
Abstract
Pioneering work in the 1990s first linked a novel microaerobic bacterium, Helicobacter hepaticus, with chronic active hepatitis and inflammatory bowel disease in several murine models. Targeted H. hepaticus infection experiments subsequently demonstrated its ability to induce colitis, colorectal cancer, and extraintestinal diseases in a number of mouse strains with defects in immune function and/or regulation. H. hepaticus is now widely utilized as a model system to dissect how intestinal microbiota interact with the host to produce both inflammatory and tolerogenic responses. This model has been used to make important advances in understanding factors that regulate both acquired and innate immune response within the intestine. Further, it has been an effective tool to help define the function of regulatory T cells, including their ability to directly inhibit the innate inflammatory response to gut microbiota. The complete genomic sequence of H. hepaticus has advanced the identification of several virulence factors and aided in the elucidation of H. hepaticus pathogenesis. Delineating targets of H. hepaticus virulence factors could facilitate novel approaches to treating microbially induced lower bowel inflammatory diseases.
Similar content being viewed by others

Initial Isolation and Characterization of H. hepaticus and H. bilis
Since the isolation and characterization of Helicobacter pylori, the etiological agent of peptic ulcer disease, in 1982 a number of other Helicobacter species have been isolated, many of which can produce serious nongastric disease in their respective hosts. Helicobacter species that colonize the lower bowel and biliary tract of mice have been associated with the development of colitis resembling human inflammatory bowel disease (IBD) in susceptible hosts. Further, H. hepaticus infection has been demonstrated to exacerbate the development of cancer at both intestinal and extraintestinal sites. The most well studied member of this group of enterohepatic Helicobacter species (EHS) is H. hepaticus. In this review, we will focus on the initial identification of H. hepaticus, the basis for H. hepaticus-induced lower bowel inflammation, and the characterization of bacterial virulence factors.
In the early 1990s, a chronic active hepatitis was detected in several inbred strains of mice including A/JCr, originating from a barrier-maintained facility. A distinctive morphological pattern of liver damage was noted, which had not been previously identified in mice. Using a special silver stain, a spiral-like organism was observed in the liver. Infected A/JCr mice were then shipped to our laboratory for characterization and diagnostic evaluation. A microaerobic bacterium was consistently isolated from the liver and colon of these mice between 3 and 18 months of age. By biochemical and molecular methodologies these bacteria were confirmed to represent a new Helicobacter sp., which we named H. hepaticus.1, 2, 3 Shortly after the isolation of H. hepaticus, we isolated a fusiform bacterium from the liver, bile, and lower intestine of aged, inbred mice. On the basis of 16S rRNA gene sequence analysis, the organism was classified as a novel Helicobacter sp., H. bilis.
Currently there are nine formally named EHS isolated from mice as well as several other novel Helicobacter spp. awaiting formal naming. Most recently, Helicobacter pullorum, a human pathogen, has been isolated from commercial, barrier maintained mice.4 Surveys conducted in 1995 and 2007 indicated that H. hepaticus and other EHS have a worldwide distribution in academic and commercial mice.5, 6 Contrary to the earlier survey, no Helicobacter spp., however, were found in mice in 2007 from advertised Helicobacter-free production areas from two US vendors. These results demonstrated that infection with EHS is widespread in most, if not all, mouse colonies used for experimental investigations.
Spontaneous IBD in Immunodeficient and Genetically Engineered Mice: Association with Helicobacter spp.
Before the isolation of EHS, it had been speculated that microflora might have an important role in the etiology of spontaneous colitis that developed in a number of immunodeficient laboratory mouse strains, including interleukin (IL)-10-deficient, scid, and Rag. Berg et al.7 also demonstrated that inbred mice on different strain backgrounds with undefined intestinal flora influenced the degree of colitis and lower bowel adenocarcinoma noted in mice; C57BL IL10−/− mice were less prone to develop colitis and carcinoma, BALBc IL10−/− mice were intermediate in susceptibility, and 129 IL10−/− mice developed the most severe disease. Once identified, we found that although H. hepaticus infection could be demonstrated in a number of immunologically normal mice without colitis, it was strongly associated with chronic proliferative typhlitis, colitis, and rectal prolapse in several immunodeficient mouse strains housed at the National Cancer Institute and Massachusetts Institute of Technology.8, 9 The specific pathogen-free health status of these colonies was verified to include lack of serum antibodies to murine viruses and negative cultures for respiratory pathogens, Salmonella sp., and Citrobacter rodentium. These findings suggested infection with H. hepaticus was strongly associated with the development of colitis in a number of laboratory mouse strains, and raised the question of whether H. hepaticus might be an etiological agent of murine colitis.
Establishing the Link Between Helicobacter spp. and IBD
To establish the link between H. hepaticus and the development of colitis, we demonstrated that H. hepaticus inoculated into Helicobacter spp. free defined flora C.B. 17 scid mice reconstituted with CD45RBhigh T cells resulted in severe IBD, similar to that noted with human disease.10 The expression of typhlitis and colitis was statistically less severe in defined flora scid mice that had been reconstituted with T cells but not infected with H. hepaticus when compared to reconstituted scid mice infected with H. hepaticus,10 demonstrating that infection with this single species could significantly exacerbate colitis in a murine disease model.11, 12 Similar results were obtained following targeted infection of C57BL IL-10-deficient mice with H. hepaticus.13, 14 Targeted infection experiments have now demonstrated that H. hepaticus is able to induce colitis and in some cases lower bowel carcinoma in a number of mouse strains with defects in immune function and/or regulation ( Table 1). Interestingly, the same strain susceptibility noted by Berg et al. in IL10−/− mice was also documented in Rag−/− mice developing colitis-associated lower bowel carcinoma when experimentally infected with H. hepaticus.7, 15
Basis of the Gastrointestinal Inflammatory Response to H. hepaticus
The experiments described indicate that H. hepaticus infection can induce colitis in certain susceptible mouse strains, but the nature of the inflammatory response and why certain strains were susceptible required further investigation. Seminal experiments from the early 1990s demonstrated that mice lacking IL-10 developed spontaneous colitis, which was significantly less severe when mice were housed in SPF conditions, suggesting that enhanced susceptibility to microbiota induced inflammation.16 In contrast to H. hepaticus monoassociated C57BL IL10−/− mice, which did not develop colitis, Kullberg and co-workers directly demonstrated that targeted infection of SPF-housed C57BL IL-10 mice with H. hepaticus significantly exacerbated the development of colitis.13, 17 The inflammatory response in the H. hepaticus-infected C57BL IL-10-deficient mice was characterized by a Th1-like cytokine profile.13, 18 More recently, these results have been replicated by demonstrating that wild-type (WT) C57BL mice treated with an antibody directed at the IL-10 receptor (anti-IL-10R), but not those treated with a control antibody, develop colitis following infection with H. hepaticus.19 Colitis in anti-IL-10R-treated mice is characterized by increased expression of mRNA coding for interferon (IFN)-γ and IL-17 within the colon. Mesenteric lymph node cells (MLNs) derived from H. hepaticus-infected mice treated with anti-IL-10R secreted higher levels of IL-17 and IFN-γ compared with control MLNs following exposure to soluble helicobacter antigen.19 Mice deficient in IL-12p40 but not those deficient in IL-12p35 were resistant to H. hepaticus-induced colitis following anti-IL-10R treatment, which was associated with decreased expression of IFN-γ and IL-17, suggesting that IL-23 rather than IL-12p70 was necessary for the generation of both Th1 and Th17 responses observed following H. hepaticus infection. These experiments indicated that H. hepaticus can induce antigen-specific Th1 and Th17 responses in mice with impaired IL-10 signaling.
Although H. hepaticus infection of mice with impaired IL-10 signaling induces CD4-mediated Th1 and Th17 responses, the exact role of both IFN-γ and IL-17 in H. hepaticus-mediated colitis has been more difficult to discern. Initial experiments with an IFN-γ-depleting antibody suggested that IFN-γ was necessary for H. hepaticus to induce colitis in IL-10-deficient mice.13 However, the severity of H. hepaticus-induced colitis in mice doubly deficient for IL-10 and IFN-γ was very similar to that in mice lacking IL-10 alone.18 Further, although H. hepaticus-induced inflammation within the colon of IFN-γ-deficient mice treated with anti-IL-10R was decreased compared with similarly treated WT mice, there was no difference in the level of inflammation in the cecum between genotypes.19 Although, to our knowledge, the role of IL-17 in regulating H. hepaticus-induced inflammation in mice with impaired IL-10 signaling has not been directly tested, there are conflicting results from experiments testing the role of IL-17 in the development of colitis that develops following the adoptive transfer of CD4+CD45RBhigh cells into lymphocyte-deficient hosts.20, 21 Further, as it has been suggested that other Th17 cell-derived cytokines such as Il-17F20 and IL-2222, 23 can modulate disease severity in T cell-dependent models of colitis, the role of these cytokines in H. hepaticus-dependent colitis requires additional study.
Although the exact role of T-helper subsets in the development of H. hepaticus-induced colitis in susceptible hosts remains to be determined, increasing evidence has focused attention on the innate inflammatory response to H. hepaticus. Early studies demonstrated that H. hepaticus could induce colitis in certain strains of lymphocyte-deficient mice including scid and 129SvEv/Rag−/−,10, 24, 25, 26, 27 and progressed to colonic carcinoma with neoplastic peritoneal invasion in some murine models.24, 28 H. hepaticus-induced innate colitis appeared to be dependent upon IL-23, as antibody depletion of both IL-12 p40 and IL-12 p19 (the components of IL-23) inhibited H. hepaticus-induced colitis in 129SvEv/Rag−/− mice.29 Antibody depletion assays have also demonstrated an important role of tumor necrosis factor (TNF) in H. hepaticus-induced innate colitis.25
Surprisingly, even in the innate colitis model, H. hepaticus infection induced robust levels of the T-helper-associated cytokines IL-17 and IFN-γ within the colon of 129SvEv/Rag−/− mice,29 and treatment of H. hepaticus-infected 129SvEv/Rag−/− mice with either IL-17 or IFN-γ-depleting antibody ameliorated colitis.30 This suggested that cells of the innate immune system express IL-17 and IFN within the colon following H. hepaticus infection. Indeed, in 129SvEv/Rag−/− mice infected with H. hepaticus, there was a marked increase in the percentage of colonic lamina propria (LP) cells that stain positively for IL-17 alone or both IL-17 and IFN-γ following overnight stimulation with IL-23.30 These IL-17+ cells lacked lineage markers (CD11b, GR1, B220), but expressed high levels of Thy1,30 and thus bear similarities to a previous described rare population of LP cells termed lymphoid tissue inducer-like (LTi-like) cells.31, 32 Some LTi-like cells express CD4 on their cell surface,31 whereas these newly described IL-17+ innate lymphoid cells do not.30 However they share a number of other cell surface markers including IL-7Rα.30, 31 Interestingly, there was a marked increase in the frequency of Thy1high innate lymphoid cells within the colonic lamina propia of 129SvEv/Rag−/− following H. hepaticus infection, and when isolated by sorting, these cells produced IFN-γ, IL-17, and IL-22 after overnight culture, which was enhanced by treatment with IL-23.30
It has previously been shown that the development of LTi and LTi-like cells, as well as classic Th17 cells require the transcription factor, retinoic acid related receptorγt (RORγt).31, 32, 33 Interestingly, H. hepaticus-induced innate-Thy1high+ LP cells expressed higher levels of RORγt than remaining Thy1− LP cells,30 and both C57BL/6Rag−/− mice treated with anti-Thy1 antibody, or C57BL/6Rag−/− mice that lacked the gene for RORγt, were resistant to anti-CD40-induced innate colitis.30 These data strongly suggest that Thy1+ RORγt-expressing innate lymphoid cells have a critical role in driving intestinal inflammation. Further, if these innate lymphoid cells require the IL-7 receptor for their survival and/or expansion in similar manner to LTi cells,34 this could explain the earlier observation that H. hepaticus is unable to induce colitis in 129SvEv/Rag−/− mice that also lack IL-7.27
These studies argue that innate immune mechanisms have a critical role in the ability of H. hepaticus to induce colitis in susceptible hosts. However, it is important to recognize that H. hepaticus infection does not induce colitis in most WT mouse strains despite persistent infection, suggesting that a lymphocyte-mediated mechanism inhibits H. hepaticus-induced innate inflammation. Consistent with this, several groups have shown that the adoptive transfer of CD4+CD25+ T-regulatory cells into 129SvEv/Rag−/− mice can inhibit the development of colitis following H. hepaticus infection.25, 28 Inhibition depends upon the ability of transferred cells to express IL-10, as IL-10-deficient CD4+CD25+ T cells are unable to inhibit H. hepaticus-induced innate inflammation, and in fact appear to exacerbate disease.24, 25 Remarkably, transfer of CD4+CD25+ T-regulatory cells into H. hepaticus-infected 129SvEv/Rag-/- mice with colitis leads to rapid resolution of disease.24, 35 Several studies revealed that potency of CD4+CD25+ cells to protect against or treat IBD can be enhanced by H. hepaticus infection of donor mice.36, 37 Thus, CD4+CD25+ T-regulatory cells can both inhibit and treat H. hepaticus-induced innate inflammation in the lower bowel, based on an IL-10-dependent mechanism. Interestingly, CD25+ cells are not the only CD4+ subtype with regulatory activity, as it has been demonstrated that CD4+CD45RBlowCD25− cells harvested from H. hepaticus-infected C57BL/10 mice are potent inhibitors of colitis induced by adoptive transfer of IL-10-deficient CD4+ T cells into H. hepaticus-infected C57BL/10Rag−/− mice.36
Experiments described above, indicating that the recognition of H. hepaticus by the innate immune system has a key role in regulating the ensuing inflammatory response in the lower bowel, suggest that understanding the innate inflammatory signaling pathways and receptors utilized by H. hepaticus could lead to important insights into the basis of microbiota-driven colonic inflammation. H. hepaticus challenge of ex vivo-cultured bone marrow-derived macrophages induces activation of both the nuclear factor (NF)-κB and ERK pathways.26, 38 Evidence for both pro- and anti-inflammatory roles of individual NF-κB subunits in the response to H. hepaticus come from experiments demonstrating that 129SvEv/Rag−/− mice that also lack the NF-κB subunit c-Rel are resistant to H. hepaticus-induced colonic inflammation,39 whereas 129SvEv mice that lack the p50/p105 subunit of NF-κB are sensitized to H. hepaticus-induced colitis, based on a defect within the innate immune system.26, 40
Identifying the innate immune receptors responsible for inducing inflammatory signaling in response to Helicobacter spp. infection is an active area of research. Whole H. hepaticus bacteria are able to induce NF-κB activation in HEK293 cells transfected with toll-like receptor 2 (TLR2), but not those transfected with TLR4.41 However, a recent study demonstrated that the absence of TLR2 in 129SvEv/Rag−/− mice does not influence the severity of the H. hepaticus-induced colitis.42 Nonetheless, there is strong evidence suggesting the involvement of TLRs in mediating the in vivo inflammatory response to H. hepaticus as irradiated 129SvEv/Rag−/− reconstituted with Myd88-deficient bone marrow cells are resistant to H. hepaticus-induced innate colitis.43
Intracellular receptors of the Nod family plan an important role in recognizing bacterial products such as peptidoglycan that are injected by bacterial secretion systems.44 It has recently been reported that the genome of H. hepaticus codes for a type VI secretion system that may function to inject substrates into host epithelial cells.45 Indeed, it has been suggested that the ability of H. muridarum, a murine EHS, to induce NF-κB reported activity in HEK293 cells depends upon Nod1.46 Although, of note, H. hepaticus is a very poor inducer of NF-κB activation in HEK293 cells,46 and a weak inducer of inflammatory gene expression in the murine intestinal crypt epithelial cells (m-ICcl2 cells),47 suggesting that H. hepaticus is unlikely to engage Nod1 in these epithelial cell lines. In contrast, it has been found that H. hepaticus, as well as LPS derived from H. hepaticus, can inhibit TLR4-induced inflammatory gene expression in m-ICcl2 cells,47 raising the possibility that H. hepaticus may have prominent anti-inflammatory properties on epithelial cells. An anti-inflammatory role of an H. hepaticus-derived product is supported by studies showing that a H. hepaticus mutant lacking the type VI secretion system colonized the intestine of C57BL/6Rag–/– recipients of CD4+CD45RBhi T cells to higher levels than the WT H. hepaticus strain, and induced elevated levels of inflammatory gene expression.45 Finally, a role of Nod2 in recognition of H. hepaticus has been suggested by studies demonstrating higher levels of H. hepaticus in infected Nod2-deficient C57BL/6 mice than in infected WT mice, although intestinal pathology was not detected following infection of either WT or Nod2-deficient mice.48
H. hepaticus Infection Modulates Tumorigenesis at Intestinal and Extraintestinal Sites
At the time of initial characterization in A/JCr mice, it was recognized that in addition to chronic active hepatitis, infection with H. hepaticus was also associated with the development of hepatocellular carcinoma in A/JCr as well as liver tumor development in AXB, B6C3F1, and a substrain of C57BL mice.1, 3, 49, 50, 51, 52 Previous studies using germ-free mice link chronic bacterial infection with chronic inflammatory diseases in the lower bowel.53 Targeted bacterial infection with H. hepaticus in 129SvEv/Rag2−/− mice lead to colitis-associated colorectal cancer (CRC) with malignancy invading the peritoneal cavity.24, 28 Gut bacteria-induced lesions in 129SvEv/Rag2 -deficient mice resembled neoplasia in human patients54 with IBD, in that carcinoma clearly arises from colitis-associated dysplastic epithelial foci that become locally invasive.55 The finding that 129SvEv/Rag2−/− mice lacking lymphocytes exhibited CRC demonstrated that innate immunity is sufficient for development of carcinoma.28 However, immunologically intact 129 WT mice infected with H. hepaticus did not develop carcinoma, showing that IL-10-dependent activities of adaptive immune cells normally serve to reinforce homeostasis and prevent cancer in the lower bowel.28, 37 Adoptive transfer studies using CD4+ cell subsets demonstrated that an imbalance between IL-6, IL-10, and tumor growth factor-β increased risk of neoplastic epithelial invasion.37 Interestingly, H. hepaticus-induced malignancy in the 129SvEv/Rag2−/− mice was readily reversible by blocking underlying bacteria-driven inflammation with antibodies directed at TNFα and IL-6.37, 56
Pathogenic gut microbial infections have also been shown to trigger development of intestinal adenomatous polyps in mice with a mutated Apc gene (ApcMin/+ or Min).57 Mice heterozygous for Apc gene (ApcMin/+) develop a large number of intestinal polyps by 3 months of age.58 This preneoplastic process has been shown to be inhibited by adoptive transfer of IL-10-dependent TReg cells, and depends on their ability to produce IL-10.59, 60 Targeted infection with H. hepaticus increases multiplicity and malignancy of intestinal polyps in the ApcMin/+ mouse model by an inflammation-associated TNF-α-dependent mechanism.57 Likewise, infection with other pathogenic enteric microbiota including C. rodentium61 and an enterotoxigenic Bacteroides fragilis62 have been shown to promote colon tumorigenesis in ApcMin/+ mice. Recent evidence points to key roles of IL-17 as well as IL-23 in colonic hyperplasia and tumor formation.62, 63 Taken together, these models may provide novel mechanistic insights in colon carcinogenesis in humans.
More recently, experiments examining the ability of gastrointestinal (GI) tract bacteria and subpopulations of immune cells to induce or inhibit IBD have revealed surprising insights into extraintestinal carcinogenesis. For example, H. hepaticus infection was found to trigger mammary tumorigenesis in ApcMin/+ in the absence of IBD.57, 64 Mammary tumors also developed rapidly in susceptible mice after transfer of CD4+ CD45RBhigh T cells.57, 63 Although WT Treg completely inhibited H. hepaticus-induced diseases in these models, IL-10-deficient Treg cells exacerbated rather than inhibited H. hepaticus-induced extraintestinal pathology.24, 37 Interestingly, previous exposures of Treg donor mice to H. hepaticus enhanced their ability to inhibit extraintestinal pathology,57, 63, 64 raising the possibility that therapies aimed toward intestinal homeostasis may offer sustained protection from systemic diseases.
Virulence Factors of Enterohepatic Helicobacters
Human IBD as well as IBD in mouse models is multifactorial, with genetic and environmental factors contributing to pathogenesis. The complexity of the system necessitates parallel investigations into bacterial virulence factors, gene expression changes in intestinal tissue, host innate and adaptive immune responses, changes in the gut microbial flora, and of course, lesion development in the intestine. Using the H. hepaticus genome, we have begun studying in detail, with the use of isogenic mutants, how different virulence genes have a role in chronic intestinal inflammation. The pathogenic potential of several virulence factors from H. hepaticus have been well characterized.
Of the known EHS, H. hepaticus strain 3B1 (ATCC 51449), the prototype of EHS, has been the most extensively investigated. This murine pathogen contains a circular genome of ∼1.8 Mb with 1875 predicted open reading frames.65 The G+C content of the H. hepaticus genome is 35.9%, which places it between the G+C content of H. pylori (39%) and Campylobacter jejuni (30.5%). In addition, ∼50% of predicted H. hepaticus genes are orthologous to those in the genome of either H. pylori or C. jejuni, both of which cause significant GI diseases in humans.65
Cytolethal distending toxin
Bacterial cytolethal distending toxins (CDTs) are present in multiple Gram-negative pathogens including strains of Salmonella spp, Shigella spp., C. jejuni, and Escherichia coli, and belong to the AB2-type of toxins. In general these toxins are comprised of subunits CdtA and CdtC, which together bind to subunit CdtB and deliver CdtB, the active subunit of the toxin, intracellularly.66 Several biological functions of the CdtB subunit have been reported, including a DNase I-like function, cell-cycle arrest, phosphatase activity, and eventual cell death.67 The cdtB ortholog and CDT activity are found in several enterohepatic helicobacters, including H. hepaticus, H. bilis, H. canis, and H. marmotae, as well as in H. cineadi and H. pullorum isolated from humans.68, 69, 70
H. hepaticus 3B1 and an isogenic mutant of the CDT gene (cdtB-deficient HhcdtBm7) were evaluated in outbred Swiss Webster mice. Although inoculation with WT 3B1 leads to persistent colonization, colonization by the mutant failed in female mice and was transient in males.71 WT 3B1 infection caused significantly increased expression of ileal and cecal IFN-γ mRNA, higher Th1-induced IgG2a responses, and increased mucosal IgA secretion compared with the isogenic mutant.13, 71 CDT-deficient isogenic H. hepaticus mutants also lose their ability to persistently colonize the large intestine of B6.129-IL10tm1Cgn (IL10−/−) mice. Importantly, the mutant-infected mice developed significantly less severe typhlocolitis when compared with WT 3B1-infected mice.71, 72, 73 Mutation of the CDT gene of H. cinaedi, a human pathogen, also reduced its ability to induce lower bowel pathology in IL-10-deficient mice.74
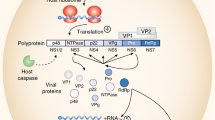
Molecular mechanisms operable in H. hepaticus CDT-induced cell death are also being actively investigated. CDT activated the ATM-dependent DNA damage check point response in a Myc-dependent manner.75 In addition, exposure of INT407 cells to H. hepaticus CDT resulted in upregulation or pro-apoptotic Bax, release of Cyt c, and activation of caspases 3 and 9.76 Events operable in H. hepaticus CDT-induced cell arrest and apoptosis are summarized in Figure 1.

ATM-dependent checkpoint responses and apoptosis triggered by H. hepaticus cytolethal distending toxin (CDT)-induced DNA damage.75, 76 Apaf-1, apoptotic peptidase-activating factor 1; ATM, ataxia telangiectasia mutated; Bax, Bcl-2-associated X protein; Bcl-2, B-cell leukemia/lymphoma 2; CHK2, checkpoint kinase 2; Cyt c, cytochrome c; DSB, double-strand breakage; Nbs 1, Nijmegen breakage syndrome 1 protein.
H. hepaticus pathogenicity island (PAI)
PAIs are distinct genetic elements on bacterial chromosomes that encode one or more virulence-associated factors and often have a G+C content distinct from the rest of the core genome.65 Several PAIs, such as LEE in enteropathogenic E. coli, cag in H. pylori, and SaPl1 in Staphylococcus aureus, significantly contribute to pathogenicity of these infectious agents.77 In the genome of H. hepaticus 3B1, a 71-kb region, HHGI1, displays genetic features characteristic of PAIs.65 HHGI1 is highly variable among certain H. hepaticus isolates. Male A/JCr mice infected with H. hepaticus strain MIT 96-1809 lacking the entire HHGI1 or strain MIT 96-284 missing ∼62 out of 71 kb of HHGI1 developed less severe hepatitis than those infected with H. hepaticus 3B1, which has an intact HHGI1.78 An isogenic mutant (HhPAId1) of H. hepaticus 3B1 containing a deletion of 19 predicted genes (HH0250 to HH0268) within HHGI1 colonized the intestine of B6.129-IL10tm1Cgn (IL10−/−) mice at higher levels but provoked less severe typhlocolitis and hyperplasia when compared with H. hepaticus 3B1.79 The magnitude of the Th1-associated IgG2c response against this isogenic mutant (HhPAId1) was less than against WT H. hepaticus and was accompanied by suppressed cecal and colonic mRNA expression levels for proinflammatory IFN-γ, TNF-α, and IL17a. These results demonstrate that HHGI1 contributes to the pathogenicity of H. hepaticus at least in part via upregulation of proinflammatory mediators.
Recently, a study indicated that HHGI1 encodes for a set of 12 genes (HH0237, HH0242 to HH0245, HH0247 to HH0252, HH0291), which are homologous to known bacterial type VI secretion systems (T6SS).45 T cell-reconstituted Rag1−/− mice infected with a T6SS-deficient ΔIcmF H. hepaticus mutant (encoded by HH0252) developed higher colonization levels of the mutant compared with WT H. hepaticus-infected mice, consistent with higher levels of mutant HhPAId1 cecal colonization.79 In addition, infection with ΔIcmF H. hepaticus induced higher expression in the colon of proinflammatory mediators, including IL-17, IL-23, IL-1β, TNFα, and IFNγ than infection with WT H. hepaticus 3B1.45 However, unlike the HhPAId1 mutant, the T6SS mutant ΔIcmF, and H. hepaticus 3B1 induced a similar degree of colitis by 2–4 weeks postinoculation.45, 79 Therefore, further investigations will be required to define the role(s) of T6SS and additional components in HHGI1 in the pathogenesis of H. hepaticus.
Urease
Gastric helicobacters produce urease, which is essential for establishing persistent colonization in the acidic milieu of the stomach.80 Approximately 40% of known EHS species, including H. hepaticus, are urease positive.81 The H. hepaticus ure gene cluster is similar to that of H. pylori; however, regulatory mechanisms of urease activity in H. hepaticus are distinct from those in H. pylori. For example, H. hepaticus urease does not confer acid resistance, transcription of ure genes is not induced by acid, and urease activity is suppressed by the transcriptional regulator Fur.82, 83
By infecting male A/JCr mice with a urease-deficient, isogenic mutant (HhureNT9) of H. hepaticus 3B1, we demonstrated that the lack of urease activity did not affect the level of cecal colonization but its presence was essential for hepatic colonization.81 Consequently, the HhureNT9-infected mice developed less-severe hepatitis and produced significantly lower hepatic mRNA levels of proinflammatory cytokines IFN-γ and TNF-α when compared with the H. hepaticus 3B1-infected mice.81
Enzymes in oxidative stress resistance
To survive and establish colonization in the host, bacteria have evolved various mechanisms to combat oxidative stress that confers bactericidal effects via reactive oxygen species. The H. hepaticus catalase enzyme KatA, which converts hydrogen peroxide into molecular oxygen and water, has a key role in this process. The amino-acid sequence of H. hepaticus KatA contains three motifs (R-F-Y-D, RERIPER, and VVHAKG) in the heme ligand domain and three surface-predicted motifs that are shared among bacterial and mammalian catalases.84 KatA-deficient H. hepaticus are more sensitive to exposure of oxygen and H2O2 and also suffer severe DNA fragmentation by H2O2 treatment compared with H. hepaticus 3B1.85
Bacterial hydrogenase, a hydrogen uptake hydrogenase, oxidizes molecular hydrogen to yield protons and electrons, which have a key role in energy conservation in bacteria. H. hepaticus genes or HH0056 to HH0059 encode subunits of hydrogenase HyaA to HyaD. Mutation in the H. hepaticus hyaB (coding for the large subunit) does not affect cecal and hepatic colonization levels when compared with H. hepaticus 3B1 in male A/JCr mice; however, liver pathology is diminished in the hyaB mutant-infected mice.86 It is unknown how hydrogenase contributes to the pathogenesis of H. hepaticus, but it is speculated that energy released by hydrogen oxidation may be utilized to promote synthesis of virulence-related proteins or enzymes that result in hepatic or intestinal inflammation and necrosis.86
Flagellar genes
All Helicobacter species possess flagella for motility; H. hepaticus is a spiral rod with bipolar sheathed flagella. In other related bacteria, flagellar genes are mainly regulated by the sigma factor FliA (σ28). H. hepaticus contains two identical copies of the gene encoding the major flagellin subunit FlaA (open reading frames HH1364 (flaA1) and HH1653 (flaA2)),65 which are regulated by the sigma factor FliA (σ28). Mutations in fliA or both copies of the flaA gene resulted in no detectable synthesis of FlaA as well as severely truncated flagella, and thus both mutants were nonmotile and unable to colonize mice.87 The flaA_1H. hepaticus mutant, which has flagella but decreased motility, also does not colonize mice, indicating that full motility is a prerequisite for intestinal colonization by H. hepaticus, and that the presence of flagella alone is not sufficient.
Summary
As described in this review, H. hepaticus has had a central role in shaping our understanding of how intestinal microbiota interact with the host to produce both inflammatory and tolerant responses. The study of immunopathology that develops following H. hepaticus infection of susceptible mice has helped to define the importance of key inflammatory mediators, including IL-23 and IL-17, and highlighted the importance of the innate immune response in the development of chronic intestinal inflammation. The use of H. hepaticus infection models has provided definitive evidence that regulatory T cells directly inhibit the innate inflammatory response to lower bowel microbiota and delineated the critical role of IL-10 in this process. Remarkably, H. hepaticus infection has powerful effects on carcinogenesis both within the intestine and at extraintestinal sites, and many of the same immunoregulatory pathways that influence the GI inflammatory response to H. hepaticus also appear to influence the development of cancer. The complete genomic sequence of H. hepaticus has advanced the identification of potential virulence factors, and evaluating the function of these factors is ongoing. In addition, genome sequencing of five additional EHS isolated from humans with GI disease (H. bilis, H. canadensis, H. cinaedi, H. pullorum, and H. winghamensis) has been completed and these results can be found at http://www.broadinstitute.org/annotation/genome/Helicobacter_group/. Comparative genome analysis of these five genomes plus H. hepaticus will provide insight into the etiopathogenesis of EHS infection in humans. For example, preliminary data indicate that both H. cinaedi and H. pullorum have PAIs with similar features to the HHGI1 found in H. hepaticus. Studies are needed to ascertain whether these PAIs impart similar in vivo characteristics in humans. Additional areas for future investigation include defining the cellular and molecular pathways that contribute to host recognition of H. hepaticus, and determining how initial recognition events lead to activation of innate and acquired inflammatory effector mechanisms. Further, clarifying how responses to H. hepaticus infection influences carcinogenesis at local and distant sites could lead to important new insights into the relationship between specific microbiota and the development of cancer. Finally, delineating the targets of H. hepaticus-associated virulence factors could lead to novel approaches to treating microbial induced inflammation in the lower bowel.
References
Fox, J.G. et al. Helicobacter hepaticus sp. nov., a microaerophilic bacterium isolated from livers and intestinal mucosal scrapings from mice. J. Clin. Microbiol. 32, 1238–1245 (1994).
Ward, J.M. et al. Chronic active hepatitis and associated liver tumors in mice caused by a persistent bacterial infection with a novel Helicobacter species. J. Natl Cancer. Inst. 86, 1222–1227 (1994).
Fox, J.G. et al. Chronic proliferative hepatitis in A/JCr mice associated with persistent Helicobacter hepaticus infection: a model of Helicobacter-induced carcinogenesis. Infect. Immun. 64, 1548–1558 (1996).
Boutin, S.R. et al. Helicobacter pullorum outbreak in C57BL/6NTac and C3H/HeNTac barrier-maintained mice. J. Clin. Microbiol. 48, 1908–1910 (2010).
Shames, B. et al. Identification of widespread Helicobacter hepaticus infection in feces in commercial mouse colonies by culture and PCR assay. J. Clin. Microbiol. 33, 2968–2972 (1995).
Taylor, N.S., Xu, S., Nambiar, P., Dewhirst, F.E. & Fox, J.G. Enterohepatic Helicobacter species are prevalent in mice from commercial and academic institutions in Asia, Europe, and North America. J. Clin. Microbiol. 45, 2166–2172 (2007).
Berg, D.J. et al. Enterocolitis and colon cancer in interleukin-10-deficient mice are associated with aberrant cytokine production and CD4(+) TH1-like responses. J. Clin. Invest. 98, 1010–1020 (1996).
Ward, J.M. et al. Inflammatory large bowel disease in immunodeficient mice naturally infected with Helicobacter hepaticus. Lab. Anim. Sci. 46, 15–20 (1996).
Foltz, C.J. et al. Spontaneous inflammatory bowel disease in multiple mutant mouse lines: association with colonization by Helicobacter hepaticus. Helicobacter 3, 69–78 (1998).
Cahill, R.J. et al. Inflammatory bowel disease: an immunity-mediated condition triggered by bacterial infection with Helicobacter hepaticus. Infect Immun 65, 3126–3131 (1997).
Powrie, F., Leach, M.W., Mauze, S., Caddle, L.B. & Coffman, R.L. Phenotypically distinct subsets of CD4+ T cells induce or protect from chronic intestinal inflammation in C. B-17 scid mice. Int. Immunol. 5, 1461–1471 (1993).
Powrie, F. et al. Inhibition of Th1 responses prevents inflammatory bowel disease in scid mice reconstituted with CD45RBhi CD4+ T cells. Immunity 1, 553–562 (1994).
Kullberg, M.C. et al. Helicobacter hepaticus triggers colitis in specific-pathogen-free interleukin-10 (IL-10)-deficient mice through an IL-12- and gamma interferon-dependent mechanism. Infect. Immun. 66, 5157–5166 (1998).
Burich, A. et al. Helicobacter-induced inflammatory bowel disease in IL-10- and T cell-deficient mice. Am. J. Physiol. Gastrointest. Liver Physiol. 281, G764–778 (2001).
Erdman, S.E., Poutahidis, T., Rogers, A.B., Tomszaki, M., Horwitz, B. & Fox, J.G. Susceptibility of three strains of Rag2-deficient mice to Helicobacter hepaticus induced colitis and colorectal cancer. Gastroenterology 124 (Suppl 1), A340 (2003).
Kuhn, R., Lohler, J., Rennick, D., Rajewsky, K. & Muller, W. Interleukin-10-deficient mice develop chronic enterocolitis. Cell 75, 263–274 (1993).
Dieleman, L.A. et al. Helicobacter hepaticus does not induce or potentiate colitis in interleukin-10-deficient mice. Infect. Immun. 68, 5107–5113 (2000).
Kullberg, M.C. et al. Helicobacter hepaticus-induced colitis in interleukin-10-deficient mice: cytokine requirements for the induction and maintenance of intestinal inflammation. Infect. Immun. 69, 4232–4241 (2001).
Kullberg, M.C. et al. IL-23 plays a key role in Helicobacter hepaticus-induced T cell-dependent colitis. J. Exp. Med. 203, 2485–2494 (2006).
Leppkes, M. et al. ROR gamma-expressing Th17 cells induce murine chronic intestinal inflammation via redundant effects of IL-17A and IL-17F. Gastroenterology 136, 257–267 (2009).
O'Connor, W. Jr et al. A protective function for interleukin 17A in T cell-mediated intestinal inflammation. Nat. Immunol. 1, 603–609 (2009).
Zenewicz, L.A. et al. Innate and adaptive interleukin-22 protects mice from inflammatory bowel disease. Immunity 29, 947–957 (2008).
Sugimoto, K. et al. IL-22 ameliorates intestinal inflammation in a mouse model of ulcerative colitis. J. Clin. Invest. 118, 534–544 (2008).
Erdman, S.E. et al. CD4(+)CD25(+) regulatory lymphocytes require interleukin 10 to interrupt colon carcinogenesis in mice. Cancer Res. 63, 6042–6050 (2003).
Maloy, K.J. et al. CD4+CD25+ T(R) cells suppress innate immune pathology through cytokine-dependent mechanisms. J. Exp. Med. 197, 111–119 (2003).
Tomczak, M.F. et al. NF-kappa B is required within the innate immune system to inhibit microflora-induced colitis and expression of IL-12 p40. J. Immunol. 171, 1484–1492 (2003).
von Freeden-Jeffry, U. et al. IL-7 deficiency prevents development of a non-T cell non-B cell-mediated colitis. J. Immunol. 161, 5673–5680 (1998).
Erdman, S.E. et al. CD4+ CD25+ regulatory T lymphocytes inhibit microbially induced colon cancer in Rag2-deficient mice. Am. J. Pathol. 162, 691–702 (2003).
Hue, S. et al. Interleukin-23 drives innate and T cell-mediated intestinal inflammation. J. Exp. Med. 203, 2473–2483 (2006).
Buonocore, S. et al. Innate lymphoid cells drive interleukin-23-dependent innate intestinal pathology. Nature 464, 1371–1375 (2010).
Eberl, G. & Littman, D.R. Thymic origin of intestinal alphabeta T cells revealed by fate mapping of RORgammat+ cells. Science 305, 248–251 (2004).
Eberl, G. et al. An essential function for the nuclear receptor RORgamma(t) in the generation of fetal lymphoid tissue inducer cells. Nat. Immunol. 5, 64–73 (2004).
Ivanov, II et al. The orphan nuclear receptor RORgammat directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell 126, 1121–1133 (2006).
Adachi, S. et al. Essential role of IL-7 receptor alpha in the formation of Peyer's patch anlage. Int. Immunol. 10, 1–6 (1998).
Maloy, K.J., Antonelli, L.R., Lefevre, M. & Powrie, F. Cure of innate intestinal immune pathology by CD4+CD25+ regulatory T cells. Immunol. Lett. 97, 189–192 (2005).
Kullberg, M.C. et al. Bacteria-triggered CD4(+) T regulatory cells suppress Helicobacter hepaticus-induced colitis. J. Exp. Med. 196, 505–515 (2002).
Poutahidis, T. et al. Rapid reversal of interleukin-6-dependent epithelial invasion in a mouse model of microbially induced colon carcinoma. Carcinogenesis 28, 2614–2623 (2007).
Tomczak, M.F. et al. Defective activation of ERK in macrophages lacking the p50/p105 subunit of NF-kappaB is responsible for elevated expression of IL-12 p40 observed after challenge with Helicobacter hepaticus. J. Immunol. 176, 1244–1251 (2006).
Wang, Y. et al. c-Rel is essential for the development of innate and T cell-induced colitis. J. Immunol. 180, 8118–8125 (2008).
Tomczak, M.F. et al. Inhibition of Helicobacter hepaticus-induced colitis by IL-10 requires the p50/p105 subunit of NF-kappa B. J. Immunol. 177, 7332–7339 (2006).
Mandell, L. et al. Intact gram-negative Helicobacter pylori, Helicobacter felis, and Helicobacter hepaticus bacteria activate innate immunity via toll-like receptor 2 but not toll-like receptor 4. Infect. Immun. 72, 6446–6454 (2004).
Boulard, O., Asquith, M.J., Powrie, F. & Maloy, K.J. TLR2-independent induction and regulation of chronic intestinal inflammation. Eur. J. Immunol. 40, 516–524 (2009).
Asquith, M.J., Boulard, O., Powrie, F. & Maloy, K.J. Pathogenic and protective roles of MyD88 in leukocytes and epithelial cells in mouse models of inflammatory bowel disease. Gastroenterology 139, 519–529 (2010).
Fritz, J.H., Ferrero, R.L., Philpott, D.J. & Girardin, S.E. Nod-like proteins in immunity, inflammation and disease. Nat. Immunol. 7, 1250–1257 (2006).
Chow, J. & Mazmanian, S.K. A pathobiont of the microbiota balances host colonization and intestinal inflammation. Cell Host Microbe 7, 265–276 (2010).
Chaouche-Drider, N. et al. A commensal Helicobacter sp. of the rodent intestinal flora activates TLR2 and NOD1 responses in epithelial cells. PLoS One 4, e5396 (2009).
Sterzenbach, T. et al. Inhibitory effect of enterohepatic Helicobacter hepaticus on innate immune responses of mouse intestinal epithelial cells. Infect. Immun. 75, 2717–2728 (2007).
Petnicki-Ocwieja, T. et al. Nod2 is required for the regulation of commensal microbiota in the intestine. Proc. Natl Acad. Sci. USA 106, 15813–15818 (2009).
Garcia, A. et al. Genetic susceptibility to chronic hepatitis is inherited codominantly in Helicobacter hepaticus-infected AB6F1 and B6AF1 hybrid male mice, and progression to hepatocellular carcinoma is linked to hepatic expression of lipogenic genes and immune function-associated networks. Infect. Immun. 76, 1866–1876 (2008).
Ihrig, M., Schrenzel, M.D. & Fox, J.G. Differential susceptibility to hepatic inflammation and proliferation in AXB recombinant inbred mice chronically infected with Helicobacter hepaticus. Am. J. Pathol. 155, 571–582 (1999).
Ward, J.M., Anver, M.R., Haines, D.C. & Benveniste, R.E. Chronic active hepatitis in mice caused by Helicobacter hepaticus. Am. J. Pathol. 145, 959–968 (1994).
Fox, J.G. et al. Gut microbes define liver cancer risk in mice exposed to chemical and viral transgenic hepatocarcinogens. Gut 59, 88–97 (2010).
Engle, S.J. et al. Elimination of colon cancer in germ-free transforming growth factor beta 1-deficient mice. Cancer Res. 62, 6362–6366 (2002).
Boivin, G.P. et al. Pathology of mouse models of intestinal cancer: consensus report and recommendations. Gastroenterology 124, 762–777 (2003).
Riddell, R.H. et al. Dysplasia in inflammatory bowel disease: standardized classification with provisional clinical applications. Hum. Pathol. 14, 931–968 (1983).
Erdman, S.E. et al. Nitric oxide and TNF-alpha trigger colonic inflammation and carcinogenesis in Helicobacter hepaticus-infected, Rag2-deficient mice. Proc. Natl Acad. Sci. USA 106, 1027–1032 (2009).
Rao, V.P. et al. Innate immune inflammatory response against enteric bacteria Helicobacter hepaticus induces mammary adenocarcinoma in mice. Cancer Res. 66, 7395–7400 (2006).
Moser, A.R., Pitot, H.C. & Dove, W.F. A dominant mutation that predisposes to multiple intestinal neoplasia in the mouse. Science 247, 322–324 (1990).
Erdman, S.E. et al. CD4+CD25+ regulatory lymphocytes induce regression of intestinal tumors in ApcMin/+ mice. Cancer Res. 65, 3998–4004 (2005).
Rao, V.P. et al. Proinflammatory CD4+ CD45RB(hi) lymphocytes promote mammary and intestinal carcinogenesis in Apc(Min/+) mice. Cancer Res. 66, 57–61 (2006).
Newman, J.V., Kosaka, T., Sheppard, B.J., Fox, J.G. & Schauer, D.B. Bacterial infection promotes colon tumorigenesis in Apc(Min/+) mice. J. Infect. Dis. 184, 227–230 (2001).
Wu, S. et al. A human colonic commensal promotes colon tumorigenesis via activation of T helper type 17T cell responses. Nat. Med. 15, 1016–1022 (2009).
Erdman, S.E. et al. Unifying roles for regulatory T cells and inflammation in cancer. Int. J. Cancer 126, 1651–1665 (2010).
Rao, V.P., Poutahidis, T., Fox, J.G. & Erdman, S.E. Breast cancer: should gastrointestinal bacteria be on our radar screen? Cancer Res. 67, 847–850 (2007).
Suerbaum, S. et al. The complete genome sequence of the carcinogenic bacterium Helicobacter hepaticus. Proc. Natl Acad. Sci. USA 100, 7901–7906 (2003).
Smith, J.L. & Bayles, D.O. The contribution of cytolethal distending toxin to bacterial pathogenesis. Crit. Rev. Microbiol. 32, 227–248 (2006).
Ge, Z., Schauer, D.B. & Fox, J.G. In vivo virulence properties of bacterial cytolethal-distending toxin. Cell Microbiol. 10, 1599–1607 (2008).
Chien, C.C. et al. Identification of cdtB homologues and cytolethal distending toxin activity in enterohepatic Helicobacter spp. J. Med. Microbiol. 49, 525–534 (2000).
Taylor, N.S., Ge, Z., Shen, Z., Dewhirst, F.E. & Fox, J.G. Cytolethal distending toxin: a potential virulence factor for Helicobacter cinaedi. J. Infect. Dis. 188, 1892–1897 (2003).
Young, V.B. et al. Cytolethal distending toxin in avian and human isolates of Helicobacter pullorum. J. Infect. Dis. 182, 620–623 (2000).
Ge, Z. et al. Cytolethal distending toxin is essential for Helicobacter hepaticus colonization in outbred Swiss Webster mice. Infect. Immun. 73, 3559–3567 (2005).
Pratt, J.S., Sachen, K.L., Wood, H.D., Eaton, K.A. & Young, V.B. Modulation of host immune responses by the cytolethal distending toxin of Helicobacter hepaticus. Infect. Immun. 74, 4496–4504 (2006).
Young, V.B. et al. In vitro and in vivo characterization of Helicobacter hepaticus cytolethal distending toxin mutants. Infect. Immun. 72, 2521–2527 (2004).
Shen, Z. et al. Cytolethal distending toxin promotes Helicobacter cinaedi-associated typhlocolitis in interleukin-10-deficient mice. Infect. Immun. 77, 2508–2516 (2009).
Guerra, L. et al. Myc is required for activation of the ATM-dependent checkpoints in response to DNA damage. PLoS One 5, e8924 (2010).
Liyanage, N.P. et al. Helicobacter hepaticus cytolethal distending toxin causes cell death in intestinal epithelial cells via mitochondrial apoptotic pathway. Helicobacter 15, 98–107 (2010).
Gal-Mor, O. & Finlay, B.B. Pathogenicity islands: a molecular toolbox for bacterial virulence. Cell Microbiol. 8, 1707–1719 (2006).
Boutin, S.R. et al. Different Helicobacter hepaticus strains with variable genomic content induce various degrees of hepatitis. Infect. Immun. 73, 8449–8452 (2005).
Ge, Z. et al. Helicobacter hepaticus HHGI1 is a pathogenicity island associated with typhlocolitis in B6.129-IL10tm1Cgn mice. Microbes. Infect. 10, 726–733 (2008).
Burne, R.A. & Chen, Y.Y. Bacterial ureases in infectious diseases. Microbes Infect. 2, 533–542 (2000).
Ge, Z. et al. Helicobacter hepaticus urease is not required for intestinal colonization but promotes hepatic inflammation in male A/JCr mice. Microb. Pathog. 45, 18–24 (2008).
Belzer, C. et al. Differential regulation of urease activity in Helicobacter hepaticus and Helicobacter pylori. Microbiology 151, 3989–3995 (2005).
Belzer, C., van Schendel, B.A., Kuipers, E.J., Kusters, J.G. & van Vliet, A.H. Iron-responsive repression of urease expression in Helicobacter hepaticus is mediated by the transcriptional regulator Fur. Infect. Immun. 75, 745–752 (2007).
Alyamani, E.J. et al. Helicobacter hepaticus catalase shares surface-predicted epitopes with mammalian catalases. Microbiology 153, 1006–1016 (2007).
Hong, Y., Wang, G. & Maier, R.J. A Helicobacter hepaticus catalase mutant is hypersensitive to oxidative stress and suffers increased DNA damage. J. Med. Microbiol. 56, 557–562 (2007).
Mehta, N.S., Benoit, S., Mysore, J.V., Sousa, R.S. & Maier, R.J. Helicobacter hepaticus hydrogenase mutants are deficient in hydrogen-supported amino acid uptake and in causing liver lesions in A/J mice. Infect. Immun. 73, 5311–5318 (2005).
Sterzenbach, T. et al. Role of the Helicobacter hepaticus flagellar sigma factor FliA in gene regulation and murine colonization. J. Bacteriol. 190, 6398–6408 (2008).
Chin, E.Y., Dangler, C.A., Fox, J.G. & Schauer, D.B. Helicobacter hepaticus infection triggers inflammatory bowel disease in T cell receptor alpha\beta mutant mice. Comp. Med. 50, 586–594 (2000).
Shomer, N.H., Dangler, C.A., Schrenzel, M.D. & Fox, J.G. Helicobacter bilis-induced inflammatory bowel disease in scid mice with defined flora. Infect. Immun. 65, 4858–4864 (1997).
Shomer, N.H., Dangler, C.A., Marini, R.P. & Fox, J.G. Helicobacter bilis/Helicobacter rodentium co-infection associated with diarrhea in a colony of scid mice. Lab. Anim. Sci. 48, 455–459 (1998).
Fox, J.G. et al. Persistent hepatitis and enterocolitis in germfree mice infected with Helicobacter hepaticus. Infect. Immun. 64, 3673–3681 (1996).
Erdman, S., Fox, J.G., Dangler, C.A., Feldman, D. & Horwitz, B.H. Typhlocolitis in NF-kappa B-deficient mice. J. Immunol. 166, 1443–1447 (2001).
Maggio-Price, L. et al. Helicobacter bilis infection accelerates and H. hepaticus infection delays the development of colitis in multiple drug resistance-deficient (mdr1a‐/‐) mice. Am. J. Pathol. 160, 739–751 (2002).
Maggio-Price, L. et al. Helicobacter infection is required for inflammation and colon cancer in SMAD3-deficient mice. Cancer Res. 66, 828–838 (2006).
Fox, J.G. et al. A novel urease-negative Helicobacter species associated with colitis and typhlitis in IL-10-deficient mice. Infect. Immun. 67, 1757–1762 (1999).
Franklin, C.L. et al. Helicobacter typhlonius sp. nov., a novel murine urease-negative Helicobacter species. J. Clin. Microbiol. 39, 3920–3926 (2001).
Whary, M.T. et al. Rapid onset of ulcerative typhlocolitis in B6.129P2-IL10tm1Cgn (IL-10‐/‐) mice infected with Helicobacter trogontum is associated with decreased colonization by altered Schaedler′s flora. Infect. Immun. 74, 6615–6623 (2006).
Acknowledgements
This work was supported by grants R01-DK052413, P01-CA026731, R01-CA067529, P30-ES02109 (to J.G.F.), R01-A1052267 (to B.H.H., S.E.E.), R01-CA108854 (to S.E.E.).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declared no conflict of interest.
PowerPoint slides
Rights and permissions
About this article
Cite this article
Fox, J., Ge, Z., Whary, M. et al. Helicobacter hepaticus infection in mice: models for understanding lower bowel inflammation and cancer. Mucosal Immunol 4, 22–30 (2011). https://doi.org/10.1038/mi.2010.61
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/mi.2010.61
