
Abstract
The intestinal tract represents the largest mucosal surface and is a major site of multifaceted interactions between the host mucosal immune system and components of the intestinal microbiota. Host immune responses to the commensal microbiota are tightly controlled and, meanwhile, the microbiota actively shapes intestinal immune responses to itself. Appreciation of these interactions during health and disease may direct therapeutic approaches to a broad range of autoimmune and inflammatory disorders in humans. In this review, we will discuss findings on how the intestinal immune system, especially adaptive immune cells, helps accommodate the large number of resident bacteria, and in turn how the microbiota shapes intestinal immune responses to achieve mutualism.
Similar content being viewed by others

Introduction
The gastrointestinal tract represents a major site of dynamic interactions between the host mucosal immune system and components of the commensal microbiota.1 One of the key features of the intestinal immune system is its ability to distinguish between pathogenic and symbiotic bacteria, and thus protect against infection while avoiding detrimental and unnecessary inflammatory responses toward the normal microbiota.2 When these intestinal immune responses are dysregulated, they can result in chronic inflammatory disorders of the gut, including inflammatory bowel disease (IBD), celiac disease, and food allergies.3 It is plausible that immunological tolerance selectively exists to the intestinal microbiota but is broken during active inflammation.4 However, accumulating evidence provided by our group and others demonstrates a scenario of systemic immunological “ignorance” rather than “tolerance” toward the intestinal microbiota.5, 6, 7
Shaped by evolution over millennia, host immune cells have developed a hierarchy of homeostatic mechanisms to ensure mucosal immune compartmentation and maintain systemic ignorance to commensal bacteria, including a layer of immunoglobulin (Ig) A- and antimicrobial peptide-containing mucus, a physical epithelial barrier, and innate and adaptive immune components. These non-redundant immune mechanisms coordinate to allow an abundant and diverse microbiota to thrive, and at the same time, to limit microbiota-driven inflammation. Understanding of the host–microbiota interactions during both steady-state homeostasis and pathological intestinal inflammation may help to direct therapeutic approaches to IBD as well as to a broad range of immune-mediated inflammatory disorders in humans. In this review, we will discuss findings interpreting how the intestinal immune system, especially the adaptive immune component, helps accommodate the rich and diverse resident microbiota, and in turn how the microbiota shapes intestinal immune responses to achieve mutualism. First, we will discuss the tight compartmentation of mucosal innate and adaptive immune responses, formed mainly by intestinal IgA reactive to microbial antigens, resulting in immune exclusion of the commensal microbiota antigens and systemic immune “ignorance”. We will also highlight recent advances in understanding the influences of regulatory T (Treg) cells and effector T cells in the context of immune homeostasis and dysregulation, and feedback of the microbiota to intestinal T-cell regulation.
A Tale of Tolerance or Ignorance?
The human intestine harbors nearly 100 trillion microorganisms composed of more than 1,000 distinct bacterial species as defined by high-throughput microbial 16S ribosomal RNA gene characterization.8 Given the size and extent of the immunological challenge represented by the microbiota, it is unlikely that the host immune system actively reacts to all of these potential antigens. Earlier work proposed that the intestinal immune system is immunologically tolerant to commensal bacterial antigens under steady-state conditions and this selective tolerance is lost during intestinal inflammation.4
In recent years, an increasing number of studies have revealed a system of immunological barriers with distinct immune mechanisms that operate to minimize exposure of enteric bacteria to the systemic immune system. First, the epithelial layer of the intestinal tract is formed by tightly connected intestinal epithelial cells and serves as a physical protective layer, separating luminal contents from the underlying immune compartments, and providing an efficient barrier to block the entry of microflora into the lamina propria.9 Specialized intestinal epithelial cells such as mucus-secreting goblet cells and antimicrobial peptide-producing Paneth cells also contribute to the constitution of the mucosal barrier.10 IgA, secreted by plasma cells and transported by intestinal epithelial cells into the lumen, is more abundant than the sum of all other Ig isotypes combined,11 and it joins the effort with bactericidal peptides in the mucus layer to form a passive defense line, which sequesters most resident bacteria in the lumen and dramatically reduces the microbial burden of the epithelium. The third layer of intestinal defense is formed by innate and adaptive immunity. Intestinal immune cells are extensively distributed throughout the intestinal mucosa, which is customarily divided into organized inductive and diffusely distributed effector sites.12 Innate and adaptive immune cells accumulate in these mucosal immune compartments and coordinate both to maintain a state of limited mucosal activation and to initiate active immune responses to invading microbes.
This accumulating evidence raised concerns about whether there is immunological unresponsiveness to the microflora. With the complexity of the commensal microbiota and difficulties of culturing anaerobes, 20 random, recombinant intestinal bacterial antigens (rIBs) were generated to probe the normal immune responses toward microbial antigens.5 There was no systemic B- or T-cell reactivity detected to any of these rIBs or to commensal flagellins in normal C3H/HeJ mice. When mice were parenterally immunized with each of the rIBs plus ovabumin (OVA), a classic exogenous antigen, the systemic immune response to each rIB was equivalent to the response to OVA, indicating that the systemic immune compartment is not tolerant but is naïve to these antigens. Consequently, the systemic immune response to these antigens found in colitic C3H/HeJBir.IL-10−/− mice represents a loss of ignorance rather than a breaking of tolerance. These findings are in line with the report by Macpherson et al. that there was no specific IgG against Enterobacter cloacae, a dominant aerobic commensal strain in normal pathogen-free mice.6 When injected with 104–106 live organisms intravenously, the recipients developed antigen-specific serum IgG response, which is consistent with a state of systemic ignorance rather than tolerance toward microbial organisms.
In contrast to the lack of concomitant systemic immune response, a strong intestinal IgA response to half of the rIBs and to two immunodominant microbiota flagellins, CBir1 and FlaX,13, 14 was identified, indicating a tight intestinal compartmentation of the active immune response to the microbiota.5 In order to probe this compartmentation of the adaptive immune response to microbiota antigens more rigorously, a T-cell receptor (TCR) transgenic mouse line was constructed in which the majority of CD4+ T cells are specific for an immunodominant microbiota flagellin, CBir1.15 Despite the documented presence of the flagellin in the intestine, CBir1 TCR transgenic (CBir1 Tg) CD4+ T cells in the spleen remained naïve and there was no detectable serum IgG to CBir1, but there was a profound IgA anti-CBir1 response in the intestine. When CBir1 Tg mice were given CBir1 antigen parenterally, the transgenic CD4+ T cells responded vigorously. These results confirm the data with the random microbiota antigens,5 and indicate that the mechanisms maintaining homeostasis with the microbiota are powerful enough to prevent activation of potentially deleterious systemic T cells even when the T-cell repertoire is skewed so dramatically to a microbiota antigen. CBir1 Tg T cells proliferated robustly also after transfer into wild type C57BL/6 mice in response to systemic immunization, but not to mucosal stimulation with CBir1 flagellin.15
What are the mechanisms maintaining the mucosal compartmentation? CBir1 Tg T cells proliferate in response to oral CBir1 flagellin when adoptively transferred into IgA−/− mice, polymeric immunoglobulin receptor (pIgR)−/− mice (which have normal systemic IgA and IgM, but no secretory IgA or IgM), as well as TCRβxδ−/− mice (which have low levels of CBir1 flagellin-specific secretory IgA), demonstrating a critical role of antigen-specific intestinal IgA in control of systemic CD4+ T-cell responses to commensal bacterial antigens by immune exclusion. IgA specific for enteric antigens is produced with the aid of dendritic cells (DCs) that sample live bacteria at various mucosal sites, including Peyer's patches and lamina propria.7, 16, 17 These bacteria-loaded DCs induce B-cell differentiation into IgA+ plasma cells in a T cell-dependent or -independent manner, and these B cells then home from lymphoid structures to the lamina propria and secrete dimeric IgA that is transcytosed across the epithelial cell layer through a process mediated by pIgR.6, 18, 19 Secreted IgA binds to luminal bacteria, limiting attachment to the intestinal epithelial surface and precluding the penetration of bacteria and their components across the gut epithelium.6, 20 Meanwhile, DCs harboring live commensal bacteria are restricted to the mucosal immune compartments by mesenteric lymph nodes, which prevent the bacteria from entering systemic secondary lymphoid tissues.7 With these non-overlapping immune exclusion mechanisms, the host innate immune system can efficiently sample the luminal microbial contents or invading organisms and induce relevant protective adaptive immune responses within the intestinal mucosa to enteric bacteria. This induction of mucosal immune responses is confined to the local mucosal sites, but the effects can be disseminated over the whole mucosal surface through recirculation and homing of primed B and T cells via the lymph and blood. Systemic immunity remains naïve (not tolerant) to commensal epitopes, which ensures prompt and robust systemic immune responses against bacterial infection.
Intestinal CD4+ T Cells in Homeostasis and Disease
Overview of CD4+ effector and regulatory T-cell subsets
Based on TCR domain structures, T cells can be divided into αβ T cells and γδ T cells. T cells expressing αβ TCR are primarily responsible for antigen-specific cellular immunity, whereas γδ T cells are non-MHC-restricted and involved in primary immune responses.21 The vast majority of thymocytes and peripheral T cells are αβ T cells; γδ T cells only represent 1–5% of circulating T cells.21 However, in the murine small intestine, a large fraction of intraepithelial lymphocytes (IELs) express γδ TCR chains.22 Those γδ-expressing IELs, in particular, regulate the continuous turnover of intestinal epithelial cells,23 and enhance epithelial cell growth through secretion of keratinocyte growth factor in vitro.24 Furthermore, γδ IELs have been shown to help maintain epithelial barrier function by restricting pathogen transmigration.25
Efficient elimination of invasive pathogens and control of symbiotic microbes at mucosal sites often involves the coordination of innate and adaptive immune systems. Upon presentation of microbial antigens by antigen-presenting cells (APCs) such as DCs, naïve CD4+ αβ T cells (hereafter called CD4+ T cells) can differentiate into interferon (IFN)-γ-producing T-helper (Th) 1 cells, interleukin (IL)-4-producing Th2 cells, IL-17-producing Th17 cells, or adaptive Treg cells. All of these subsets are present in the intestinal lamina propria. The lineage commitment of CD4+ T cells is dependent on the cytokine cues from APCs and other microenvironmental factors, and key cytokine products and transcription factors of each lineage are usually crucial for lineage self-maintenance through positive feedback or active cross-inhibition of other lineage fates.26
Treg–IgA axis reinforces intestinal homeostasis
Both Treg cells and IgA are enriched in the intestine. CD4+Foxp3+ Treg cells and IL-10-producing Tr1 cells carry out non-redundant functions to regulate a large array of innate and adaptive immune responses both in the context of physiological and pathological settings, thus reinforcing intestinal immune homeostasis and limiting potential collateral damage to host tissue and beneficial organisms.27 The regulatory function of Treg and Tr1 cells is best exemplified in animal models of colitis induced by the transfer of naive CD45RBhiCD4+ T cells28 or C3H/HeJBir memory Th1 cells29 into recombination-activating gene (RAG) deficient or severe combined immunodeficient mice, in which the recipients are rescued by the co-transfer of CD25+ Treg or Tr1 cells, probably through the actions of transforming growth factor (TGF)-β or IL-10, respectively.30, 31, 32
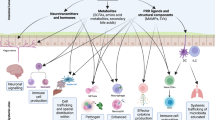
TGF-β is a multifunctional and pleiotropic cytokine. Its major immunological effects include maintenance of both Foxp3+ Treg and IL-10+ Tr1 cells and inhibition of cell proliferation.33, 34, 35 TGF-β is also essential for the induction of T cell-dependent and -independent IgA class switching and production in B cells.36 In support of this concept, B cell-specific TGF-βRII-deficient mice37, 38 and mice deficient in TGF-β downstream signaling molecules SMAD2 and SMAD339, 40 have reduced IgA responses at both systemic and mucosal sites. On the contrary, B cells from mice with mutant SMAD7, thus lacking a negative regulator of the TGF-β pathway, exhibit increased Ig class switch to IgA.41 TGF-β is abundant in the intestine, where it has multiple cellular sources, including epithelial cells, DCs, and Treg cells. It has long been speculated that TGF-β produced by Treg cells may stimulate B-cell IgA class switching.42, 43 Recently, we demonstrated that Treg cells are the major cellular source of TGF-β needed for intestinal IgA B-cell responses to microbiota antigens.15 Depletion of CD4+CD25+ Treg cells by anti-CD25 monoclonal antibody decreased lamina propria T-cell dependent CD5−IgA+ B-2 cells and total intestinal IgA and IgA anti-flagellin production. Conversely, repletion of either CD25+ or Foxp3+ Treg cells promoted intestinal IgA+ B cells as well as total and antigen-specific IgA in TCRβxδ−/− mice. The role of TGF-β in this system was determined by studies in which blockade of TGF-β inhibited Treg cell stimulation of IgA responses. These data indicate that Treg cells are the major helper cells for microbiota antigen (such as flagellin)-specific IgA responses. The main function of IgA is frequently stated to be protection against pathogens. It seems counterintuitive that, in this context, Treg cells are induced and in turn stimulate IgA as a protective strategy against intestinal bacteria. Instead, we propose that the major purpose of IgA and Treg cells in the intestine is to reinforce mutualism between the host and the commensal microbiota, and to restrict intestinal inflammation that can perturb such commensalism and impair colonization resistance to pathogens provided by the microbiota (Figure 1). The microbiota is also well adapted to compete for luminal nutrients, minimizing colonization by pathogens. We hereby propose a homeostatic model unit composed of Treg cells, IgA, and the microbiota, all of which act together to protect the host from pathogen infections, which is likely more effective than IgA would be alone (Figure 1).

Treg-immunoglobulin A (IgA)-microbiota as a functional unit. Treg cells are the major helper cells for microbiota antigen-specific IgA responses.15 This finding leads to the concept that the overall purpose of IgA and Treg cells in the intestine is to maintain mutualism with the microbiota and restrict intestinal inflammation. Intestinal inflammation reduces anaerobic microbiota such as the Firmicutes, and allows oxygen-tolerant organisms and pathogens such as the Proteobacteria to bloom.77, 78 The microbiota is well adapted to compete with pathogens for luminal nutrients, minimizing colonization of the pathogens. Treg cells, IgA, and the microbiota act together as a functional triad to protect the host from pathogens, which is likely to be more effective than IgA would be alone. DC, dendritic cell; IEC, intestinal epithelial cell; IEL, intraepithelial lymphocyte; TGF-β transforming growth factor-β; TLR, Toll-like receptor; Treg, regulatory T cells.
Lamina propria effector T cells in intestinal immune homeostasis and IBD
Th1 cells provide protection against intracellular bacteria and viruses. Th2 responses are essential to fight off parasitic helminthes, which are rare or absent in the normal intestine in the Western world. Th17 cells mediate immune responses against extracellular bacteria and fungi, and recently have been appreciated for their role in “prototypical” Th1 models of autoimmune diseases such as experimental autoimmune encephalomyelitis and IBD.44 The discovery of IL-23, which consists of the IL-12p40 and IL-23p19 subunits rather than IL-12p40 and IL-12p35 for IL-12, has led to a reconsideration of the relative roles of IL-12 and IL-23 in autoimmune diseases.45, 46 IL-12-driven excessive Th1 responses against commensal antigens have been widely accepted as a major pathway for IBD pathogenesis, but recent studies have uncovered a novel role of the IL-23-Th17 cell axis in experimental IBD and indicate divergent roles for IL-12 and -23 in local and systemic inflammation.46, 47 Clinical data and a genome-wide association study suggest that IL-23 receptor (IL-23R) is associated with susceptibility to Crohn's disease and ulcerative colitis.48 Anti-IL-23p19 monoclonal antibody prevents, as well as treats, colitis in an experimental model induced by adoptive transfer of microbiota antigen-specific T cells, confirming a crucial role of the IL-23/Th17 pathway in the pathogenesis of chronic intestinal inflammation.49 In addition to conventional IFN-γ single-positive Th1 cells and IL-17 single-positive Th17 cells, large numbers of IFN-γ+IL-17+ double-positive cells have been found in the lamina propria of colitic RAG−/− recipients of CBir1 flagellin-specific T cells but not in non-colitic recipients.50 More studies need to be carried out to identify the role of these double-positive cells, which may be associated with, or arise as a consequence of, severe intestinal inflammation.
Additional components of the Th17 pathway in IBD pathogenesis have also been examined. IL-6, produced by innate cells in response to microbiota stimulation, is crucial for Th17 cell development. Antagonism of IL-6 receptor signaling inhibits colitis development in an adoptive CD8+ T-cell transfer model.51 Anti-IL-6 monoclonal antibody has also been used in the treatment of active Crohn's disease clinically.52 In a CD4+ T-cell transfer model of colitis, blockade of IL-6 signaling inhibited T cell spontaneous proliferation (discussed below), abolished colitis development, and restrained Th17 cell development and generation of IFN-γ+IL-17+ double-positive cells.50 IL-6 may thus enhance colitis progression through promotion of Th17 cell clonal expansion in the lamina propria. The involvement of multiple factors in various effector cell pathways may contribute to the heterogeneity of human IBD, and will require better characterization of ongoing disease-related immune responses in order to apply appropriate treatment.
Plasticity of intestinal T cells in health and disease
Plasticity of CD4+ T cells has been an active area of research, which challenges the current concept of stable CD4+ T-cell lineage commitment. There is substantial late developmental plasticity in the Th17 program, which allows committed Th17 cells to transition to IFN-γ-producing Th1-like cells in a process driven by innate production of IL-12 and -23 in a STAT4- and T-bet-dependent manner.53, 54, 55, 56 These data provide the basis for understanding the relationship between Th17- and Th1-mediated pathophysiology. In recent years, a series of studies have also shown that Treg cells can differentiate into Th17 or T follicular helper cells in the intestine.57, 58, 59 Two recent reports further show that Foxp3+ Treg cells can acquire an effector T-cell phenotype in highly virulent or inflammatory settings.60, 61 We speculate that Treg cell acquisition of effector T-cell activity may allow control of enteric microbial infection, and should provide the host more flexibility of response to invading microorganisms. Others speculate that Treg cells may acquire Th effector characteristics to more efficiently repress those T effectors.62 In either case, these data raise concerns that Treg therapy may not be beneficial in the setting of established inflammation. Factors to be considered for Treg therapy include the optimal time course and co-administration of inhibitors of inflammatory cytokines, such as anti-IL-12p40. It should be noted that CD4+ T-cell plasticity has been demonstrated only in states of inflammation or in vitro. It is not clear at present whether CD4+ T-cell subset plasticity has a role in normal intestinal homeostasis in vivo.
Commensal Microbiota Shapes Intestinal T-Cell Responses
The intestinal microbiota is regulated by the host immune cells; and in turn, the microbiota actively shapes host adaptive immune responses. Symbiotic bacteria benefit the host by extracting dietary nutrients, preventing colonization by opportunistic pathogens and contributing to the development of the intestinal architecture.63 The influence of the microbiota on developmental aspects of adaptive immunity is best exemplified in germ-free animals. Germ-free mice have been shown to lack maturation of gut-associated lymphoid tissues and exhibit extensive defects in humoral antibody responses.1, 64 Understanding how the microbiota shapes the intestinal innate and adaptive immune responses during health and disease is an active area of research, which may provide potential therapeutic strategies to modulate host immune homeostasis.
Among the commensal microbiota, some organisms appear to have a greater impact on the mucosal T-cell responses than others. For example, segmented filamentous bacteria (SFB) are potent stimulators of small intestinal Th17 cells, as witnessed by the dearth of Th17 cells in germ-free mice and their restoration when such germ-free mice are colonized with SFB.65, 66 Retinoic acid, which is abundant in the gut and induces lymphocyte gut-homing molecules, inhibits Th17 cell development,67 resulting in a question of what controls Th17 cell gut tropism.68 The intestinal lamina propria is a natural site for Th17 cell development,69 possibly due to SFB colonization65, 66 and the innate IL-23 production in the intestinal microenvironment.47 Therefore, there is perhaps no need for Th17 cells to home to the intestinal lamina propria.
The microbiota influences not only effector T-cell (Th17) responses, but also Treg cells, which are also induced by intestinal bacteria29, 34 or their products. Of interest here, intestinal colonization of germ-free mice with a non-toxigenic form of Bacteroides fragilis that produces polysaccharide A (PSA), or treatment of germ-free mice with purified PSA, can protect against the development of, and even cure, experimental colitis through PSA-induced IL-10-producing Foxp3+-regulatory cells via a Toll-like receptor (TLR)2-dependent mechanism.70, 71
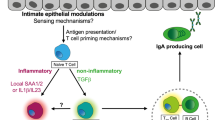
The commensal microbiota has also been implicated in T-cell homeostatic proliferation, in that adoptively transferred polyclonal CD4+ T cells do not undergo spontaneous proliferation in germ-free severe combined immunodeficient mice.72 This raises questions of whether microbiota-driven T-cell spontaneous proliferation is due to the microbiota antigenic stimulation or microbiota-derived innate signals. Reconstitution with cecal bacterial lysate-pulsed wild-type DCs, but not with IL-6−/− or MyD88−/− DCs, restored T-cell spontaneous proliferation in germ-free RAG−/− recipients,50 indicating that the microbiota initiates T-cell spontaneous proliferation by activating innate DCs to produce IL-6 via a TLR/MyD88-dependent pathway independent of TCR activation. Microbiota activation of T-cell responses have long been known to have a crucial role in the pathogenesis of IBD, particularly the microbiota stimulation of bacterial antigen-specific T cells that induce colitis, in that the mice develop colitis when raised with a conventional microbiota but fail to develop colitis when housed under germ-free conditions.73, 74, 75 It seems logical to reason that microbiota-induced T-cell spontaneous proliferation is necessary and may be the prerequisite for colitis development in lymphoreplete hosts as well. In our recent report,50 we demonstrate that co-transfer of large numbers of OT-II T cells inhibited spontaneous proliferation of CBir1 flagellin-specific T cells, and also abrogated colitis development induced by CBir1 Tg T cells in RAG−/− mice, possibly due to competition for DC occupancy or cytokine availability in limited space. These data provide a novel model for experimental colitis pathogenesis, which involves both microbiota stimulation of innate immune cells with subsequent T-cell “spontaneous” proliferation and microbiota antigen-induced TCR activation for the induction of experimental colitis (Figure 2).

“Two-hit model” of microbiota for the pathogenesis of colitis. Once intestinal immune homeostasis breaks, the microbiota stimulates mucosal dendritic cells (DCs) to produce interleukin (IL)-6 via Toll-like receptor (TLR) ligation. IL-6 promotes spontaneous proliferation of naïve T cells, which are in T0 status. T0 cells, in response to microbial antigens in the lamina propria, proliferate and, upon T-cell receptor (TCR) stimulation, differentiate into Th1 and Th17 cells under the influence of IL-12 and -6/transforming growth factor (TGF)-β, respectively, which eventually leads to intestinal inflammation. (Adapted from Feng et al.50) IEC, intestinal epithelial cell; IEL, intraepithelial lymphocyte; IgA, immunoglobulin A.
Concluding Remarks
The adaptive immune system evolved concomitantly with development of a complex microbiota and it has been argued that the two are integrally linked.76 Over the past decades, much has been learned about the organization and function of the adaptive and innate immune system in the gut in general, but how these systems relate to the microbiota has lagged considerably, because little was known about the intestinal microbiota that is changing rapidly with the massive genome sequencing of microbiota bacteria underway around the world. The tools necessary to define the mechanisms of the immune–microbiota interaction are now available. The host's dialog with the microbiota is a major purpose of the mucosal immune system and is critical to the overall health of the host. The studies discussed in this review are just a glimpse into this fascinating and emerging area of science.
References
Round, J.L. & Mazmanian, S.K. The gut microbiota shapes intestinal immune responses during health and disease. Nat. Rev. Immunol. 9, 313–323 (2009).
Coombes, J.L. & Powrie, F. Dendritic cells in intestinal immune regulation. Nat. Rev. Immunol. 8, 435–446 (2008).
Rescigno, M. & Di Sabatino, A. Dendritic cells in intestinal homeostasis and disease. J. Clin. Invest. 119, 2441–2450 (2009).
Duchmann, R. et al. Tolerance exists towards resident intestinal flora but is broken in active inflammatory bowel disease (IBD). Clin. Exp. Immunol. 102, 448–455 (1995).
Konrad, A., Cong, Y., Duck, W., Borlaza, R. & Elson, C.O. Tight mucosal compartmentation of the murine immune response to antigens of the enteric microbiota. Gastroenterology 130, 2050–2059 (2006).
Macpherson, A.J. et al. A primitive T cell-independent mechanism of intestinal mucosal IgA responses to commensal bacteria. Science 288, 2222–2226 (2000).
Macpherson, A.J. & Uhr, T. Induction of protective IgA by intestinal dendritic cells carrying commensal bacteria. Science 303, 1662–1665 (2004).
Hooper, L.V. & Macpherson, A.J. Immune adaptations that maintain homeostasis with the intestinal microbiota. Nat. Rev. Immunol. 10, 159–169 (2010).
Strober, W. The multifaceted influence of the mucosal microflora on mucosal dendritic cell responses. Immunity 31, 377–388 (2009).
Artis, D. Epithelial-cell recognition of commensal bacteria and maintenance of immune homeostasis in the gut. Nat. Rev. Immunol. 8, 411–420 (2008).
Fagarasan, S. & Honjo, T. Intestinal IgA synthesis: regulation of front-line body defences. Nat. Rev. Immunol. 3, 63–72 (2003).
Newberry, R.D. & Lorenz, R.G. Organizing a mucosal defense. Immunol. Rev. 206, 6–21 (2005).
Duck, L.W. et al. Isolation of flagellated bacteria implicated in Crohn's disease. Inflamm. Bowel Dis. 13, 1191–1201 (2007).
Lodes, M.J. et al. Bacterial flagellin is a dominant antigen in Crohn disease. J. Clin. Invest. 113, 1296–1306 (2004).
Cong, Y., Feng, T., Fujihashi, K., Schoeb, T.R. & Elson, C.O. A dominant, coordinated T regulatory cell-IgA response to the intestinal microbiota. Proc. Natl Acad. Sci. USA 106, 19256–19261 (2009).
Niess, J.H. et al. CX3CR1-mediated dendritic cell access to the intestinal lumen and bacterial clearance. Science 307, 254–258 (2005).
Rescigno, M. et al. Dendritic cells express tight junction proteins and penetrate gut epithelial monolayers to sample bacteria. Nat. Immunol. 2, 361–367 (2001).
Kaetzel, C.S., Robinson, J.K., Chintalacharuvu, K.R., Vaerman, J.P. & Lamm, M.E. The polymeric immunoglobulin receptor (secretory component) mediates transport of immune complexes across epithelial cells: a local defense function for IgA. Proc. Natl. Acad. Sci. USA 88, 8796–8800 (1991).
Johansen, F.E. & Brandtzaeg, P. Transcriptional regulation of the mucosal IgA system. Trends Immunol. 25, 150–157 (2004).
Suzuki, K. et al. Aberrant expansion of segmented filamentous bacteria in IgA-deficient gut. Proc. Natl. Acad. Sci. USA 101, 1981–1986 (2004).
Pennington, D.J., Silva-Santos, B. & Hayday, A.C. Gammadelta T cell development—having the strength to get there. Curr. Opin. Immunol. 17, 108–115 (2005).
van Wijk, F. & Cheroutre, H. Intestinal T cells: facing the mucosal immune dilemma with synergy and diversity. Semin. Immunol. 21, 130–138 (2009).
Komano, H. et al. Homeostatic regulation of intestinal epithelia by intraepithelial gamma delta T cells. Proc. Natl Acad. Sci. USA 92, 6147–6151 (1995).
Boismenu, R. & Havran, W.L. Modulation of epithelial cell growth by intraepithelial gamma delta T cells. Science 266, 1253–1255 (1994).
Dalton, J.E. et al. Intraepithelial gammadelta+ lymphocytes maintain the integrity of intestinal epithelial tight junctions in response to infection. Gastroenterology 131, 818–829 (2006).
Ouyang, W., Kolls, J.K. & Zheng, Y. The biological functions of T helper 17 cell effector cytokines in inflammation. Immunity 28, 454–467 (2008).
Sakaguchi, S., Yamaguchi, T., Nomura, T. & Ono, M. Regulatory T cells and immune tolerance. Cell 133, 775–787 (2008).
Mottet, C., Uhlig, H.H. & Powrie, F. Cutting edge: cure of colitis by CD4+CD25+ regulatory T cells. J. Immunol. 170, 3939–3943 (2003).
Cong, Y., Weaver, C.T., Lazenby, A. & Elson, C.O. Bacterial-reactive T regulatory cells inhibit pathogenic immune responses to the enteric flora. J. Immunol. 169, 6112–6119 (2002).
Asseman, C., Mauze, S., Leach, M.W., Coffman, R.L. & Powrie, F. An essential role for interleukin 10 in the function of regulatory T cells that inhibit intestinal inflammation. J. Exp. Med. 190, 995–1004 (1999).
Asseman, C., Read, S. & Powrie, F. Colitogenic Th1 cells are present in the antigen-experienced T cell pool in normal mice: control by CD4+ regulatory T cells and IL-10. J. Immunol. 171, 971–978 (2003).
Li, M.O., Wan, Y.Y. & Flavell, R.A. T cell-produced transforming growth factor-beta1 controls T cell tolerance and regulates Th1- and Th17-cell differentiation. Immunity 26, 579–591 (2007).
Marie, J.C., Letterio, J.J., Gavin, M. & Rudensky, A.Y. TGF-beta1 maintains suppressor function and Foxp3 expression in CD4+CD25+ regulatory T cells. J. Exp. Med. 201, 1061–1067 (2005).
Maynard, C.L. et al. Regulatory T cells expressing interleukin 10 develop from Foxp3+ and Foxp3- precursor cells in the absence of interleukin 10. Nat. Immunol. 8, 931–941 (2007).
Barnes, M.J. & Powrie, F. Regulatory T cells reinforce intestinal homeostasis. Immunity 31, 401–411 (2009).
Cerutti, A. The regulation of IgA class switching. Nat. Rev. Immunol. 8, 421–434 (2008).
Borsutzky, S., Cazac, B.B., Roes, J. & Guzman, C.A. TGF-beta receptor signaling is critical for mucosal IgA responses. J. Immunol. 173, 3305–3309 (2004).
Cazac, B.B. & Roes, J. TGF-beta receptor controls B cell responsiveness and induction of IgA in vivo. Immunity 13, 443–451 (2000).
Ashcroft, G.S. et al. Mice lacking Smad3 show accelerated wound healing and an impaired local inflammatory response. Nat. Cell. Biol. 1, 260–266 (1999).
Klein, J. et al. B cell-specific deficiency for Smad2 in vivo leads to defects in TGF-beta-directed IgA switching and changes in B cell fate. J. Immunol. 176, 2389–2396 (2006).
Li, R. et al. Deletion of exon I of SMAD7 in mice results in altered B cell responses. J. Immunol. 176, 6777–6784 (2006).
Cerutti, A. & Rescigno, M. The biology of intestinal immunoglobulin A responses. Immunity 28, 740–750 (2008).
Mestecky, J., Russell, M.W. & Elson, C.O. Perspectives on mucosal vaccines: is mucosal tolerance a barrier? J. Immunol. 179, 5633–5638 (2007).
Zhu, J. & Paul, W.E. CD4T cells: fates, functions, and faults. Blood 112, 1557–1569 (2008).
Cua, D.J. et al. Interleukin-23 rather than interleukin-12 is the critical cytokine for autoimmune inflammation of the brain. Nature 421, 744–748 (2003).
McGovern, D. & Powrie, F. The IL23 axis plays a key role in the pathogenesis of IBD. Gut 56, 1333–1336 (2007).
Uhlig, H.H. et al. Differential activity of IL-12 and IL-23 in mucosal and systemic innate immune pathology. Immunity 25, 309–318 (2006).
Duerr, R.H. et al. A genome-wide association study identifies IL23R as an inflammatory bowel disease gene. Science 314, 1461–1463 (2006).
Elson, C.O. et al. Monoclonal anti-interleukin 23 reverses active colitis in a T cell-mediated model in mice. Gastroenterology 132, 2359–2370 (2007).
Feng, T., Wang, L., Schoeb, T.R., Elson, C.O. & Cong, Y. Microbiota innate stimulation is a prerequisite for T cell spontaneous proliferation and induction of experimental colitis. J. Exp. Med. 207, 1321–1332 (2010).
Tajima, M. et al. IL-6-dependent spontaneous proliferation is required for the induction of colitogenic IL-17-producing CD8+ T cells. J. Exp. Med. 205, 1019–1027 (2008).
Ito, H. et al. A pilot randomized trial of a human anti-interleukin-6 receptor monoclonal antibody in active Crohn's disease. Gastroenterology 126, 989–996 (2004); discussion 947.
Lee, Y.K. et al. Late developmental plasticity in the T helper 17 lineage. Immunity 30, 92–107 (2009).
Lexberg, M.H. et al. Th memory for interleukin-17 expression is stable in vivo. Eur. J. Immunol. 38, 2654–2664 (2008).
Martin-Orozco, N., Chung, Y., Chang, S.H., Wang, Y.H. & Dong, C. Th17 cells promote pancreatic inflammation but only induce diabetes efficiently in lymphopenic hosts after conversion into Th1 cells. Eur. J. Immunol. 39, 216–224 (2009).
Nurieva, R., Yang, X.O., Chung, Y. & Dong, C. Cutting edge: in vitro generated Th17 cells maintain their cytokine expression program in normal but not lymphopenic hosts. J. Immunol. 182, 2565–2568 (2009).
Osorio, F. et al. DC activated via dectin-1 convert Treg into IL-17 producers. Eur. J. Immunol. 38, 3274–3281 (2008).
Tsuji, M. et al. Preferential generation of follicular B helper T cells from Foxp3+ T cells in gut Peyer's patches. Science 323, 1488–1492 (2009).
Yang, X.O. et al. Molecular antagonism and plasticity of regulatory and inflammatory T cell programs. Immunity 29, 44–56 (2008).
Oldenhove, G. et al. Decrease of Foxp3+ Treg cell number and acquisition of effector cell phenotype during lethal infection. Immunity 31, 772–786 (2009).
Zhou, X. et al. Instability of the transcription factor Foxp3 leads to the generation of pathogenic memory T cells in vivo. Nat. Immunol. 10, 1000–1007 (2009).
Wohlfert, E. & Belkaid, Y. Plasticity of T reg at infected sites. Mucosal Immunol. 3, 213–215 (2010).
Hooper, L.V. & Gordon, J.I. Commensal host-bacterial relationships in the gut. Science 292, 1115–1118 (2001).
Smith, K., McCoy, K.D. & Macpherson, A.J. Use of axenic animals in studying the adaptation of mammals to their commensal intestinal microbiota. Semin. Immunol. 19, 59–69 (2007).
Ivanov, I.I. et al. Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell 139, 485–498 (2009).
Gaboriau-Routhiau, V. et al. The key role of segmented filamentous bacteria in the coordinated maturation of gut helper T cell responses. Immunity 31, 677–689 (2009).
Mucida, D. et al. Reciprocal TH17 and regulatory T cell differentiation mediated by retinoic acid. Science 317, 256–260 (2007).
Maynard, C.L. & Weaver, C.T. Intestinal effector T cells in health and disease. Immunity 31, 389–400 (2009).
Ivanov, I.I. et al. The orphan nuclear receptor RORgammat directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell 126, 1121–1133 (2006).
Mazmanian, S.K., Round, J.L. & Kasper, D.L. A microbial symbiosis factor prevents intestinal inflammatory disease. Nature 453, 620–625 (2008).
Round, J.L. & Mazmanian, S.K. Inducible Foxp3+ regulatory T-cell development by a commensal bacterium of the intestinal microbiota. Proc. Natl Acad. Sci. USA 107, 12204–12209 (2010).
Kieper, W.C. et al. Recent immune status determines the source of antigens that drive homeostatic T cell expansion. J. Immunol. 174, 3158–3163 (2005).
Rath, H.C. et al. Normal luminal bacteria especially Bacteroides species, mediate chronic colitis, gastritis, and arthritis in HLA-B27/human beta2 microglobulin transgenic rats. J. Clin. Invest. 98, 945–953 (1996).
Sellon, R.K. et al. Resident enteric bacteria are necessary for development of spontaneous colitis and immune system activation in interleukin-10-deficient mice. Infect. Immun. 66, 5224–5231 (1998).
Taurog, J.D. et al. The germfree state prevents development of gut and joint inflammatory disease in HLA-B27 transgenic rats. J. Exp. Med. 180, 2359–2364 (1994).
McFall-Ngai, M. Adaptive immunity: care for the community. Nature 445, 153 (2007).
Lupp, C. et al. Host-mediated inflammation disrupts the intestinal microbiota and promotes the overgrowth of Enterobacteriaceae. Cell Host Microbe 2, 119–129 (2007).
Stecher, B. et al. Salmonella enterica serovar typhimurium exploits inflammation to compete with the intestinal microbiota. PLoS Biol. 5, 2177–2189 (2007).
Acknowledgements
We thank Dr Yingzi Cong for helpful comments and discussions. This work was supported by the NIH grants DK071176, DK64400, and CO6RR20136.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declared no conflict of interest.
PowerPoint slides
Rights and permissions
About this article
Cite this article
Feng, T., Elson, C. Adaptive immunity in the host–microbiota dialog. Mucosal Immunol 4, 15–21 (2011). https://doi.org/10.1038/mi.2010.60
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/mi.2010.60
This article is cited by
-
Impact of gut Microbiome alteration in Ulcerative Colitis patients on disease severity and outcome
Clinical and Experimental Medicine (2022)
-
Helicobacter genus in the intestine and liver of stray cats: the molecular, histopathological, and immunohistochemical study
Brazilian Journal of Microbiology (2020)
-
Oral immune therapy: targeting the systemic immune system via the gut immune system for the treatment of inflammatory bowel disease
Clinical & Translational Immunology (2016)
-
Symbiont-derived sphingolipids modulate mucosal homeostasis and B cells in teleost fish
Scientific Reports (2016)
-
Candida utilis and Cyberlindnera (Pichia) jadinii: yeast relatives with expanding applications
Applied Microbiology and Biotechnology (2016)
