
Abstract
Background/Objectives:
Impaired energy metabolism is the defining characteristic of obesity-related heart failure. The adipocyte-derived peptide apelin has a role in the regulation of cardiovascular and metabolic homeostasis and may contribute to the link between obesity, energy metabolism and cardiac function. Here we investigate the role of apelin in the transition from metabolic adaptation to maladaptation of the heart in obese state.
Methods:
Adult male C57BL/6J, apelin knock-out (KO) or wild-type mice were fed a high-fat diet (HFD) for 18 weeks. To induce heart failure, mice were subjected to pressure overload after 18 weeks of HFD. Long-term effects of apelin on fatty acid (FA) oxidation, glucose metabolism, cardiac function and mitochondrial changes were evaluated in HFD-fed mice after 4 weeks of pressure overload. Cardiomyocytes from HFD-fed mice were isolated for analysis of metabolic responses.
Results:
In HFD-fed mice, pressure overload-induced transition from hypertrophy to heart failure is associated with reduced FA utilization (P<0.05), accelerated glucose oxidation (P<0.05) and mitochondrial damage. Treatment of HFD-fed mice with apelin for 4 weeks prevented pressure overload-induced decline in FA metabolism (P<0.05) and mitochondrial defects. Furthermore, apelin treatment lowered fasting plasma glucose (P<0.01), improved glucose tolerance (P<0.05) and preserved cardiac function (P<0.05) in HFD-fed mice subjected to pressure overload. In apelin KO HFD-fed mice, spontaneous cardiac dysfunction is associated with reduced FA oxidation (P<0.001) and increased glucose oxidation (P<0.05). In isolated cardiomyocytes, apelin stimulated FA oxidation in a dose-dependent manner and this effect was prevented by small interfering RNA sirtuin 3 knockdown.
Conclusions:
These data suggest that obesity-related decline in cardiac function is associated with defective myocardial energy metabolism and mitochondrial abnormalities. Furthermore, our work points for therapeutic potential of apelin to prevent myocardial metabolic abnormalities in heart failure paired with obesity.
Similar content being viewed by others


Introduction
Cardiac metabolic reprogramming is a defining characteristic of obesity and cardiovascular diseases. In obesity, the heart displays an impaired metabolic phenotype, characterized by the dysregulation of energy substrate utilization in the myocardium.1 The alterations in myocardial energy metabolism are indicative of metabolic reprogramming, a change in how energy is generated and how fuel is utilized. In the healthy adult heart, fatty acid (FA) oxidation contributes up to 70% of the total energy production, while much of the rest is generated from carbohydrates via glucose oxidation.2, 3, 4 In disease states such as ischemia and hypertrophy, the balance between FA oxidation and glucose oxidation is perturbed and the heart becomes more glucose-dependent.5, 6, 7 This initial adaptive response is beneficial, in that it maintains adenosine triphosphate (ATP) levels in the face of diminished mitochondrial oxidative phosphorylation.8 Recent studies linking specific metabolic alterations to cardiac performance have strengthened the concept that altered energy metabolism is more than a primary effect of cardiac remodeling, but may in fact be a powerful driver of cardiac dysfunction.9, 10, 11, 12
Adipose tissue is a highly active metabolic and endocrine organ and has a substantial role in the pathogenesis of obesity-related cardiovascular complications.13 Altered levels of adipocyte-derived factors, termed adipokines, may be particularly related with heart diseases and metabolic disorders.14,15 Among novel adipokines, apelin has emerged as an important regulator of cardiovascular and metabolic homeostasis. Apelin, an endogenous ligand for the G-protein-coupled receptor APJ (angiotensin receptor-like 1), exerts strong inotropic actions and increases coronary blood flow by vascular dilation.16 In response to pathological stress, apelin-APJ axis regulates cardiac hypertrophy, myocardial remodeling and heart contractility.17 Apelin-deficient mice demonstrate age-related progressive cardiac dysfunction, which is prevented by apelin infusion,18 suggesting an essential role of apelinergic system in maintaining cardiac performance. In humans, circulating and cardiac levels of apelin are reduced in patients with chronic heart failure indicating a potential role for diminished apelinergic system in the pathophysiology of heart failure. On the other hand, plasma apelin concentrations are increased in obese subjects 19 and apelin treatment in obese conditions stimulates glucose utilization in adipose tissue and skeletal muscle leading to increasing insulin sensitivity.20 Moreover, our recent study suggests that apelin enhances muscle mitochondrial biogenesis and citrate synthase activity, a marker of mitochondria content, culminating in amelioration of both lipid and glucose metabolism in insulin-resistant mice.21 More recently, it has been reported that apelin ameliorates diabetic cardiomyopathy via stimulation of sirtuin 3, a mitochondrial sirtuin deacetylase.22 Altogether, these studies suggest that apelinergic system may have a role in obesity-related mitochondrial abnormalities and cardiometabolic manifestations. However, the role of apelin in myocardial metabolic adaptation or maladaptation of the heart in obese state remains to be determined.
Herein we report that transition from hypertrophy to heart failure in obese conditions is characterized by defective myocardial FA utilization and mitochondrial defects. Furthermore, we demonstrate that apelin regulates metabolic reprogramming and preserves mitochondrial integrity in heart failure paired with increased adiposity.
Materials and methods
Animals
The investigation conforms to the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication No. 85-23, revised 1985) and was performed in accordance with the recommendations of the French Accreditation of the Laboratory Animal Care (approved by the local Centre National de la Recherche Scientifique ethics committee). At 12 weeks, C57BL/6J mice were fed a high-fat diet (HFD, 45% fat) for 18 weeks or a normal diet (ND, 4% fat) and then subjected to aortic banding (AB) for 4 weeks as previously described.17 Sham-operated animals underwent a similar procedure without AB.
Experimental protocols
After 18 weeks of HFD, mice were randomly divided into four groups: (1) sham vehicle, (2) AB vehicle, (3) sham apelin and (4) AB apelin. Mice received apelin (0.1 μmol kg–1 day–1) or vehicle (phosphate-buffered saline) intraperitoneally 10 min before AB and then every 24 h for 4 weeks.
Body fat mass composition
To determine total body fat and lean mass, mice were placed in a plastic holder inserted into the EchoMRI-3-in-1 system from Echo Medical Systems (Houston, TX, USA).
Apelin KO mice
Apelin knock-out (KO) mice were generated by homologous recombination of a targeting vector in embryonic stem cells (Genoway, Lyon, France). Recombined embryonic stem cell clones were injected into C57BL/6J derived blastocysts to generate chimeric mice. Constitutive KO heterozygous females and homozygous males were characterized by PCR and Southern blot. As cardiac phenotype in 3-month-old apelin KO mice appeared normal,18 we examined cardiometabolic phenotype of these animals after 18 weeks of HFD.
Glucose tolerance test and plasma measures
Mice were fasted for 6 h and then were injected with glucose (1 g kg–1, intraperitoneally). Blood glucose levels from the tail vein were monitored over time using a glucometer as previously described.20 Insulinemia (Mercodia, Uppsala, Sweden) and glycemia (Accu-check, Roche Diagnostics, Boulogne-Billancourt, France) were measured in fasted state.
Echocardiographic studies
Echocardiography was performed in isoflurane anesthetized mice using a Vivid7 imaging system (General Electric Healthcare, Toulouse, France) equipped with a 14-MHz sectorial probe. Two-dimensional images were recorded in parasternal long- and short-axis projections, with guided M-mode recordings at the midventricular level in both views. Left ventricular (LV) dimensions and wall thickness were measured in at least five beats from each projection and averaged. Interventricular septum (IVS) and posterior wall (PW) thickness and internal dimensions at diastole and systole (LVIDd and LVIDs, respectively) were measured. Fractional shortening and ejection fraction were calculated from the two-dimensional images.
Primary cultures of cardiomyocytes and transfection
Primary cultures of cardiomyocytes were prepared from heart ventricles of C57BL/6J mice exposed to a HFD for 18 weeks as described previously.23 Briefly, mice were intraperitoneally heparinized with 200 U heparin and pentobarbital sodium (100 mg kg–1, intraperitoneal). The heart was quickly removed, cannulated via the aorta and perfused at constant pressure (100 cm H2O) at 37 °C for ∼3 min with a Ca2+-free bicarbonate-based buffer. The enzymatic digestion was initiated by adding collagenase type B (0.5 mg ml–1; Boehringer, Mannheim, Germany), collagenase type D (0.5 mg ml–1; Boehringer), and protease type XIV (0.02 mg ml–1; Sigma, Saint-Quentin Fallavier, France) to the perfusion solution. When the heart became swollen after ∼3 min of digestion, 50 μM Ca2+ was added to the enzyme solution. After 6–7 min, LV was removed, cut into several chunks and digested for 10 min at 37 °C in the same enzyme solution. The supernatant containing the dispersed myocytes was filtered. The cell pellet was resuspended in Ca solution I (125 μM Ca2+). After the myocytes were pelleted by gravity for ∼10 min, the supernatant was aspirated and the myocytes were resuspended in Ca solution II (250 μM Ca2+). The final cell pellet was suspended in Ca solution III (500 μM Ca2+). The isolated cells were stored in HEPES-buffered solution consisting of (in mM) 1 CaCl2, 137 NaCl, 5.4 KCl, 15 dextrose, 1.3 MgSO4, 1.2 NaH2PO4 and 20 HEPES, adjusted to pH 7.4 with NaOH.
Freshly isolated cardiomyocytes were suspended in minimal essential medium (Sigma M1018) containing 1.2 mM Ca2+, 2.5% fetal bovine serum and 1% penicillin–streptomycin. The myocytes were then plated at 1 × 104 cells cm–2 and substrate oxidations were measured after 24 h of cell culture. For small interfering RNA (siRNA) transfection, after 1 h of culture, cardiomyocytes were treated with siRNA negative control (Scramble) or siRNAs to sirtuin 3 (Sirt3) according to the manufacturer’s recommendations (Thermoscientific, Illkirch, France). Following 24 h of siRNA incubation, cardiomyocytes were treated with apelin (10−7 M) for 1 h for FA oxidation measurements. The effects of siRNAs were evaluated at 24 and 48 h after transfection by reverse transcriptase-PCR analysis for Sirt3 mRNA expression.
Measurements of FA and glucose oxidation
FA oxidation was measured using [1-14C]-palmitate in heart tissue and isolated cardiomyocytes as previously described.21 The heart tissue samples were incubated in modified Krebs–Henseleit buffer containing 1.5% FA-free bovine serum albumin, 5 mmol l–1 glucose, 1 mmol l–1 palmitate and 0.5 μCi ml–1 [14C]palmitate (Perkin Elmer, Courtaboeuf, France) for 60 min. Isolated cardiomyocytes were incubated in the same modified Krebs–Henseleit buffer, but without glucose. After incubation, tissues were homogenized in 800 μl lysis buffer. Complete oxidation was determined by acidifying the incubation buffer with 1 ml of 1 mol l–1 H2SO4, and the 14CO2 was trapped by benzethonium hydroxide (Sigma) placed in a 0.5 ml microtube. After 120 min, the radioactivity was counted (Cytoscint; MP Biomedicals, Strasbourg, France). Similarly, glucose oxidation was assessed by measuring the [14C]-glucose oxidation in cardiac tissue as previously described. Sample incubation for 1 h was performed using a modified Krebs–Henseleit buffer containing 0.2% bovine serum albumin, 20 mM Hepes, 10 mM glucose, 0.8 μCi ml–1 [14C]glucose (Perkin Elmer). Results were normalized for mg of proteins.
Electron microscopy
Briefly, excised hearts were fixed in 2.5% glutaraldehyde/1% paraformaldehyde, post-fixed in 2% osmium tetroxide, embedded in resin and sectioned. Cardiac mitochondrial number relative to the section area was determined from electron micrographs as described previously.21
Mitochondrial DNA analysis
Total DNA was extracted from heart tissue using a commercial kit (DNeasy; Qiagen, Marseille, France). The content of mitochondrial (mt)DNA was calculated using real-time quantitative PCR by measuring the threshold cycle ratio of a mitochondrial encoded gene (COX1) and a nuclear-encoded gene (cyclophilin A) as previously described.21
Real-time PCR
Total RNAs were isolated from heart mouse and isolated cardiomyocytes using the RNeasy mini kit (Qiagen). Total RNAs (500 ng) were reverse transcribed using random hexamers and Superscript II reverse transcriptase (Invitrogen, Paisley, UK). Real-time PCR was performed as previously described.21 Analysis of the 18S ribosomal RNA was performed using the ribosomal RNA control Taqman Assay Kit (Applied Biosystems, Saint Aubin, France) to normalize gene expression. The sequences of the primers are listed in Supplementary Table S1 in the Data Supplement.
Citrate synthase assay
Citrate synthase activity was measured in heart homogenates by quantification of oxaloacetate utilization as previously described.24 In brief, heart tissue was homogenized (10 mg tissue per 500 μl buffer) on ice in buffer containing 0.1 mol l–1 KH2PO4 and 0.05% bovine serum albumin. Results were expressed as μmol min–1 per mg proteins.
Protein assay
Protein amount of samples was determined using the DC protein assay kit (Bio-Rad, Marnes-la-Coquette, France) according to the manufacturer’s instructions.
Western blot analysis
Isolated cardiomyocytes were treated with apelin (10−7 M) for 24h and then lysed and loaded (40 μg of protein) on a 10% sodium dodecyl sulfate–polyacrylamide gel electrophoresis gel and transferred to polyvinylidene fluoride membrane. Antibodies against SIRT3 and GAPDH were purchased from Cell Signaling Technology (Lyon, France).
Statistical analysis
Data are expressed as mean±s.e.m. Comparison of multiple groups was performed by two-way analysis of variance followed by a Bonferroni’s post hoc test for in vivo studies using GraphPad Prism version 4.00 for Windows (Pully, Switzerland). All other statistical analyses were performed by one-way analysis of variance followed by the Bonferroni post hoc test. Statistical significance was defined as P<0.05.
Results
Cardiometabolic adaptation to HFD-induced obesity
To determine the consequences of obesity on cardiac function and metabolism, male C57BL/6J mice were fed a ND (4% fat) or HFD (45% fat) for 18 weeks. As shown in Table 1, in comparison with ND-fed mice, HFD-fed animals had increased body weight, fat mass, plasma glucose and insulin levels. Moreover, HFD-fed mice exhibited impaired glucose tolerance (Figure 1a). Subsequent echocardiographic analysis revealed increased LV posterior wall thickness and interventricular septum thickness in HFD-fed mice as compared with ND-fed animals (Figures 1b and c) without significant changes in fractional shortening and ejection fraction (Figures 1d and e) between groups suggesting cardiac adaptation to HFD. In agreement with echocardiographic data, the expression levels of two well-known marker genes for impaired LV function, atrial natriuretic peptide and brain natriuretic peptide, were unchanged in HFD-fed mice (Figure 1f). As shown in Figures 1g and h, obesity-related cardiac hypertrophy was accompanied by increased FA oxidation and by reduced glucose oxidation.

Cardiometabolic profile of HFD-induced obese mice. (a) Glucose tolerance test (GTT) in mice after 18 weeks of exposure to ND (n=8) or HFD (n=10). (b) Interventricular septum thickness (IVST), (c) LV posterior wall thickness (LVPWT) and (d) fractional shortening (FS) and (e) ejection fraction (EF) were analyzed by two-dimensional guided M-mode echocardiography in ND-fed and HFD-fed mice. (f) Real-time reverse transcriptase (RT)-PCR analysis of atrial natriuretic peptide (ANP) and brain natriuretic peptide (BNP) expression levels in heart tissue from ND-fed (n=6) and HFD-fed (n=7) mice. (g) Myocardial FA oxidation and (h) glucose oxidation in ND-fed (n=7) and HFD-fed (n=8) mice. Results are means±s.e.m. **P<0.01; ***P<0.001 vs ND.
Cardiometabolic reprogramming in a murine model combining obesity and heart failure: effects of apelin
In order to study obesity-related transition from compensated hypertrophy to heart failure, mice were fed a HFD for 18 weeks and then subjected to chronic pressure overload generated by AB. Echocardiographic analyses revealed a significant increase in interventricular septum thickness and LV posterior wall thickness after 4 weeks of AB in HFD-fed mice compared with sham-operated mice (Figures 2a and b). Importantly, cardiac contractile function was severely impaired in HFD-fed AB mice as measured by the decrease in fractional shortening and ejection fraction compared with sham-operated HFD-fed mice (Figures 2a and b). In HFD-fed AB mice, cardiac dysfunction was accompanied by concomitant increase in atrial natriuretic peptide and brain natriuretic peptide expression levels (Figure 2f). In contrast, daily treatment of HFD-fed AB mice with apelin for 4 weeks reversed cardiac hypertrophy (Figure 2a and c) and contractile dysfunction (Figures 2d and e) as compared with vehicle-treated HFD-fed AB mice. In addition, AB-induced increase in atrial natriuretic peptide and brain natriuretic peptide expression levels was significantly reduced by apelin treatment (Figure 2c). At the metabolic level, apelin treatment prevented AB-induced reduction in myocardial FA utilization (Figure 2d) and an increase in glucose oxidation (Figure 2e). In addition, apelin treatment attenuated the increase in fasting plasma glucose levels (Figure 2f) and improved glucose tolerance as compared with vehicle-treated HFD-fed AB mice (Figure 2g).

Cardiometabolic reprogramming in obesity-related heart failure: effects of apelin. (a) Representative two-dimensional and M-Mode echocardiographic images of mice subjected to 4 weeks of AB (n=7) or sham operation (Sham, n=8) and received 0.1 μmol kg–1 day–1 intraperitoneal apelin or vehicle for 4 weeks. (b) Echocardiographic measurements of interventricular septum thickness in diastole (IVSTd), posterior wall thickness in diastole (PWTd), LV internal diameter in diastole (LVIDd), fractional shortening (FS) and ejection fraction (EF) in sham or AB HFD-fed mice treated with vehicle or apelin. (c) Real-time reverse transcriptase (RT)-PCR analysis of atrial natriuretic peptide (ANP) and brain natriuretic peptide (BNP) expression levels in left ventricles of sham or AB HFD-fed mice treated with vehicle or apelin. (d) Myocardial FA oxidation, (e) glucose oxidation, (f) plasma glucose level and (g) glucose tolerance test (GTT) in vehicle- or apelin-treated HFD-fed subjected to sham or AB surgery. Results are means±s.e.m. *P<0.05; **P<0.01; ***P<0.001 vs sham-vehicle; §P<0.05; §§P<0.01; §§§P<0.001 vs AB-vehicle.
Mitochondrial abnormalities in obesity-related heart failure: effects of apelin
To study mitochondrial changes in obesity-related heart failure, we examined mitochondrial ultrastructure, density, mtDNA content and expression of key regulators of mitochondrial biogenesis in HFD-fed mice subjected to AB. Electron microscopic examination revealed mitochondrial damage including swelling and structural disruption in HFD-fed mice after 4 weeks of AB as compared with sham mice (Figure 3a). As shown in Figure 3b, tissue sections from AB hearts exhibited a 41% decrease in mitochondrial density as compared with sham mice. In addition, the ratio of mtDNA to nuclear DNA in the left ventricles was lower in HFD-fed AB mice than in the sham mice (Figure 3c). Consistent with a decline in cardiac mtDNA content, myocardial activity of the mitochondrial enzyme citrate synthase was markedly decreased in hearts from HFD-fed mice after 4 weeks of AB as compared with sham mice (Figure 3d). Importantly, myocardial expression of mitochondrial biogenesis regulatory factors including peroxisome proliferator-activated receptor-γ coactivator-1α (PGC-1α), nuclear respiratory factor-1 (NRF-1), mitochondrial transcription factor A (TFAM) was markedly reduced in HFD-fed AB mice as compared with sham mice (Figures 3e and g). Quantitative real-time PCR (Figure 3i, P<0.05) and western blot (Figure 3k) analysis showed higher Sirt3 mRNA expression in HFD-fed AB mice as compared with sham mice. Importantly, treatment with apelin attenuated AB-induced changes in mitochondrial ultrastructure and density (Figures 3a and b), mtDNA (Figure 3c), activity of citrate synthase (Figure 3d) and expression of PGC-1α, NRF-1, TFAM (Figures 3e and g). As shown in Figure 3i, abundant Sirt3 mRNA expression was observed in apelin-treated sham or AB mice as compared with vehicle-treated sham or AB mice, respectively. Immunoblot analysis confirmed increased Sirt3 expression in apelin-treated AB mice as compared with vehicle-treated AB mice (Figure 3k).

Mitochondrial abnormalities in heart failure linked to obesity: effects of apelin. (a) Representative electron micrographs of cardiac tissues from vehicle- or apeline-treated HFD-fed mice subjected to sham or AB for 4 weeks: M, mitochondria; Myo, myofilaments; L, lipid droplets; arrow, swelling; asterisks, structural disruption, original magnifications × 3000 or × 10 000. The ultrastructural injury in cardiac tissue from sham and AB mice treated with vehicle or apelin was evaluated by electron microscopy. (b) Quantitative analysis of mitochondrial density in heart tissues based on analysis of electron micrographs (n=5 animals per group). (c) Real-time PCR mtDNA/nDNA ratios (n=5), (d) myocardial citrate synthase activity, (e–g) myocardial expression of mitochondrial biogenesis-related genes. (i) Real-time reverse transcriptase (RT)-PCR analysis and (k) western blot analysis of Sirt3 expression in vehicle- (n=6) or apeline-treated (n=6) HFD-fed mice after 4 weeks of AB. *P<0.05; ***P<0.001 vs sham-vehicle; §P<0.05; §§§P<0.001 vs AB-vehicle.
Cardiometabolic phenotype of HFD-fed apelin KO mice
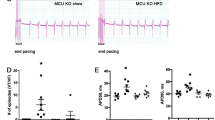
We next investigated the cardiometabolic profile of apelin KO and wild-type (WT) mice exposed to a HFD for 18 weeks. Echocardiographic assessment of LV function revealed that HFD resulted in an increased nterventricular septum thickness and LV posterior wall thickness and significantly reduced fractional shortening and ejection fraction, suggesting impaired cardiac function, as compared with WT mice (Figures 4a and d). Analysis of myocardial metabolic changes in HFD-fed apelin KO mice revealed decreased FA oxidation and increased glucose oxidation compared with HFD-fed WT mice (Figures 4e and f). As shown in Figures 5a and c, downregulation of FA metabolism in HFD-fed apelin KO mice was associated with a reduction in expression of PGC-1α, NRF-1 and TFAM. Importantly, myocardial expression of CPT1, a key enzyme of mitochondrial FA uptake in mitochondria, was significantly decreased in HFD-fed apelin KO mice as compared with in HFD-fed WT mice (Figure 5d). In contrast to HFD-fed WT animals, HFD-fed apelin KO mice exhibited lower Sirt3 mRNA expression (Figure 5e) and Sirt3 protein level (Figure 5f).

Cardiometabolic phenotype of apelin KO mice. Echocardiographic evaluation of (a) interventricular septum thickness (IVST), (b) LV posterior wall thickness (LVPWT), (c) fractional shortening (FS), (d) ejection fraction (EF) in WT (n=6) or apelin KO (n=7) mice. (e) Myocardial FA oxidation and (f) glucose oxidation in WT or apelin KO mice. Results are means±s.e.m. *P<0.05; **P<0.01; ***P<0.001 vs WT mice.

Mitochondrial biogenesis-related gene expression levels in apelin KO mice. (a–e) Real-time reverse transcriptase (RT)-PCR analysis of myocardial expression of PGC-1α, NRF-1, TFAM, CPT1 and Sirt3 in apelin KO or WT mice. (f) Western blot analysis and densitometric evaluation of Sirt3 level in apelin KO (n=5) or WT (n=5) mice. Results are means±s.e.m. *P<0.05; **P<0.01 vs WT mice.
Apelin-induced stimulation of FA oxidation via Sirt3 activation in cardiomyocytes
To further explore the mechanism by which apelin regulates energy metabolism in obese conditions, we examined whether Sirt3 knockdown affects apelin-dependent stimulation of FA oxidation in cardiomyocytes isolated from HFD-fed mice. In agreement with in vivo results, treatment of cardiomyocytes with increasing apelin concentrations (10−6–10−9M) resulted in stimulation of FA oxidation (Figure 6a). We further showed that apelin treatment significantly increased Sirt3 mRNA expression in a dose-dependent manner (Figure 6b) and Sirt3 protein level (Figure 6c). As shown in Figures 6d and e, knockdown of Sirt3 by siRNA abolished apelin-mediated FA oxidation in cardiomyocytes. Consistent with the in vivo results, we also observed increased expression of PGC-1α, TFAM and NRF-1 in cardiomyocytes after 24 h of apelin (10−7M) treatment (Figures 6f and h).

Apelin-induced stimulation of Sirt3-dependent FA oxidation in isolated cardiomyocytes. (a) Effect of increasing doses of apelin (10−9–10−6 M) on FA oxidation in cardiomyocytes isolated from HFD-fed mice. (b) Dose-dependent effect of apelin (10−9–10−7 M) on Sirt3 mRNA expression at 24 h. (c) Western blot analysis of Sirt3 protein level after apelin treatment (10−7M, 24 h) in isolated cardiomyocytes. (d) Representative images of cultured cardiomyocytes after 24 h transfection with Sirt3 siRNA or siRNA negative control (Scramble) with or without apelin stimulation for 1 h. (e) Effect of apelin (10−7 M) on FA oxidation in cultured cardiomyocytes after 24 h transfection with Sirt3 siRNA or Scramble. Data are representative of three independent experiments. (f–h) Quantitative reverse transcriptase (RT)-PCR analysis of PGC-1α, TFAM and NRF-1 expression levels in cardiomyocytes after apelin stimulation (10−7 M) for 24 h. Results are means±s.e.m. (n=3). *P<0.05; **P<0.01 vs control; §P<0.05 vs apelin-treated Scramble.
Discussion
This study demonstrates that transition from cardiometabolic adaptation to maladaptation in obese state is associated with reprogrammed myocardial energy metabolism and impaired mitochondrial integrity. Furthermore, our study provides a novel insight into the role of apelin in the regulation of cardiac energy metabolism during the transition from hypertrophy to heart failure in obesity. Using mouse model combining obesity and heart failure, we show that apelin treatment promotes myocardial FA oxidation, reduces glucose utilization and improves glucose tolerance. In addition, apelin treatment prevents mitochondrial damage and cardiac dysfunction in obesity-related heart failure. Finally, we show that apelin-dependent regulation of cardiomyocyte FA oxidation involves Sirt3 activation.
Impaired energy metabolism is the defining characteristic of acute or chronic cardiac events.10, 11, 12 Although the adult heart uses FA for >70% of its energy supply, the cardiac metabolic machinery is highly flexible, allowing rapid switch of substrate utilization in response to a variety of stresses.11,12,25 It has been shown that a shift toward glucose preference is favorable for sustaining energy production in acute ischemic conditions.9,26 Indeed, in terms of oxygen requirement, myocardial ATP production from glucose is less oxygen consuming than from FA.23 This initial adaptive response is beneficial for maintaining ATP levels in the face of diminished mitochondrial oxidative phosphorylation.24,27 However, the role of metabolic reprogramming as an adaptive versus a maladaptive response in obesity-related heart failure remains obscure. This study shows that transition from hypertrophy to heart failure is associated with changes in energy substrate utilization. We found that mice exposed to a HFD for 18 weeks exhibit cardiac hypertrophy with preserved contractile function suggesting cardiac adaptation to increased adiposity. In these conditions, FA is a major substrate for energy homeostasis. However, pressure overload-induced cardiac dysfunction leads to decreased capacity for FA oxidation and accelerated glucose metabolism in HFD-fed mice. Our results suggest that increased glucose metabolism was unable to maintain cardiac function in response to chronic pressure overload in obese state. These data are consistent with the general concept that downregulation of FA oxidation pathway is linked to decline in cardiac performance in the advanced heart failure.25,28,29 Importantly, we demonstrate that apelin has a key role in the maladaptive cardiac and metabolic responses. Indeed, chronic treatment with apelin stimulates FA utilization and decreases glucose oxidation in concert with preservation of cardiac contractile function in HFD-fed mice subjected to pressure overload. In contrast, cardiac dysfunction in apelin KO obese mice is associated with decreased myocardial FA utilization and increased glucose oxidation. Recent studies have reported that apelinergic system contributes to maintaining contractile function in failing heart. Apelin-deficient mice develop severe heart failure in response to pressure overload and manifest progressive cardiac dysfunction with ageing.18 We also found that apelin treatment reduces fasting plasma glucose and improves glucose intolerance in obese heart failure mice. These data suggest that in pathologic conditions combining heart failure and increased adiposity, the promotion of FA oxidation is able to maintain contractile function and metabolic homeostasis. This notion is compatible with previous data demonstrating that prolonged decrease in FA utilization and accelerated glucose oxidation is associated with deleterious cardiac remodeling and function.29 Interestingly, Iwanaga et al.30 reported unchanged apelin expression in the compensatory hypertrophy; however, both apelin and APJ were decreased in the progression of cardiac dysfunction. Recently, Koguchi et al.31 also reported downregulation of apelin/APJ expression in end-stage of heart failure. Moreover, exogenous apelin ameliorates cardiac dysfunction and myocardial remodeling and restores apelin/APJ expression in chronic heart failure.31 Similarly, in apelin KO mice, a continuous infusion of apelin reversed pressure overload-induced decreased contractility18 suggesting that beneficial effects of apelin may at least in part be explained by restoration of apelin/APJ signaling pathways. In a recent important report, Japp et al.32 demonstrated that short-term apelin infusion caused peripheral and coronary vasodilatation, reduced cardiac preload and afterload, and increased cardiac output in humans. These hemodynamic effects appeared to be preserved in patients with heart failure. They concluded that APJ agonism may have potential therapeutic benefits in patients with heart failure. However, recently Scimia et al.33 have reported that APJ may act as a dual receptor in failing heart. Genetic loss of APJ confers resistance to chronic pressure overload by markedly reducing hypertrophy and heart failure. In contrast, mice lacking apelin remain sensitive, suggesting an apelin-independent function of APJ. Thus, the beneficial effect may be obtained not by general apelin receptor agonism, but rather by selective inhibiting the ability of APJ to respond to mechanical stretch or by blocking its interaction with molecules that initiate pathological signaling cascades.33 Further studies are required to determine whether the downregulation of APJ expression selectively in cardiomyocytes could contribute to myocardial energy metabolism.
The mechanisms underlying the development of obesity-related heart failure are complex, and not well understood. Several studies have provided convincing evidence that mitochondrial dysfunction may be an important event in the development of heart failure.34,35 Our data show that transition from hypertrophy to heart failure is associated with mitochondrial damage and reduced expression of the key regulators of mitochondrial biogenesis including PGC-1α, NRF-1 and TFAM. As a consequence, these mitochondrial defects could lead to impaired ATP production and contractile dysfunction. One important determinant of the capacity to oxidize energy substrates is the ability to utilize oxygen in failing heart. The maximal capacity to utilize oxygen is determined by the mass of mitochondria and by their intrinsic activity. Our study shows that apelin treatment for 4 weeks markedly increases mtDNA content and citrate synthase activity, an enzyme marker of mitochondrial mass. In addition, apelin treatment prevents pressure overload-induced mitochondrial damage and decreases in PGC-1α, NRF-1 and TFAM mRNA expression levels in HFD-fed mice, suggesting an important role of apelin in the mitochondrial integrity in heart failure paired with obesity. Recent study has demonstrated that treatment of rats with apelin for 2 weeks increases mitochondrial content through PGC-1β activation, but not PGC-1α in skeletal muscle.36 It should be noted, however, that this study evaluated the effects of apelin in rats in physiological conditions. Our study show that in pathological situations apelin regulates cardiac metabolism via SIRT3-dependent mechanism. Among seven members of the sirtuin family, Sirt3 is of particular interest with regard to mitochondrial function because it is localized primarily in mitochondria.37 In HFD-fed mice subjected to pressure overload, treatment with apelin increases mitochondrial SIRT3 expression. In contrast, apelin KO mice exhibited lower Sirt3 mRNA and protein levels as compared with WT mice. Sirt3 has been shown to regulate multiple metabolic processes, including FA oxidation and ATP production.38 Importantly, we show that knockdown of Sirt3 by siRNA abolishes apelin-mediated FA oxidation in isolated cardiomyocytes suggesting that apelin regulates cellular metabolism via SIRT3-dependent pathway. These data are in line with a recent study demonstrating apelin-dependent amelioration of diabetic cardiomyopathy via Sirt3 signaling pathways.22 Activation of Sirt3 pathways may be particularly important in the context of obesity-related cardiac remodeling, as transition to heart failure in overweight is associated with profound metabolic alterations. In conclusion, our studies highlight the importance of cardiac energy metabolism in the transition from hypertrophy to heart failure in obese state and suggest that apelin may represent a potential therapeutic target for obesity-related cardiometabolic complications.
References
Taegtmeyer H . Energy metabolism of the heart: from basic concepts to clinical applications. Curr Probl Cardiol 1994; 19: 59–113.
Lopaschuk GD, Belke DD, Gamble J, Itoi T, Schonekess BO . Regulation of fatty acid oxidation in the mammalian heart in health and disease. Biochim Biophys Acta 1994; 1213: 263–276.
Van der Vusse GJ, van Bilsen M, Glatz JF . Cardiac fatty acid uptake and transport in health and disease. Cardiovasc Res 2000; 45: 279–293.
Visser FC . Imaging of cardiac metabolism using radiolabelled glucose, fatty acids and acetate. Coron Artery Dis 2001; 12 (Suppl 1): S12–S18.
Hendrickson SC St, Louis JD, Lowe JE, Abdel-aleem S . Free fatty acid metabolism during myocardial ischemia and reperfusion. Mol Cell Biochem 1997; 166: 85–94.
Katz AM . Metabolism of the failing heart. Cardioscience 1993; 4: 199–203.
Lopaschuk GD, Stanley WC . Glucose metabolism in the ischemic heart. Circulation 1997; 95: 313–315.
Tian R, Abel ED . Responses of GLUT4-deficient hearts to ischemia underscore the importance of glycolysis. Circulation 2001; 103: 2961–2966.
Essop MF, Opie LH . Metabolic therapy for heart failure. Eur Heart J 2004; 25: 1765–1768.
Lopaschuk GD, Ussher JR, Folmes CD, Jaswal JS, Stanley WC . Myocardial fatty acid metabolism in health and disease. Physiol Rev 2010; 90: 207–258.
Stanley WC, Lopaschuk GD, Hall JL, McCormack JG . Regulation of myocardial carbohydrate metabolism under normal and ischaemic conditions. Potential for pharmacological interventions. Cardiovasc Res 1997; 33: 243–257.
Abel ED, Litwin SE, Sweeney G . Cardiac remodeling in obesity. Physiol Rev 2008; 88: 389–419.
Romacho T, Elsen M, Röhrborn D, Eckel J . Adipose tissue and its role in organ crosstalk. Acta Physiol 2014; 210: 733–753.
Nakamura K, Fuster JJ, Walsh K . Adipokines: a link between obesity and cardiovascular disease. J Cardiol 2014; 63: 250–259.
Blüher M . Adipose tissue dysfunction contributes to obesity related metabolic diseases. Best Pract Res Clin Endocrinol Metab 2013; 27: 163–177.
Kleinz MJ, Davenport AP . Emerging roles of apelin in biology and medicine. Pharmacol Ther 2005; 107: 198–211.
Foussal C, Lairez O, Calise D, Pathak A, Guilbeau-Frugier C, Valet P et al. Activation of catalase by apelin prevents oxidative stress-linked cardiac hypertrophy. FEBS Lett 2010; 584: 2363–2370.
Kuba K, Zhang L, Imai Y, Arab S, Chen M, Maekawa Y et al. Impaired heart contractility in apelin gene-deficient mice associated with aging and pressure overload. Circ Res 2007; 101: e32–e42.
Boucher J, Masri B, Daviaud D, Gesta S, Guigne C, Mazzucotelli A et al. Apelin, a newly identified adipokine up-regulated by insulin and obesity. Endocrinology 2005; 146: 1764–1771.
Dray C, Knauf C, Daviaud D, Waget A, Boucher J, Buleon M et al. Apelin stimulates glucose utilization in normal and obese insulin-resistant mice. Cell Metab 2008; 8: 437–445.
Attane C, Foussal C, Le Gonidec S, Benani A, Daviaud D, Wanecq E et al. Apelin treatment increases complete Fatty Acid oxidation, mitochondrial oxidative capacity, and biogenesis in muscle of insulin-resistant mice. Diabetes 2012; 61: 310–320.
Zeng H, He X, Hou X, Li L, Chen JX . Apelin gene therapy increases myocardial vascular density and ameliorates diabetic cardiomyopathy via upregulation of sirtuin 3. Am J Physiol Heart Circ Physiol 2014; 306: H585–H597.
Sinatra ST . Metabolic cardiology: an integrative strategy in the treatment of congestive heart failure. Altern Ther Health Med 2009; 15: 44–52.
Tian R, Abel ED . Responses of GLUT4-deficient hearts to ischemia underscore the importance of glycolysis. Circulation 2001; 103: 2961–2966.
Yan J, Young ME, Cui L, Lopaschuk GD, Liao R, Tian R . Increased glucose uptake and oxidation in mouse hearts prevent high fatty acid oxidation but cause cardiac dysfunction in diet-induced obesity. Circulation 2009; 119: 2818–2828.
Nagoshi T, Yoshimura M, Rosano GM, Lopaschuk GD, Mochizuki S . Optimization of cardiac metabolism in heart failure. Curr Pharm Des 2011; 17: 3846–3853.
Allard MF, Emanuel PG, Russell JA, Bishop SP, Digerness SB, Anderson PG . Preischemic glycogen reduction or glycolytic inhibition improves postischemic recovery of hypertrophied rat hearts. Am J Physiol 1994; 267: H66–H74.
Stanley WC, Recchia FA, Lopaschuk GD . Myocardial substrate metabolism in the normal and failing heart. Physiol. Rev. 2005; 85: 1093–1129.
Augustus AS, Buchanan J, Park TS, Hirata K, Noh HL, Sun J et al. Loss of lipoprotein lipase-derived fatty acids leads to increased cardiac glucose metabolism and heart dysfunction. J Biol Chem 2006; 281: 8716–8723.
Iwanaga Y, Kihara Y, Takenaka H, Kita T . Down-regulation of cardiac apelin system in hypertrophied and failing hearts: Possible role of angiotensin II-angiotensin type 1 receptor system. J Mol Cell Cardiol 2006; 41: 798–806.
Koguchi W, Kobayashi N, Takeshima H, Ishikawa M, Sugiyama F, Ishimitsu T . Cardioprotective effect of apelin-13 on cardiac performance and remodeling in end-stage heart failure. Circ J 2012; 76: 137–144.
Japp AG, Cruden NL, Barnes G, van Gemeren N, Mathews J, Adamson J et al. Acute cardiovascular effects of apelin in humans:Potential role in patients with chronic heart failure. Circulation 2010; 121: 1818–1827.
Scimia MC, Hurtado C, Ray S, Metzler S, Wei K, Wang J et al. APJ acts as a dual receptor in cardiac hypertrophy. Nature 2012; 488: 394–398.
Rosca MG, Vazquez EJ, Kerner J, Parland W, Chandler MP, Stanley W et al. Cardiac mitochondria in heart failure: decrease in respirasomes and oxidative phosphorylation. Cardiovasc Res 2008; 80: 30–39.
Sharov VG, Todor AV, Silverman N, Goldstein S, Sabbah HN . Abnormal mitochondrial respiration in failed human myocardium. J Mol Cell Cardiol 2000; 32: 2361–2367.
Frier BC, Williams DB, Wright DC . The effects of apelin treatment on skeletal muscle mitochondrial content. Am J Physiol Regul Integr Comp Physiol 2009; 297: R1761–R1768.
Schwer B, North BJ, Frye RA, Ott M, Verdin E . The human silent information regulator (Sir)2 homologue hSIRT3 is a mitochondrial nicotinamide adenine dinucleotide-dependent deacetylase. J Cell Biol 2002; 158: 647–657.
Ahn BH, Kim HS, Song S, Lee IH, Liu J, Vassilopoulos A, Deng CX, Finkel T . A role for the mitochondrial deacetylase Sirt3 in regulating energy homeostasis. Proc Natl Acad Sci USA 2008; 105: 14447–14452.
Acknowledgements
This work was supported by grants from the National Institute of Health and Medical Research (INSERM), Fondation Lefoulon-Delalande, Fondation de France, Région Midi-Pyrénées and Association Française d'Etudes et de Recherches sur l’Obésité.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on International Journal of Obesity website
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/
About this article

Cite this article
Alfarano, C., Foussal, C., Lairez, O. et al. Transition from metabolic adaptation to maladaptation of the heart in obesity: role of apelin. Int J Obes 39, 312–320 (2015). https://doi.org/10.1038/ijo.2014.122
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ijo.2014.122
This article is cited by
-
Potential clinical biomarkers and perspectives in diabetic cardiomyopathy
Diabetology & Metabolic Syndrome (2023)
-
Protamine as a barrier against the angiogenic effect of insulin: a possible role of apelin
Scientific Reports (2023)
-
Apelin expression deficiency in mice contributes to vascular stiffening by extracellular matrix remodeling of the aortic wall
Scientific Reports (2021)
-
Epicardial adipose tissue as a metabolic transducer: role in heart failure and coronary artery disease
Heart Failure Reviews (2017)
-
Betaine Down Regulates Apelin Gene Expression in Cardiac and Adipose Tissues of Insulin Resistant Diabetic Rats Fed by High-Calorie Diet
International Journal of Peptide Research and Therapeutics (2017)
