
Abstract
Inhibition of anaerobic digestion through accumulation of volatile fatty acids occasionally occurs as the result of unbalanced growth between acidogenic bacteria and methanogens. A fast recovery is a prerequisite for establishing an economical production of biogas. However, very little is known about the microorganisms facilitating this recovery. In this study, we investigated the organisms involved by a novel approach of mapping protein-stable isotope probing (protein-SIP) onto a binned metagenome. Under simulation of acetate accumulation conditions, formations of 13C-labeled CO2 and CH4 were detected immediately following incubation with [U-13C]acetate, indicating high turnover rate of acetate. The identified 13C-labeled peptides were mapped onto a binned metagenome for improved identification of the organisms involved. The results revealed that Methanosarcina and Methanoculleus were actively involved in acetate turnover, as were five subspecies of Clostridia. The acetate-consuming organisms affiliating with Clostridia all contained the FTFHS gene for formyltetrahydrofolate synthetase, a key enzyme for reductive acetogenesis, indicating that these organisms are possible syntrophic acetate-oxidizing (SAO) bacteria that can facilitate acetate consumption via SAO, coupled with hydrogenotrophic methanogenesis (SAO-HM). This study represents the first study applying protein-SIP for analysis of complex biogas samples, a promising method for identifying key microorganisms utilizing specific pathways.
Similar content being viewed by others

Introduction
Anaerobic digestion (AD) of organic materials to biogas has several environmental benefits with the methane-rich biogas as a source of renewable energy, as a wastewater treatment technology for the removal of pathogens, as well as odor and pollution reductions from agricultural, industrial and municipal wastes.
The AD of organic matter to biogas involves the coordinated activity of diverse subgroups of highly specialized microbial organisms, hence the stability of AD is highly dependent on the microbial community structure and interactions within the community. Failure to maintain the balance between these groups can lead to reactor inhibitions and breakdown (Demirel and Yenigün, 2002). Accumulation of volatile fatty acids (VFAs) may cause acidification and result in reduced process performance or worst-case scenario: complete reactor failure (Angelidaki and Ahring, 1993; Chen et al., 2008; Krakat et al., 2011). Accumulation of acetate, an important intermediate VFAs in the anaerobic decomposition of organic matter, has been observed under various operational conditions, and the effects on the microbial community have been addressed in several studies (Palatsi et al., 2011; Fotidis et al., 2013; Lü et al., 2013; Rajagopal et al., 2013; Labatut et al., 2014). Although methanogenesis from acetate (aceticlastic methanogenesis) is fairly well described, less is known about the organisms and metabolisms involved in oxidation of acetate to hydrogen and carbon dioxide (syntrophic acetate oxidation (SAO)) catalyzed by SAO bacteria. The formyltetrahydrofolate synthetase-encoding gene, fthfs, is key in the reductive acetogenesis (acetyl-CoA pathway). The enzyme encoded by this gene is also able to catalyze the reverse reaction, oxidizing acetate into H2 and CO2 (Xu et al., 2009; Hori et al., 2011). The gene has therefore been widely used as a biomarker for acetogenesis, although also present in a variety of non-acetogenic bacteria (Lovell and Leaphart, 2005).
Only a few SAO bacterial species have been isolated, and only their genomic potential has been determined (Manzoor et al., 2013; Müller et al., 2013). Isolated synthrophic acetate-oxidizing bacteria (SAOB) cover several bacterial phyla and represent both thermophiles (Thermacetogenium phaeum (Hattori et al., 2000); and Thermotoga lettingae (Balk et al., 2002)), thermotolerant (Tepidanaerobacter acetatoxydans (Westerholm et al., 2011)); and mesophiles (Clostridium ultunense (Schnürer et al., 1996); and Synthrophaceticus schinkii (Westerholm et al., 2010)). The oxidation of VFAs by syntrophic bacteria is not a thermodynamically favorable process and requires that they are closely associated with methanogens or with non-methanogenic hydrogenotrophs.
Culture-independent approaches using stable isotopes under high levels of acetate revealed high utilization activity of Methanosarcina from the archaeal population and putative SAO bacteria affiliating with the Clostridia (Hao et al., 2014). Other studies have identified the Synergistes group 4 as a major group of acetate-utilizing bacteria in anaerobic sludge batch reactors fed with 2.5–10 mM acetate (Ito et al., 2011).
Protein-stable isotope probing (protein-SIP) can be used to determine not only the identity, but also the activity of active key microorganisms (Jehmlich et al., 2010). Although protein-SIP has been used on other less complex systems (Jehmlich et al., 2008; Bastida et al., 2010; Taubert et al., 2011, 2012; Herbst et al., 2013), it has, as far as we are aware, not been applied on complex environmental samples such as those present in an AD.
In this study, a combination of metagenomics, amplicon sequencing and protein-SIP was used for characterizing acetate-consuming communities in AD. The proteins from actively incorporating organisms were mapped onto a binned metagenome for identification of the bacteria involved. The results obtained in this study indicate SAO bacterial species are important factors in the recovery after acid accumulation, demonstrating the potential of the combined use of metagenomics and protein-SIP as a tool for linking identity and function, as well as providing an understanding of the underlying biology in AD.
Materials and methods
Sources of inoculum
Inoculum was obtained from a commercial full-scale biogas digester at Research Centre Foulum, Denmark. The digester works with a mixture of pig and cattle manure, maize silage and deep litter manure. It is operated under thermophilic conditions (approximately 52 °C). The total solid, volatile solid, pH value and total ammonia nitrogen of the inoculum were 50.2, 40.2, 7.64 and 1.54 g l–1, respectively. The inoculum was starved under anaerobic conditions at 52 °C for 2 weeks before the main tracer experiment to reduce the background contribution of carbon dioxide and methane from the original substrates. A digestate sample was collected at Lynggård biogas plant (11-01-2012), Denmark. Lynggård is operated at 52 °C and runs with pig manure, and grass and maize silage.
Operation of AD
Glass serum bottles (500 ml) were used for preparing the anaerobic incubation assay. Aliquots (195 ml) of inoculum were transferred into the 500 ml serum bottles, which were then sealed with butyl rubber stoppers and aluminum crimps. Five treatments were prepared with [U-13C] and [12C] acetate; detailed information of the reactor setup is summarized in Table 1. For the high-acetate concentration condition, substrate was added at the beginning of the experiment, whereas for the low-concentration condition, substrate was added on a daily basis. All experiments were carried out in triplicate, with the exception of the controls (blank reactors), which were carried out in duplicate. The experiment was run under static incubation conditions for 9 days at 52 °C using strict anaerobic techniques.
Basic analytical methods
The volume of a produced biogas was measured using an acidified water displacement method at room temperature and atmospheric pressure. Headspace biogas was collected using a gas-tight syringe with a needle through a septum and transferred into a 20 ml headspace vial. The compositions of CH4 and CO2 in the biogas samples were analyzed by a gas chromatograph 7890A (Agilent Technologies, Horsholm, Denmark) equipped with a thermal conductivity detector and a GC sampler 80 (Agilent Technologies).
Liquid samples for VFA analysis were collected periodically and the concentration of VFA determined on a 7890A gas chromatograph (Agilent Technologies), equipped with flame ionization detector. For details of the analysis, refer to Supplementary Methods.
Membrane introduction mass spectrometry measurement
Membrane introduction mass spectrometry was used to monitor the incorporation of 13C into the produced methane and carbon dioxide during the degradation of 13C fully labeled acetate as described elsewhere (Mulat et al., 2014). The membrane introduction mass spectrometry data are reported in terms of atom percent as follows:

where 13X represents 13CO2 or 13CH4, and 12X represents 12CO2 or 12CH4.
Protein-SIP and amplicon analysis
Liquid samples (5 ml) for protein-SIP and amplicon analysis were collected from all reactors periodically and stored at −20 °C until analysis. Sampling was performed at 8, 24, 32, 48, 96, 144 and 192 h following the beginning of the experiment. Protein extraction was performed on samples collected at 8, 24, 48 and 192 h using a protocol as previously described (Hansen et al., 2014). Cell debris was removed by centrifugation at 14 500 xg for 10 min at 4 °C.
Proteins were acetone precipitated as previously described (Botelho et al., 2010). Precipitated proteins were resuspended in sodium dodecyl sulfate–polyacrylamide gel electrophoresis sample buffer, supplemented with dithiothreitol to a final concentration of 40 mM and denatured by boiling at 95 °C for 10 min before being loaded onto a pre-cast 4–15% gradient sodium dodecyl sulfate-gel (Bio-Rad, Sundbyberg, Sweden) and separated for 10 min at 160 V.
Liquid chromatography-tandem mass spectrometry
In-gel digestion of proteins was performed as previously described (Shevchenko et al., 2006) and tryptic peptides were analyzed by an automated liquid chromatograph-electrospray ionization tandem mass spectrometer, consisting of an UltiMate 3000 RSLCnano system (Thermo Scientific, Bremen, Germany) coupled to a Q Exactive mass spectrometer (Thermo Scientific) via a Nanospray Flex ion source (Thermo Scientific). The analytical conditions were as previously described (Kjeldal et al., 2014), with the following modifications; analytes were eluted during a 120 min linear gradient, ranging from 12 to 40% (V/V) of solvent B (100% acetonitrile supplemented with 0.1% (V/V) formic acid) followed by a final step gradient to 90% solvent B which was maintained for 20 min.
Protein analysis
A six-frame translation (every region of DNA has six possible reading frames) and prediction of open reading frames in the in-house constructed metagenome of the anaerobic reactor of Foulum was carried out in MaxQuant v. 1.5.1.2 (Martinsried, Germany). A two-search strategy as previously described was utilized (Seifert et al., 2013). Briefly, an initial survey search was performed, searching against the non-redundant NCBI database, restricting the taxonomy to prokaryota. Sequences that were identified in the survey search were exported from NCBI and merged with the six-frame translation of the metagenome, and the merged database was used in the subsequent main search. For details, refer to Supplementary Methods.
Raw files were analyzed using OpenMS (https://www.openms.de) and the TOPP tools (Kohlbacher et al., 2007; Sturm et al., 2008). RIA and protein LR were, as described elsewhere (Kleindienst et al., 2014). determined using the opensource software OpenMS and the MetaproSIP tool (Sachsenberg et al., 2015).
DNA extraction
DNA extraction was conducted on the liquid biological triplicates collected at 8, 32 and 192 h of incubation periods, using the FastDNA Spin Kit for Soil (MP Biomedicals, Taastrup, Denmark) with minor modifications (for details, refer to Supplementary Methods).
Metagenome preparation
Two metagenomes were prepared from samples collected at the Foulum (13-12-2011) and Lynggård (11-01-2012) biogas plants. DNA was extracted, following a cetyltrimethylammonium bromide and enzyme-based method as described elsewhere (Klocke et al., 2007). DNA was paired-end sequenced (2 × 150 bp) on a Illumina HiSeq2000 (Illumina, Carlsbad, CA, USA), and metagenome reads following analyzed in CLC Genomics Workbench v. 7.03 (CLC Bio, Aarhus, Denmark). For details, refer to Supplementary Methods.
The analysis and binning were performed exactly as described elsewhere (Albertsen et al., 2013). Briefly, the binning of scaffolds to population genomes was performed by plotting the coverage estimates of the metagenomes of Foulum and Lynggard against each other for all scaffolds. Completeness and contamination of six genome bins was evaluated through calculation of GC content, tetranucleotide frequency and by the identification of conserved essential single-copy marker genes. A detailed description of the approach has been published elsewhere (Albertsen et al., 2013).
Core genes of the Wood–Ljungdahl pathway were searched against the metagenome using Hidden Markov model profiles of these genes downloaded from protein family domains (Pfams) homepage (http://pdam.xfam.org/; Finn et al., 2014) and HMMER3 package (http://hmmer.janelia.org/). For details, refer to Supplementary Methods.
Amplicon sequencing
Materials and methods of amplicon sequencing are provided in Supplementary Methods.
Data availability
All amplicon data are available at European Nucleotide Archive (ENA) under accession number PRJEB10871. Metagenomes and assemblies, and bin genomes are available at ENA under accession number PRJEB10932. The mass spectrometry proteomics data have been deposited at ProteomeXchange Consortium (Vizcaíno et al., 2014) via the PRIDE partner repository with the data set identifier PXD002996.
Results
Degradation of acetate and methane production
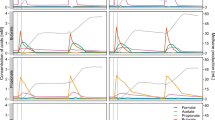
Batch reactors inoculated with anaerobic digester sludge were fed two different concentrations of labeled acetate, and the concentration of acetate as a function of time was monitored (Figure 1). The degradation of acetate in the blank reactor (inoculum only) and in the reactors fed with low (4 mM) and high (100 mM) concentrations of [U-13C]acetate were significantly different; the background concentration of acetate in the blank reactor remained at ~2 mM for the first 48 h and declined to below the detection limit between 48 and 72 h. No acetate accumulation was observed in reactors fed with low acetate concentration, and the total turnover of acetate was approximately 2.95 g l–1 during the 9 days of incubation.

Temporal change of the residual acetate in (a) the blank reactors (▴); reactors fed with low concentration (4 mM) of [U-13C]acetate (▪); and (b) reactors fed with high concentrations (100 mM) of [U-13C]acetate (•). The lines represent mean values (n=3), and error bars denote the standard deviation. Arrows indicate addition of acetate.
A continuous turnover of acetate occurred in the high acetate concentration-fed reactor, and the total turnover of acetate was around 8.2 g l–1 (acetate was spiked in at the beginning of the experiment only). The degradation of acetate in the high concentration-fed reactor started immediately and followed a linear trend during the first 120 h (corresponding to a consumption of ~83% of the amended [U-13C]acetate). After 120 h, the turnover of [U-13C]acetate decreased and finally reached a stationary level during the last 68 h of the incubation. Following 9 days of incubation, the concentration of acetate reached ~1.6 mM.
Besides the degradation of acetate, the production of labeled and unlabeled methane and carbon dioxide, respectively, was also monitored (Figure 2). In the low acetate concentration-fed reactor, the proportion of the 13CO2 produced to total carbon dioxide increased almost linearly from 6 atom% up to 32 atom% during the 9 days of incubation, whereas the proportion of the 13CH4 produced to total methane increased gradually from 45 atom% up to 76 atom%.

Temporal change of the atom percent of 13CO2 and 13CH4 in the reactors fed with low concentrations of [U-13C]-acetate (▪ 13CH4; □ 13CO2); and in the reactors fed with high concentrations of [U-13C]acetate (• 13CH4; ○ 13CO2). The lines represent mean values (n=3), and error bars denote the standard deviation.
In the high acetate concentration-fed reactor, the production of 13CO2 increased from 20 atom% to about 45 atom% at 96 h and later stabilized at this value. The production of 13CH4 reached the peak value (80 atom%) at 24 h and remained almost stable for 96 h before starting to decline. During the time at which the levels of 13CH4 appeared to remain constant (Figure 2), almost 83% of the acetate was degraded (Figure 1). The isotopic abundance of 13C-labeled CO2 and CH4 at both low and high concentrations of 13C-labeled acetate incubations indicate the uptake and turnover of the added 13C-labeled acetate.
Phylogenetic microbial community composition of acetate-fed reactors
The microbial community compositions were evaluated by amplicon sequencing of triplicate reactors fed with low and high concentrations of acetate, respectively, see Supplementary Information. Samples at three time points were chosen for amplicon sequencing (8, 32 and 192 h). At least 29 428 sequences per sample passed filtering. From the archaeal population, the most abundant genera were affiliated with Methanobacterium, Methanosarcina, Methanobrevibacter and Methanoculleus. However, Methanomassiliicoccus and Methanothermobacter were also detected. The five most abundant classes of bacteria belonged to Clostridia, Bacteroidia, Bacilli, Thermotogae and Anaerolineae (Supplementary Figure 1).
Among abundant genera (>0.1% of the total reads), several genera from the bacterial population were seen to either decrease or increase from 8 to 192 h (Supplementary Figure 1A). The most noticeable change in the microbial community seems to occur among the two orders of Firmicutes, Bacillales (increasing), and Clostridiales (decreasing). The differences in the microbial communities in the high and low acetate time series, as well as the three replicates are illustrated by non-metric multidimensional scaling analysis (Supplementary Figure 1B). The findings revealed high reproducibility of the biological replicates and changes between the time series of each experiment.
Protein-SIP analysis
Time-resolved protein-SIP analysis showed that 13C was incorporated into peptides for the reactor fed with 100 mM [U-13C] acetate, starting from 48 h (Table 2, Figure 3). No 13C labeling was detected in the reactor fed with 4 mM [U-13C]acetate, control (fed with unlabeled acetate) and blank reactors at any time point (data not shown).

(a) RIA as a function of LR for peptides, showing incorporation of 13C at 192 h in the reactor fed with 100 mM of [U-13C]acetate (colors represent highest possible taxonomic rank with names in brackets corresponding to extracted genome bins). Functionally annotated proteins are indicated in the figure. The peptide SVAVNLAGIQGALASKGK, which showed a high degree of 13C incorporation, was identified belonging to Methanosarcina barkeri. (b) Time resolved analysis of the incorporation of 13C in the peptide SVAVNLAGIQGALASKGK belonging to the methanogenic archaea Methanosarcina barkeri. Incorporation of 13C in the peptide was evident after 48 h (the arrows indicate the 13C-labeled isotopologue). The peptide was identified as a methyl coenzyme M reductase, which catalyzes anaerobic oxidation of methane. Besides the methyl coenzyme M reductase, proteins with functions related to transport of substrate and sugars, signal transduction, translation as well as a transcriptional repressor were also identified (Figure 3 and Table 2).
In the reactor fed with high concentration of acetate, a total of five peptides incorporated 13C at 48 h (Table 2) after the start of the incubation. From these 13C-labeled peptides, one of them were assigned to the domain of bacteria and the other two to the domain of archaea. The last two peptides could not be assigned to any sequence present in either of the two metagenomes. A functional annotation was given to three of the peptides. The two peptides belonging to Archaea were identified as different subunits of the methyl coenzyme M reductase, a key enzyme involved in methane formation from methanogens (Ermler, 1997). The third peptide, which was unassigned, was identified as flagellin subunit B and is thus related to motility.
Following 192 h, a total of 56 peptides could be identified that showed incorporation of 13C (Figure 3 and Table 2). These peptides were dominated by bacterial species (Clostridia). Three labeled peptides originated from the domain of Archaea (two from the genus of Methanoculleus and one peptide from Methanosarcina barkeri). The proportion of the 13C-labeled peptides, known as relative isotope abundance (RIA), as well as how much of a peptide population is labeled, LR, is noted in Table 2. An example of the development of labeling from 24 to 192 h is given in Figure 3b, for a methyl coenzyme M reductase (chain B) from Methanosarcina barkeri.
A functional annotation could be assigned for 6 out of the 56 peptides found at 192 h (Figure 3a,Table 2). The remaining five peptides came from proteins from Clostridia (four peptides) and an unassigned peptide and had household functions, for example, initiation of RNA synthesis, signal transduction and transport substrate binding. However, one peptide belonged to putative TetR family, which is associated with antibiotic resistance.
The two metagenomes from Foulum and Lynggård biogas plants were sequenced to yield a total of 130 190 434 and 79 999 986 reads, respectively. When assembled, this amounted to a size of 165 123 257 bp for the Foulum metagenome, whereas the Lynggård metagenome was 82 793 162 bp. Both of the metagenomes represent approximately 98% Bacteria and 2% Archaea (Supplementary Figure 2). The two metagenomes were binned, based on the coverage and scaffold length, and the phylogeny from BLAST of each scaffold was superimposed (Figure 4a). The two metagenomes are relatively similar and were used as a reference database for the identified and 13C-labeled peptides (Figure 4b). The phylogenetic identification in the two metagenomes resembles the phylogenetic identification found by amplicon sequencing (Supplementary Figure 2). Protein-SIP data were superimposed onto the metagenome and all scaffolds belonging to a genome bin for which labeling of proteins were observed were extracted. The scaffolds of a total of six genome bins were extracted and completeness and potential contamination of each genome bin was evaluated (Table 3). The details of the assembly can be found in the Supplementary File 1.

The scaffold coverage from two metagenomes are plotted. The dot sizes indicate the scaffold length. (a) Coloring is according to phylogeny. (b) Coloring is observed in the protein-SIP analysis according to identified and 13C-labeled peptides. Labeling was seen in six clusters of scaffolds belonging to five subspecies of Clostridia and Methanoculleus.
The Wood–Ljungdahl pathway has four marker genes, acsB (carbon monooxide dehydrogenase), the two subunits of the corrinoid iron sulfur protein (acsC and acsD), and ftfhs (formyltetrahydrofolate synthatase) (Can et al., 2014). Searching Hidden Markov model profiles of each of the functional domains of these marker proteins against the six-frame translated metagenome, revealed that all of the six Clostridia bin genomes contained the ftfhs gene, whereas genes acsC and acsD were only found in one of the bin genomes (Clostridia (C4). All of the six genome bins tested positive for all the genes of the Wood–Ljungdahl pathway (Supplementary File 2).
Eleven proteins from a syntrophic glutamate-oxidizing bacterium of the genus Gelria (C2) were labeled with 13C during the SIP experiment (Table 2). The genus Gelria (phylum Clostridia) (C1) was the most heavily labeled in the proteome with 35 13C-labeled peptides.
Gelria belongs to the family Thermoanaerobacteraceae and is closely related to the genus of two of the known SAOB, namely Thermacetogenium phaeum and Tepidanaerobacter acetatoxydans, which are also part of the Thermoanaerobacteraceae family. Thermacetogenium had a relative read abundance of 0.01–0.11% in the amplicon data. Overall, the Thermoanaerobacteraceae family was found with an abundance of 0.8–2.0% in the different batch samples. Several ftfhs genes from Clostridia were found in the six-frame translated metagenome of the Foulum plant (Supplementary File 2).
Thermotoga lettingae is another known SAOB in the family Thermotogaceae. We did not detect any labeling in the proteins of this organism. A closely related genus, S1, was highly abundant in the amplicon data with a relative abundance of 2.1–7.2%. FTFHS was observed in Thermotogae in the six-frame translated metagenome (Supplementary Figure 1).
Six proteins from Clostridia were 13C labeled, but not found in any of the bins and could not be identified at a higher taxonomic level. Amplicon sequencing revealed that the genus Clostridium had a relative abundance of 0.6–1.4%. However, the known SAOB, Clostidium ultunese, was not detected with 13C incorporation, nor in the amplicon and metagenome data. Furthermore, Syntrophaceticus schinkii, from the family Thermoanaerobacterales Family III, order Thermoanaerobacterales, which is the last of the known SAOB, was not identified from the SIP experiment, nor was this family detected with amplicon sequencing.
Discussion
Our aim was to apply protein-SIP to describe the complex system of AD. Peptides showing incorporation of 13C were mapped onto a binned metagenome in order to identify key microorganisms involved in recovery of acetate accumulation under conditions simulating normal running conditions (low acetate levels) and stressed conditions with high levels of acetate.
The 13C isotopic profiling during the start of incubation in both the low and high concentrations of [U-13C]acetate showed that CO2 and CH4 are the main products of acetate degradation. As both the methyl and carboxyl groups are 13C labeled in the [U-13C]acetate, it is difficult to estimate the proportion of methane produced through SAO-HM, using the values of atom percent of 13CO2 and 13CH4. The 13CO2 formed can be produced either through acetoclastic methanogenesis or through SAO during the degradation of [U-13C]acetate. In a previous study, performed on sludge from the same plant as that analyzed in this study, methyl 13C-labeled acetate ([2-13C]acetate) was used to trace incorporation of 13C into CO2 and CH4 in real time during the degradation of [2-13C]acetate. The results showed that SAO-HM had a key role in the conversion of acetate to methane as 13CO2 can be produced from the degradation of [2-13C]acetate through SAO-HM alone (Mulat et al., 2014). Similar observations have been reported after amendment of [2-14C]acetate in batch incubations inoculated with manure and food waste (Karakashev et al., 2006).
A qualitative interpretation of the measured atom percent of 13CH4 and CO2 during the degradation of [U-13C]acetate revealed that the atom% of 13CO2 was generally lower than that of the 13CH4 in both the low and high concentrations of [U-13C]acetate. This is most likely due to the high background pool of unlabeled CO2 in the system.
This is corroborated by the levels of 13CH4, which reached a maximum of 75 atom% and 80 atom% in the low- and high-acetate-fed reactors, respectively. The production of unlabeled CH4 is possibly caused by a reduction of the background pool of unlabeled CO2 by hydrogenotrophs in syntrophy with SAOB. This assumption is in line with previous findings from the same AD, where SAO-HM was found to have a key role for the production of methane during the degradation of high concentrations (100 mM) of [2-13C]acetate (Mulat et al., 2014). Experimental conditions in this study and the previous study (Mulat et al., 2014) were very similar with the exception of the inoculum, which were a few months apart from the same full-scale biogas plant (Foulum). Although fluctuations in community dynamics are expected over time, the community structure between the time of the present and previous studies remained relatively stable (Mulat et al., 2014). The dominant microbial communities are almost similar in both studies (see the discussion below), indicating that SAO-HM had a significant role in the reactor fed with the high concentration of [U-13C]acetate.
The effect of the experimental design on the microbial community composition was investigated by amplicon sequencing of the 16S ribosomal RNA gene in both low- and high-acetate-fed reactors (see Supplementary Information). In general, the microbial communities observed in both batch reactors are in accordance with previous studies of AD communities (Lee et al., 2012; Wirth et al., 2012; Sundberg et al., 2013; St-Pierre and Wright, 2014). The majority of the organisms identified were affiliated to Clostridia. Clostridia participate in various stages in AD, including hydrolysis of cellulosic plant biomass and acetate oxidation (Hattori, 2008; Schlüter et al., 2008; Wirth et al., 2012). The bacterial classes of Bacteroidia, Bacilli, Thermotogae, Anaerolineae, Synergistia, as well as several Proteobacteria were also highly abundant. This composition is also in accordance with our metagenome constructed from the Foulum and Lynggaard biogas plants. Methanobacterium, Methanosarcina, Methanobrevibacter and Methanoculleus, representing hydrogenotrophic and acetoclastic methanogens, were present in the four most abundant genera of Archaea detected in our reactors, which is in accordance with previous studies from similarly operated AD (Blume et al., 2010; Sasaki et al., 2011; Hagen et al., 2014; Tuan et al., 2014; Ziganshina et al., 2014).
The abundance of Archaea (1–2%) was relatively low, compared with Bacteria (98–99%). Several previous studies of the microbial diversity in AD are based on separate analysis for Archaea and Bacteria, and the abundance of the two is thus not directly comparable (Patil et al., 2010; Kim et al., 2011; Lee et al., 2012; Rodríguez et al., 2012). Although we applied a universal primer set targeting both kingdoms, the abundance count of Archaea can be biased because of extraction and PCR. The approach used in this study does not take account of the 16S ribosomal RNA gene copy number, which has previously been shown to be generally lower in Archaea than Bacteria (Lee et al., 2009). However, the abundance ratio of Archaea and Bacteria is in accordance with the metagenome.
Time, rather than the two different concentrations of acetate tested, was the cause of the observed fluctuations in the community structures. A significant increase in Bacillales, specifically the genera Ureibacillus, was observed. Although the abundance of the genera of Ureibacillus can potentially be attributed to the presence of acetate, no incorporation of 13C from acetate was observed at protein level.
The abundances of Methanobrevibacter and Methanoculleus were constant during the incubation, the abundance of Methanobacterium decreased, and the abundance of Methanosarcina increased. The small increase of Methanosarcina is interesting because this organism is capable of both acetoclastic (Liu et al., 1985) and hydrogenotrophic (Thauer et al., 2008) pathways. Members of Methan osarcina are favored at acetate concentrations higher than 1 mM (Karakashev et al., 2006; Hori et al., 2011), although the biochemical reasoning remains unknown.
The microorganisms directly involved in acetate turnover were investigated with protein-SIP, not only identifying the microorganisms but also determining their level of activity. 13C-labeled peptides were detected only in the reactors fed with high concentrations of acetate; a reason for this could be a higher turnover rate of acetate of fewer specialized organisms and/or the relative high levels of labeling required for isotopical profiling. The focus hereafter will be on samples from the 100 mM reactor. 13C isotopically labeled peptides of the microorganisms were observed after 48 h of incubation (five peptides). The number of identified labeled peptides increased over time (56 peptides after 192 h), indicating that an adaptation period, for example, protein synthesis, for the microorganisms toward a change in environmental conditions was required.
The number of 13C-labeled peptides identified increased by using the Foulum metagenome as a reference database. Still, the number of 13C-labeled peptides detected in our reactors is low and can be explained by the very dense nature of the samples and the fact that protein extractions are often contaminated with humic substances, which, in correlation with a bias of mass spectrometry toward the most abundant proteins, challenges subsequent isotopical profiling (Doherty and Beynon, 2006; Heyer et al., 2013). Furthermore, the microbial communities in the AD are very complex, hence difficult to analyze using mass spectrometry. However, we detected between 1000 and 2100 unlabeled peptides in our reactors, which is a much greater number than those labeled with 13C, the incubation for a total of 9 days may have been too short for the slow-growing methanogens to adjust and synthesize new proteins, causing insufficient labeling.
Peptides from five subspecies of Clostridia (C1–C5), as well as Methanosarcina and Methanoculleus (M), incorporated 13C from the reactors fed with high levels of 13C-labeled acetate. Methyl coenzyme M reductase from Methanosarcina barkeri was 13C labeled, which is involved in methanogenesis, both acetoclastic and hydrogenotrophic. However, many of the peptides came from household proteins and were not directly involved in the SAO pathway. Formyltetrahydrofolate synthetase catalyzes the formation of acetate from H2 and CO2 (Xu et al., 2009; Hori et al., 2011), but has previously been associated with SAO in AD (Hori et al., 2011). The gene encoding the enzyme formyltetrahydrofolate synthetase, fthfs, is an ecological biomarker for reductive acetogenesis (Xu et al., 2009; Hori et al., 2011). Thus, we searched for fthfs in the six-frame translated Foulum metagenome, which was present in all of the Bacteria detected with the 13C-labeling approach. However, only two of the remaining marker genes, acsC and acsD, encoding CO dehydrogenase/acetyl-CoA synthase subunits.
Despite the absence of these genes, protein-SIP data suggest that these phylotypes are responsible for SAO. Most of the 13C-labeled proteins affiliated with the genus Gelria (c1). The enzymes of the Wood–Ljungdahl pathway are generally poorly understood for SAOB, and it has previously been suggested that other metabolic strategies than the Wood–Ljungdahl are being used by SAOB (Müller et al., 2013).
Most of the 13C-labeled peptides had a RIA (proportion of the peptide 13C labeled) of around 13–36%, however, four peptides had higher RIA values of 49.6% and 51.7% (Methanoculleus), 58.1% (Methanosarcina), and 93.8% (genus Gelria in the class Clostridia), respectively. The LR (how much of a peptide population is labeled) of these four peptides were 2.3% and 2.8% (Methanoculleus), and 39.2% (Methanosarcina) and 35.0% (Gelria). These data clearly confirm that these microorganisms are part of acetate degradation.
We detected unlabeled peptides from several methanogens, both aceticlastic and hydrogenotrophic. Among the methanogens, Methanosarcina had the highest 13C labeling with a RIA of 58.1% and an LR of 39.2%. The labeled peptide came from the methyl coenzyme M reductase β subunit, which is a common intermediate reaction of all metabolic pathways leading to methane formation (Grabarse et al., 2001). Thus, methyl coenzyme M reductase takes part in both methanogenic pathways (Rademacher et al., 2012). The highest 13C labeling of Methanosarcina is in accordance with the measured acetate consumption rate and the observed increase in the relative abundance of Methanosarcina during the course of incubation, as shown by the amplicon sequencing analysis. This methanogen participates in acid recovery in ADs by transformation of acetate in accordance with previous studies (Petersen, 1991). Methanosarcina was also highly abundant in the reactor fed with 100 mM [2-13C]acetate presented in our previous study, and the results of the isotope analysis demonstrated the key role of SAO-HM in the degradation of acetate (Mulat et al., 2014).
Adjusting the parameters of the AD to favor the presence and activity, Methanosarcina can possibly improve the stability of the reactor as this organism is able to carry out both acetoclastic and hydrogenotrophic methanogenesis and is more tolerant of several inhibitors such as ammonium, low retention times and high organic loading rates (De Vrieze et al., 2012). Methanosaeta, which are strictly acetoclastic methanogens, were not detected with amplicon sequencing. This could point in the direction of SAO-HM being the dominant process. This pathway is similarly dominant at thermophilic temperatures and in plants with increased levels of acetate (De Vrieze et al., 2012). However, as Methanosarcina is capable of both methanogenesis pathways, it is not possible to define the dominant pathway in the digester under high acetate levels.
Methanoculleus was the other methanogen found with labeled peptides. This particular methanogen is hydrogenotrophic and grows on H2 and CO2 (Mikucki et al., 2003). Applying [1-13C]acetate and [2-13C]acetate would allow to differentiate between acetoclastic or hydrogenotrophic methanogenesis. Furthermore, the use of primers targeting the fthfs gene would allow us to identify the specific SAOB present in our reactors. As Methanoculleus incorporated 13C into their peptides while growing on 100 mM [2-13C]acetate, they likely use the SAO-HM pathway in syntrophy with SAOB.
Conclusion
In this study, we showed that protein-SIP is a method that can be used to detect the active microorganisms incorporating 13C into their proteins in complex samples from AD batch reactors. We conclude that the mapping of protein-SIP onto a binned metagenome is highly applicable for identifying members of functional groups in complex microbial ecosystems. Peptides from Clostridia, Methanosarcina and Methanoculleus were labeled with 13C and therefore confirmed that these microorganisms were involved in the recovery after inhibitory events with high levels of acetate. The 13C-labeled and identified Clostridia are most likely oxidizing acetate as part of a synthrophy as they all contain the fthfs gene coding for formyltetrahydrofolate synthetase, a key enzyme in reductive acetogenesis. The findings therefore strongly indicate that these cells are new SAOB that can facilitate acetate consumption via SAO, coupled with hydrogenotrophic methanogenesis (SAO-HM). As Methanosarcina is a mixotrophic methanogen, its exact role as acetoclastic or hydrogenotrophic methanogenesis was not verified. Methanoculleus are hydrogenotrophic and thus likely involved in SAO-HM pathway under conditions with high concentrations of acetate.
Accession codes
References
Albertsen M, Hugenholtz P, Skarshewski A, Nielsen KL, Tyson GW, Nielsen PH . (2013). Genome sequences of rare, uncultured bacteria obtained by differential coverage binning of multiple metagenomes. Nat Biotechnol 31: 533–538.
Angelidaki I, Ahring BK . (1993). Thermophilic anaerobic digestion of livestock waste: the effect of ammonia. Appl Microbiol Biotechnol 38: 560–564.
Balk M, Weijma J, Stams AJM . (2002). Thermotoga lettingae sp. nov., a novel thermophilic, methanol-degrading bacterium isolated from a thermophilic anaerobic reactor. Int J Syst Evol Microbiol 52: 1361–1368.
Bastida F, Rosell M, Franchini AG, Seifert J, Finsterbusch S, Jehmlich N et al. (2010). Elucidating MTBE degradation in a mixed consortium using a multidisciplinary approach. FEMS Microbiol Ecol 73: 370–384.
Blume F, Bergmann I, Nettmann E, Schelle H, Rehde G, Mundt K et al. (2010). Methanogenic population dynamics during semi-continuous biogas fermentation and acidification by overloading. J Appl Microbiol 109: 441–450.
Botelho D, Wall MJ, Vieira DB, Fitzsimmons S, Liu F, Doucette A . (2010). Top-down and bottom-up proteomics of SDS-containing solutions following mass-based separation. J Proteome Res 9: 2863–2870.
Can M, Armstrong FA, Ragsdale SW . (2014). Structure, function, and mechanism of the nickel metalloenzymes, CO dehydrogenase, and acetyl-CoA synthase. Chem Rev 114: 4149–4174.
Chen Y, Cheng JJ, Creamer KS . (2008). Inhibition of anaerobic digestion process: a review. Bioresource Technol 99: 4044–4064.
De Vrieze J, Hennebel T, Boon N, Verstraete W . (2012). Methanosarcina: the rediscovered methanogen for heavy duty biomethanation. Bioresource Technol 112: 1–9.
Demirel B, Yenigün O . (2002). Two-phase anaerobic digestion processes: a review. J Chem Technol Biotechnol 77: 743–755.
Doherty MK, Beynon RJ . (2006). Protein turnover on the scale of the proteome. Exp Rev Proteomics 3: 97–110.
Ermler U . (1997). Crystal structure of methyl-coenzyme M reductase: the key enzyme of biological methane formation. Science 278: 1457–1462.
Finn RD, Bateman A, Clements J, Coggill P, Eberhardt RY, Eddy SR et al. (2014). Pfam: the protein families database. Nucl Acids Res 42: D222–D230.
Fotidis IA, Karakashev D, Angelidaki I . (2013). Bioaugmentation with an acetate-oxidising consortium as a tool to tackle ammonia inhibition of anaerobic digestion. Bioresource Technol 146: 57–62.
Grabarse W, Mahlert F, Duin EC, Goubeaud M, Shima S, Thauer RK et al. (2001). On the mechanism of biological methane formation: structural evidence for conformational changes in methyl-coenzyme M reductase upon substrate binding. J Mol Biol 309: 315–330.
Hagen LH, Vivekanand V, Linjordet R, Pope PB, Eijsink VGH, Horn SJ . (2014). Microbial community structure and dynamics during co-digestion of whey permeate and cow manure in continuous stirred tank reactor systems. Bioresource Technol 171: 350–359.
Hansen SH, Stensballe A, Nielsen PH, Herbst F-A . (2014). Metaproteomics: evaluation of protein extraction from activated sludge. Proteomics 14: 2535–2539.
Hao L, Lü F, Mazéas L, Desmond-Le Quéméner E, Madigou C, Guenne A et al. (2014). Stable isotope probing of acetate fed anaerobic batch incubations shows a partial resistance of acetoclastic methanogenesis catalyzed by Methanosarcina to sudden increase of ammonia level. Water Res 69C: 90–99.
Hattori S, Kamagata Y, Hanada S, Shoun H . (2000). Thermacetogenium phaeum gen. nov., sp. nov., a strictly anaerobic, thermophilic, syntrophic acetate-oxidizing bacterium. Int J Syst Evol Microbiol 50 (Pt 4): 1601–1609.
Hattori S . (2008). Syntrophic acetate-oxidizing microbes in methanogenic environments. Microbes Environ 23: 118–127.
Herbst F-A, Bahr A, Duarte M, Pieper DH, Richnow H-H, von Bergen M et al. (2013). Elucidation of in situ polycyclic aromatic hydrocarbon degradation by functional metaproteomics (protein-SIP). Proteomics 13: 2910–2920.
Heyer R, Kohrs F, Benndorf D, Rapp E, Kausmann R, Heiermann M et al. (2013). Metaproteome analysis of the microbial communities in agricultural biogas plants. New Biotechnol 30: 614–622.
Hori T, Sasaki D, Haruta S, Shigematsu T, Ueno Y, Ishii M et al. (2011). Detection of active, potentially acetate-oxidizing syntrophs in an anaerobic digester by flux measurement and formyltetrahydrofolate synthetase (FTHFS) expression profiling. Microbiology (Reading, England) 157: 1980–1989.
Ito T, Yoshiguchi K, Ariesyady HD, Okabe S . (2011). Identification of a novel acetate-utilizing bacterium belonging to Synergistes group 4 in anaerobic digester sludge. ISME J 5: 1844–1856.
Jehmlich N, Schmidt F, Taubert M, Seifert J, Bastida F, von Bergen M et al. (2010). Protein-based stable isotope probing. Nat Protocols 5: 1957–1966.
Jehmlich N, Schmidt F, von Bergen M, Richnow H-H, Vogt C . (2008). Protein-based stable isotope probing (Protein-SIP) reveals active species within anoxic mixed cultures. ISME J 2: 1122–1133.
Karakashev D, Batstone DJ, Trably E, Angelidaki I . (2006). Acetate oxidation is the dominant methanogenic pathway from acetate in the absence of Methanosaetaceae. Appl Environ Microbiol 72: 5138–5141.
Kim J, Shin SG, Han G, O’Flaherty V, Lee C, Hwang S . (2011). Common key acidogen populations in anaerobic reactors treating different wastewaters: molecular identification and quantitative monitoring. Water Res 45: 2539–2549.
Kjeldal H, Pell L, Pommerening-Röser A, Nielsen JL . (2014). Influence of p-cresol on the proteome of the autotrophic nitrifying bacterium Nitrosomonas eutropha C91. Arch Microbiol 196: 497–511.
Kleindienst S, Herbst F-A, Stagars M, von Netzer F, von Bergen M, Seifert J et al. (2014). Diverse sulfate-reducing bacteria of the Desulfosarcina/Desulfococcus clade are the key alkane degraders at marine seeps. ISME J 8: 2029–2044.
Klocke M, Mähnert P, Mundt K, Souidi K, Linke B . (2007). Microbial community analysis of a biogas-producing completely stirred tank reactor fed continuously with fodder beet silage as mono-substrate. Syst Appl Microbiol 30: 139–151.
Kohlbacher O, Reinert K, Gröpl C, Lange E, Pfeifer N, Schulz-Trieglaff O et al. (2007). TOPP—the OpenMS proteomics pipeline. Bioinforma Oxf Engl 23: e191–e197.
Krakat N, Schmidt S, Scherer P . (2011). Potential impact of process parameters upon the bacterial diversity in the mesophilic anaerobic digestion of beet silage. Bioresource Technol 102: 5692–5701.
Labatut RA, Angenent LT, Scott NR . (2014). Conventional mesophilic vs. thermophilic anaerobic digestion: a trade-off between performance and stability? Water Res 53: 249–258.
Lee S-H, Kang H-J, Lee YH, Lee TJ, Han K, Choi Y et al. (2012). Monitoring bacterial community structure and variability in time scale in full-scale anaerobic digesters. J Environ Monitor 14: 1893–1905.
Lee ZM-P, Bussema C, Schmidt TM . (2009). rrnDB: documenting the number of rRNA and tRNA genes in bacteria and archaea. Nucleic Acids Res 37: D489–D493.
Liu Y, Boone DR, Sleat R, Mah RA . (1985). Methanosarcina mazei LYC, a new methanogenic isolate which produces a disaggregating enzyme. Appl Environ Microbiol 49: 608–613.
Lovell CR, Leaphart AB . (2005). Community-level analysis: key genes of CO2-reductive acetogenesis. Methods Enzymol 397: 454–469.
Lü F, Hao L, Guan D, Qi Y, Shao L, He P . (2013). Synergetic stress of acids and ammonium on the shift in the methanogenic pathways during thermophilic anaerobic digestion of organics. Water Res 47: 2297–2306.
Manzoor S, Müller B, Niazi A, Bongcam-Rudloff E, Schnürer A . (2013). Draft genome sequence of Clostridium ultunense strain esp, a syntrophic acetate-oxidizing bacterium. Genome Announcements 1: e0010713.
Mikucki JA, Liu Y, Delwiche M, Colwell FS, Boone DR . (2003). Isolation of a methanogen from deep marine sediments that contain methane hydrates, and description of Methanoculleus submarinus sp. nov. Appl Environ Microbiol 69: 3311–3316.
Mulat DG, Ward AJ, Adamsen APS, Voigt NV, Nielsen J, Feilberg A . (2014). Quantifying contribution of synthrophic acetate oxidation to methane production in thermophilic anaerobic reactors by membrane inlet mass spectrometry. Environ Sci Technol 48: 2505–2511.
Müller B, Sun L, Schnürer A . (2013). First insights into the syntrophic acetate-oxidizing bacteria—a genetic study. Microbiol Open 2: 35–53.
Palatsi J, Viñas M, Guivernau M, Fernandez B, Flotats X . (2011). Anaerobic digestion of slaughterhouse waste: main process limitations and microbial community interactions. Bioresource Technol 102: 2219–2227.
Patil SS, Kumar MS, Ball AS . (2010). Microbial community dynamics in anaerobic bioreactors and algal tanks treating piggery wastewater. Appl Microbiol Biotechnol 87: 353–363.
Petersen S . (1991). Acetate oxidation in a thermophilic anaerobic sewage-sludge digestor: the importance of non-aceticlastic methanogenesis from acetate. FEMS Microbiol Lett 86: 149–152.
Rademacher A, Zakrzewski M, Schlüter A, Schönberg M, Szczepanowski R, Goesmann A et al. (2012). Characterization of microbial biofilms in a thermophilic biogas system by high-throughput metagenome sequencing. FEMS Microbiol Ecol 79: 785–799.
Rajagopal R, Massé DI, Singh G . (2013). A critical review on inhibition of anaerobic digestion process by excess ammonia. Bioresource Technol 143: 632–641.
Rodríguez E, Lopes A, Fdz-Polanco M, Stams AJM, García-Encina PA . (2012). Molecular analysis of the biomass of a fluidized bed reactor treating synthetic vinasse at anaerobic and micro-aerobic conditions. Appl Microbiol Biotechnol 93: 2181–2191.
Sachsenberg T, Herbst F-A, Taubert M, Kermer R, Jehmlich N, von Bergen M et al. (2015). MetaProSIP: automated inference of stable isotope incorporation rates in proteins for functional metaproteomics. J Proteome Res 14: 619–627.
Sasaki D, Hori T, Haruta S, Ueno Y, Ishii M, Igarashi Y . (2011). Methanogenic pathway and community structure in a thermophilic anaerobic digestion process of organic solid waste. J Biosci Bioeng 111: 41–46.
Schlüter A, Bekel T, Diaz NN, Dondrup M, Eichenlaub R, Gartemann K-H et al. (2008). The metagenome of a biogas-producing microbial community of a production-scale biogas plant fermenter analysed by the 454-pyrosequencing technology. J Biotechnol 136: 77–90.
Schnürer A, Schink B, Svensson BH . (1996). Clostridium ultunense sp. nov., a mesophilic bacterium oxidizing acetate in syntrophic association with a hydrogenotrophic methanogenic bacterium. Int J Syst Bacteriol 46: 1145–1152.
Seifert J, Herbst F-A, Halkjaer Nielsen P, Planes FJ, Jehmlich N, Ferrer M et al. (2013). Bioinformatic progress and applications in metaproteogenomics for bridging the gap between genomic sequences and metabolic functions in microbial communities. Proteomics 13: 2786–2804.
Shevchenko A, Tomas H, Havlis J, Olsen J V, Mann M . (2006). In-gel digestion for mass spectrometric characterization of proteins and proteomes. Nat Protocols 1: 2856–2860.
St-Pierre B, Wright A-DG . (2014). Comparative metagenomic analysis of bacterial populations in three full-scale mesophilic anaerobic manure digesters. Appl Microbiol Biotechnol 98: 2709–2717.
Sturm M, Bertsch A, Gröpl C, Hildebrandt A, Hussong R, Lange E et al. (2008). OpenMS - an open-source software framework for mass spectrometry. BMC Bioinformatics 9: 163.
Sundberg C, Al-Soud WA, Larsson M, Alm E, Yekta SS, Svensson BH et al. (2013). 454 pyrosequencing analyses of bacterial and archaeal richness in 21 full-scale biogas digesters. FEMS Microbiol Ecol 85: 612–626.
Taubert M, Jehmlich N, Vogt C, Richnow HH, Schmidt F, von Bergen M et al. (2011). Time resolved protein-based stable isotope probing (protein-SIP) analysis allows quantification of induced proteins in substrate shift experiments. Proteomics 11: 2265–2274.
Taubert M, Vogt C, Wubet T, Kleinsteuber S, Tarkka MT, Harms H et al. (2012). Protein-SIP enables time-resolved analysis of the carbon flux in a sulfate-reducing, benzene-degrading microbial consortium. ISME J 6: 2291–2301.
Thauer RK, Kaster A-K, Seedorf H, Buckel W, Hedderich R . (2008). Methanogenic archaea: ecologically relevant differences in energy conservation. Nat Rev Microbiol 6: 579–591.
Tuan NN, Chang Y-C, Yu C-P, Huang S-L . (2014). Multiple approaches to characterize the microbial community in a thermophilic anaerobic digester running on swine manure: a case study. Microbiol Res 169: 717–724.
Vizcaíno JA, Deutsch EW, Wang R, Csordas A, Reisinger F, Ríos D et al. (2014). ProteomeXchange provides globally coordinated proteomics data submission and dissemination. Nat Biotechnol 32: 223–226.
Westerholm M, Roos S, Schnürer A . (2010). Syntrophaceticus schinkii gen. nov., sp. nov., an anaerobic, syntrophic acetate-oxidizing bacterium isolated from a mesophilic anaerobic filter. FEMS Microbiol Lett 309: 100–104.
Westerholm M, Roos S, Schnürer A . (2011). Tepidanaerobacter acetatoxydans sp. nov., an anaerobic, syntrophic acetate-oxidizing bacterium isolated from two ammonium-enriched mesophilic methanogenic processes. Syst Appl Microbiol 34: 260–266.
Wirth R, Kovács E, Maróti G, Bagi Z, Rákhely G, Kovács KL . (2012). Characterization of a biogas-producing microbial community by short-read next generation DNA sequencing. Biotechnol Biofuels 5: 41.
Xu K, Liu H, Du G, Chen J . (2009). Real-time PCR assays targeting formyltetrahydrofolate synthetase gene to enumerate acetogens in natural and engineered environments. Anaerobe 15: 204–213.
Ziganshina EE, Bagmanova AR, Khilyas I V, Ziganshin AM . (2014). Assessment of a biogas-generating microbial community in a pilot-scale anaerobic reactor. J Biosci Bioeng 117: 730–736.
Acknowledgements
This study was part of the project HYdrogen CONtrol for Optimization of Methane Production from Livestock Waste (HYCON) funded by the Danish Strategic Research Council (grant no. 10-093944).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on The ISME Journal website
Rights and permissions
About this article

Cite this article
Mosbæk, F., Kjeldal, H., Mulat, D. et al. Identification of syntrophic acetate-oxidizing bacteria in anaerobic digesters by combined protein-based stable isotope probing and metagenomics. ISME J 10, 2405–2418 (2016). https://doi.org/10.1038/ismej.2016.39
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ismej.2016.39
This article is cited by
-
Energetically exploiting lignocellulose-rich residues in anaerobic digestion technologies: from bioreactors to proteogenomics
Biotechnology for Biofuels and Bioproducts (2023)
-
Diverse electron carriers drive syntrophic interactions in an enriched anaerobic acetate-oxidizing consortium
The ISME Journal (2023)
-
Metabolic Functional Profiles of Microbial Communities in Methane Production Systems Treating Winery Wastewater
BioEnergy Research (2023)
-
Biomethane production and microbial strategies corresponding to high organic loading treatment for molasses wastewater in an upflow anaerobic filter reactor
Bioprocess and Biosystems Engineering (2023)
-
Functional and molecular approaches for studying and controlling microbial communities in anaerobic digestion of organic waste: a review
Reviews in Environmental Science and Bio/Technology (2023)
