence factors, hyaluronic acid capsule, streptolysin S, and pyrogenic exotoxin B. Infect. Immun. 67, 5298–5305 (1999).
Beckert, S., Kreikemeyer, B. & Podbielski, A. Group A streptococcal rofA gene is involved in the control of several virulence genes and eukaryotic cell attachment and internalization. Infect. Immun. 69, 534–537 (2001).
Kreikemeyer, B., Boyle, M. D., Buttaro, B. A., Heinemann, M. & Podbielski, A. Group A streptococcal growth phase-associated virulence factor regulation by a novel operon (Fas) with homologies to two-component type regulators requires a small RNA molecule. Mol. Microbiol. 39, 392–406 (2001).
Molinari, G. et al. The role played by the group A streptococcal negative regulator Nra on bacterial interactions with epithelial cells. Mol. Microbiol. 40, 99–114 (2001).
Shelburne, S. A. et al. A combination of independent transcriptional regulators shapes bacterial virulence gene expression during infection. PLoS Pathog. 6, (2010).
Li, Z., Sledjeski, D. D., Kreikemeyer, B., Podbielski, A. & Boyle, M. D. Identification of pel, a Streptococcus pyogenes locus that affects both surface and secreted proteins. J. Bacteriol. 181, 6019–6027 (1999).
Mangold, M. et al. Synthesis of group A streptococcal virulence factors is controlled by a regulatory RNA molecule. Mol. Microbiol. 53, 1515–1527 (2004).
Biswas, I., Germon, P., McDade, K. & Scott, J. R. Generation and surface localization of intact M protein in Streptococcus pyogenes are dependent on sagA. Infect. Immun. 69, 7029–7038 (2001).
Cotter, P. D. et al. Listeriolysin S, a novel peptide haemolysin associated with a subset of lineage I Listeria monocytogenes. PLoS Pathog. 4 e1000144 (2008). The discovery of LLS, the first non-streptococcal SLS-like peptide to be described. This discovery confirms the existence of an extended family of SLS-like disease-promoting toxins within the TOMM family.
Melby, J. O., Nard, N. J. & Mitchell, D. A. Thiazole/oxazole-modified microcins: ribosomal templates for complex natural products. Curr. Opin. Chem. Biol. 15, 369–378 (2011).
Sudek, S., Haygood, M. G., Youssef, D. T. & Schmidt, E. W. Structure of trichamide, a cyclic peptide from the bloom-forming cyanobacterium Trichodesmium erythraeum, predicted from the genome sequence. Appl. Environ. Microbiol. 72, 4382–4387 (2006).
Haft, D. H. A strain-variable bacteriocin in Bacillus anthracis and Bacillus cereus with repeated Cys-Xaa-Xaa motifs. Biol. Direct 4, 1–5 (2009).
Donia, M. S. et al. Natural combinatorial peptide libraries in cyanobacterial symbionts of marine ascidians. Nature Chem. Biol. 2, 729–735 (2006).
Li, B. et al. Catalytic promiscuity in the biosynthesis of cyclic peptide secondary metabolites in planktonic marine cyanobacteria. Proc. Natl Acad. Sci. USA 107, 10430–10435 (2010).
Schmidt, E. W. Trading molecules and tracking targets in symbiotic interactions. Nature Chem. Biol. 4, 466–473 (2008).
Altschul, S. F., Gish, W., Miller, W., Myers, E. W. & Lipman, D. J. Basic local alignment search tool. J. Mol. Biol. 215, 403–410 (1990).
Schmidt, E. W. et al. Patellamide A and C biosynthesis by a microcin-like pathway in Prochloron didemni, the cyanobacterial symbiont of Lissoclinum patella. Proc. Natl Acad. Sci. USA 102, 7315–7320 (2005).
Onaka, H., Tabata, H., Igarashi, Y., Sato, Y. & Furumai, T. Goadsporin, a chemical substance which promotes secondary metabolism and morphogenesis in streptomycetes. I. Purification and characterization. J. Antibiot. (Tokyo) 54, 1036–1044 (2001).
McIntosh, J. A., Donia, M. S. & Schmidt, E. W. Ribosomal peptide natural products: bridging the ribosomal and nonribosomal worlds. Nature Prod. Rep. 26, 537–559 (2009).
Lecuit, M. Human listeriosis and animal models. Microbes Infect. 9, 1216–1225 (2007).
Linnen, M. J. et al. Epidemic listeriosis associated with Mexican-style cheese. N. Engl. J. Med. 319, 823–828 (1988).
Wiedmann, M. et al. Ribotypes and virulence gene polymorphisms suggest three distinct Listeria monocytogenes lineages with differences in pathogenic potential. Infect. Immun. 65, 2707–2716 (1997).
Jeffers, G. T. et al. Comparative genetic characterization of Listeria monocytogenes isolates from human and animal listeriosis cases. Microbiology 147, 1095–1104 (2001).
Gray, M. J. et al. Listeria monocytogenes isolates from foods and humans form distinct but overlapping populations. Appl. Environ. Microbiol. 70, 5833–5841 (2004).
Schnupf, P. & Portnoy, D. A. Listeriolysin O: a phagosome-specific lysin. Microbes Infect. 9, 1176–1187 (2007).
Gaillard, J. L., Berche, P. & Sansonetti, P. Transposon mutagenesis as a tool to study the role of hemolysin in the virulence of Listeria monocytogenes. Infect. Immun. 52, 50–55 (1986).
Clayton, E. M., Hill, C., Cotter, P. D. & Ross, R. P. Real-time PCR assay to differentiate listeriolysin S-positive and -negative strains of Listeria monocytogenes. Appl. Environ. Microbiol. 77, 163–171 (2011).
Pei, J. & Grishin, N. V. Type II CAAX prenyl endopeptidases belong to a novel superfamily of putative membrane-bound metalloproteases. Trends Biochem. Sci. 26, 275–277 (2001).
Lopez, S., Marco, A. J., Prats, N. & Czuprynski, C. J. Critical role of neutrophils in eliminating Listeria monocytogenes from the central nervous system during experimental murine listeriosis. Infect. Immun. 68, 4789–4791 (2000).
Fitzgerald, J. R. et al. Characterization of a putative pathogenicity island from bovine Staphylococcus aureus encoding multiple superantigens. J. Bacteriol. 183, 63–70 (2001).
Smith, L. A. Botulism and vaccines for its prevention. Vaccine 27 (Suppl. 4), D33–D39 (2009).
Judicial Commission of the International Committee on Systematic Bacteriology. Rejection of Clostridium putrificum and conservation of Clostridium botulinum and Clostridium sporogenes – Opinion 69. Int. J. Syst. Bacteriol. 49, 339 (1999).
Sebaihia, M. et al. Genome sequence of a proteolytic (Group I) Clostridium botulinum strain Hall A and comparative analysis of the clostridial genomes. Genome Res. 17, 1082–1092 (2007).
Severinov, K., Semenova, E., Kazakov, A., Kazakov, T. & Gelfand, M. S. Low-molecular-weight post-translationally modified microcins. Mol. Microbiol. 65, 1380–1394, (2007).
Morris, R. P. et al. Ribosomally synthesized thiopeptide antibiotics targeting elongation factor Tu. J. Am. Chem. Soc. 131, 5946–5955 (2009).
Wang, J. et al. Identification and analysis of the biosynthetic gene cluster encoding the thiopeptide antibiotic cyclothiazomycin in Streptomyces hygroscopicus 10-22. Appl. Environ. Microbiol. 76, 2335–2344 (2010).
McIntosh, J. A. & Schmidt, E. W. Marine molecular machines: heterocyclization in cyanobactin biosynthesis. Chembiochem 11, 1413–1421 (2010).
Onaka, H., Nakaho, M., Hayashi, K., Igarashi, Y. & Furumai, T. Cloning and characterization of the goadsporin biosynthetic gene cluster from Streptomyces sp. TP-A0584. Microbiology 151, 3923–3933 (2005).
Igarashi, Y. et al. Goadsporin, a chemical substance which promotes secondary metabolism and morphogenesis in streptomycetes. II. Structure determination. J. Antibiot. (Tokyo) 54, 1045–1053 (2001).
Pestka, S. & Brot, N. Studies on the formation of transfer ribonucleic acid-ribosome complexes. IV. Effect of antibiotics on steps of bacterial protein synthesis: some new ribosomal inhibitors of translocation. J. Biol. Chem. 246, 7715–7722 (1971).
Bagley, M. C., Dale, J. W., Merritt, E. A. & Xiong, X. Thiopeptide antibiotics. Chem. Rev. 105, 685–714 (2005).
Hughes, R. A. & Moody, C. J. From amino acids to heteroaromatics—thiopeptide antibiotics, nature's heterocyclic peptides. Angew. Chem. Int. Ed. Engl. 46, 7930–7954 (2007).
Williams, A. B. & Jacobs, R. S. A marine natural product, patellamide D, reverses multidrug resistance in a human leukemic cell line. Cancer Lett. 71, 97–102 (1993).
Long, P. F., Dunlap, W. C., Battershill, C. N. & Jaspars, M. Shotgun cloning and heterologous expression of the patellamide gene cluster as a strategy to achieving sustained metabolite production. Chembiochem 6, 1760–1765 (2005).
Chen, X. H. et al. Genome analysis of Bacillus amyloliquefaciens FZB42 reveals its potential for biocontrol of plant pathogens. J. Biotechnol. 140, 27–37 (2009).
Scholz, R. et al. Plantazolicin, a novel microcin B17/streptolysin S-like natural product from Bacillus amyloliquefaciens FZB42. J. Bacteriol. 193, 215–224 (2010).
Schmidt, E. W. The hidden diversity of ribosomal peptide natural products. BMC Biol. 8, 83 (2010).
Dale, J. B., Chiang, E. Y., Hasty, D. L. & Courtney, H. S. Antibodies against a synthetic peptide of SagA neutralize the cytolytic activity of streptolysin S from group A streptococci. Infect. Immun. 70, 2166–2170 (2002).
Bernheimer, A. W. Formation of a bacterial toxin (streptolysin S) by resting cells. J. Exp. Med. 90, 373–392 (1949).
Koyama, J. Biochemical studies on Streptolysin S'. II. Properties of a polypeptide component and its role in the toxin activity. J. Biochem. 54, 146–151 (1963).
Koyama, J. Biochemical studies on Streptolysin S'. IV. Properties of the oligoribonucleotide portion. J. Biochem. 56, 355–360 (1964).
Koyama, J. & Egami, F. Biochemical studies on streptolysin S' formed in the presence of yeast ribonucleic acid. I. The purification and some properties of the toxin. J. Biochem. 53, 147–154 (1963).
Bernheimer, A. W. Physical behavior of streptolysin S. J. Bacteriol. 93, 2024–2025 (1967).
Kelleher, N. L., Hendrickson, C. L. & Walsh, C. T. Posttranslational heterocyclization of cysteine and serine residues in the antibiotic microcin B17: distributivity and directionality. Biochemistry 38, 15623–15630 (1999).
Roy, R. S., Allen, O. & Walsh, C. T. Expressed protein ligation to probe regiospecificity of heterocyclization in the peptide antibiotic microcin B17. Chem. Biol. 6, 789–799 (1999).
Acknowledgements
Related work in the authors' laboratories is supported by the Irish Government under the National Development Plan; by the Irish Research Council for Science Engineering; by Enterprise Ireland; and by Science Foundation Ireland (SFI), through the Alimentary Pharmabiotic Centre (APC) at University College Cork, Ireland, which is supported by the SFI-funded Centre for Science, Engineering and Technology (SFI-CSET) and provided P.D.C., C.H. and R.P.R. with SFI Principal Investigator funding. E.M.M. has received travel-related funding from SFI and the UK Society for General Microbiology. D.A.M. is supported by the Department of Chemistry and the Institute for Genomic Biology (University of Illinois at Urbana-Champaign, USA).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Related links
Glossary
- Impetigo
-
An acute and highly contagious infection of the surface layers of the skin, characterized by blisters, pustules and yellowish crusts.
- Pharyngitis
-
Inflammation of the pharynx that can be caused by group A, C and G streptococci; also known as strep throat.
- Necrotizing fasciitis
-
A rare but severe type of soft-tissue infection that can be caused by group A Streptococcus and can destroy the muscles, skin and underlying tissue. It develops when the bacteria enter the body, usually through a minor cut or as a complication of surgery. The mortality rate is high, even with aggressive treatment and powerful antibiotics.
- Streptococcal toxic shock syndrome
-
A rare but extremely severe infection that usually presents in people who have pre-existing skin infections with group A Streptococcus. It has a high mortality rate and is characterized by hypotension and shock. Other symptoms can include kidney impairment, abnormality in blood-clotting ability, acute respiratory distress syndrome, rash and local tissue destruction.
- β-haemolysis
-
A phenotype of complete red blood cell lysis (which appears as yellowing and transparency around and under colonies grown on blood agar medium) that is routinely used as a diagnostic tool for the identification of group A Streptococcus. It is primarily dependent on streptolysin S, with streptolysin O making a minimal contribution.
- Streptolysin O
-
(SLO). A thiol-activated, ∼57-kDa cytolysin that is produced by group A, C and G streptococci and is inhibited by small amounts of cholesterol. As a result of its oxygen-labile nature, SLO is most often responsible for haemolysis under the surface of blood agar, whereas the oxygen-stable streptolysin S results in a zone of clearing surrounding colonies on the surface of blood agar. SLO is antigenic, resulting in SLO-specific antibodies that are useful for documenting recent exposure to group A Streptococcus.
- Heterocyclic compound
-
A compound that has atoms of at least two different elements within its ring structure or structures. With respect to bioorganic chemistry, heterocycles contain one or more carbon atoms and at least one ring member other than carbon.
- Carrier molecules
-
High-molecular-mass molecules, such as non-ionic detergents, albumin, α-lipoprotein, lipoteichoic acid and the RNase-resistant fraction of yeast RNA (RNA core), that can stabilize the haemolytic activity from a streptolysin S (SLS)-producing growing culture or resting cell suspension. SLS is irreversibly inactivated on separation from the carrier or on destruction of the carrier.
- Trypan blue
-
A vital stain that is usually used to selectively colour dead tissue or cells blue. A defining feature of streptolysin S-like peptides is the fact that they are completely inactivated by trypan blue.
- Thiazole/oxazole-modified microcins
-
(TOMMS). A structurally and functionally diverse family of ribosomally produced peptides with post-translationally installed heterocycles derived from Cys, Ser and Thr residues. These modifications rigidify the precursor peptide to endow biological function on the mature natural product.
- Chromosome-walking studies
-
Studies using a technique to identify and characterize regions of DNA by the sequential isolation of overlapping DNA sequences, starting with a known fragment of DNA.
- Bacteriocin
-
A small ribosomally synthesized, heat-stable peptide that is produced by one bacterium and is active against other, either in the same species (narrow spectrum) or across genera (broad spectrum).
- Class I bacteriocins
-
Antimicrobial peptides that are extensively post-translationally modified in their active form, including the lantibiotic (lanthionine-containing) family of bacteriocins.
- Microcin B17
-
(MccB17). A class I bacteriocin that is produced by strains of Escherichia coli and is active against closely related bacterial species, targeting the essential enzyme DNA gyrase.
- Lowry-type
-
A designation for atypical, completely non-haemolytic group A Streptococcus strains, named after the initial isolation of such a strain (by James and McFarland in 1971) from an outbreak of rheumatic fever at Lowry Air Force Base, Colorado, USA.
- Quorum sensing
-
A mechanism of communication between bacteria that requires the production and secretion of a signalling molecule which, when present at or above a critical threshold concentration, induces changes in gene expression in neighbouring cells.
- Myositis
-
A general term for inflammation of the skeletal muscles.
- Paracellular invasion
-
The translocation of pathogens across an epithelial barrier by passing between the host cells.
- Natural combinatorial biosynthesis
-
The production of a library of related compounds by an organism. In the case of some thiazole/oxazole-modified microcin biosynthesis pathways, a single enzyme complex processes numerous substrates that have a common recognition motif but variable structural carboxyl termini. One striking example is the cyanobactin family of natural products.
Rights and permissions
About this article
Cite this article
Molloy, E., Cotter, P., Hill, C. et al. Streptolysin S-like virulence factors: the continuing sagA. Nat Rev Microbiol 9, 670–681 (2011). https://doi.org/10.1038/nrmicro2624
Published:
Issue Date:
DOI: https://doi.org/10.1038/nrmicro2624
This article is cited by
-
Pathogenesis, epidemiology and control of Group A Streptococcus infection
Nature Reviews Microbiology (2023)
-
Streptolysin S induces pronounced calcium-ion influx-dependent expression of immediate early genes encoding transcription factors
Scientific Reports (2023)
-
Commensal production of a broad-spectrum and short-lived antimicrobial peptide polyene eliminates nasal Staphylococcus aureus
Nature Microbiology (2023)
-
Tailored liposomal nanotraps for the treatment of Streptococcal infections
Journal of Nanobiotechnology (2021)
-
The microbiome-shaping roles of bacteriocins
Nature Reviews Microbiology (2021)
