
Abstract
RASopathies comprise a group of disorders clinically characterized by short stature, heart defects, facial dysmorphism, and varying degrees of intellectual disability and cancer predisposition. They are caused by germline variants in genes encoding key components or modulators of the highly conserved RAS-MAPK signalling pathway that lead to dysregulation of cell signal transmission. Germline changes in the genes encoding members of the RAS subfamily of GTPases are rare and associated with variable phenotypes of the RASopathy spectrum, ranging from Costello syndrome (HRAS variants) to Noonan and Cardiofaciocutaneous syndromes (KRAS variants). A small number of RASopathy cases with disease-causing germline NRAS alterations have been reported. Affected individuals exhibited features fitting Noonan syndrome, and the observed germline variants differed from the typical oncogenic NRAS changes occurring as somatic events in tumours. Here we describe 19 new cases with RASopathy due to disease-causing variants in NRAS. Importantly, four of them harbored missense changes affecting Gly12, which was previously described to occur exclusively in cancer. The phenotype in our cohort was variable but well within the RASopathy spectrum. Further, one of the patients (c.35G>A; p.(Gly12Asp)) had a myeloproliferative disorder, and one subject (c.34G>C; p.(Gly12Arg)) exhibited an uncharacterized brain tumour. With this report, we expand the genotype and phenotype spectrum of RASopathy-associated germline NRAS variants and provide evidence that NRAS variants do not spare the cancer-associated mutation hotspots.
Similar content being viewed by others

Introduction
More than three decades ago, the Rat Sarcoma Virus (RAS) genes were first described and identified as key players in tumorigenesis. Harvey-, Kirsten- and neuroblastoma-RAS (HRAS, KRAS, and NRAS) encode for the three highly homologous members of the mammalian RAS subfamily of small monomeric GTPases.1 These membrane-bound proteins bind to guanine nucleotides and function as molecular switches by cycling between the active, GTP-bound state and inactive, GDP-bound state, and control processes ranging from cell growth and differentiation to cell survival and energetic metabolism.1, 2
RAS genes are frequently altered in malignancies. While changes in individual RAS genes have different prevalence in the diverse human cancers, their oncogenic variants almost invariably affect codons 12, 13, and 61, leading to constitutive activation of the transducer and upregulation of downstream signalling pathways.3 Germline alterations in RAS genes and genes encoding for proteins controlling RAS function or participating in the mitogen-activated protein kinase (MAPK) pathway, a major signalling cascade activated by RAS proteins, have been implicated in a group of developmental disorders. These disorders are collectively called ‘RASopathies’ based on their common pathogenetic mechanism and broadly overlapping phenotypic features.4, 5 Noonan syndrome (NS, OMIM 163950), the most common RASopathy, is characterized by distinctive craniofacial features, heart defects, short stature, and variable developmental delay. Furthermore, feeding difficulties, cryptorchidism in males, easy bruising, lymphatic dysplasia, ectodermal anomalies, chest deformities, and other skeletal abnormalities are also frequently associated. Intellectual abilities range from normal to mild impairment.6 To date, germline variants in more than ten genes have been causally linked to NS and clinically related phenotypes.6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16 Cardiofaciocutaneous syndrome (CFCS, OMIM 115150) and Costello syndrome (CS, OMIM 218040) are rarer RASopathies. Both disorders are genetically distinct from NS, and are characterized by a more severe health impact such as more pronounced developmental and intellectual deficits.5, 17 Ectodermal involvement (for example, hyperkeratotic skin and thin sparse hair) is generally more florid in CFCS,18 while dermal features (loose skin with deep palmar creases and cutaneous papilloma) and coarse facial appearance are distinctive of CS, which also exhibit an increased risk of neoplasia, both benign and malignant.19, 20
RASopathy-associated germline variants in the HRAS, KRAS, and NRAS genes are relatively rare. Specific alterations in HRAS cause CS, while changes in KRAS are associated with a variable phenotype and account for ~2-3% of cases with NS and 3–5% of cases with CFCS.21 In 2010, heterozygous germline variants in NRAS were identified as a rare cause of NS.22 A total number of 18 patients from twelve unrelated families have been reported, to date.22, 23, 24, 25, 26, 27 Similar to what had been observed for RASopathy-associated germline alterations in KRAS, the reported NRAS alterations causing NS do not affect the oncogenic hotspots and appear to be functional hypomorphs compared to the cancer-associated somatic variants.22, 23, 25 In contrast, changes in HRAS causing CS typically occur at two well-known oncogenic hotspots (that is, Gly12, Gly13), which has been postulated to explain the high incidence of tumours in patients with this disorder.28
Individuals with NS have a slightly increased risk to develop neoplasias, depending on the specific gene variant. Recurrently observed NS-associated neoplasias include juvenile myelomonocytic leukemic (JMML), giant cell lesions of the jaws, rhabdomyosarcoma and brain tumours.4, 21, 29, 30 JMML, a severe myeloproliferative disorder of infancy, is particularly associated with a dysregulated RAS-MAPK pathway, with somatic changes in PTPN11, KRAS and NRAS, and loss of Neurofibromin function accounting for a majority of cases.31 Somatic PTPN11, KRAS, and NRAS variants found in JMML are assumed to have a stronger impact on cellular physiology, which implies their incompatibility with life, if these variants occur in germline.5 Interestingly, there is one report about an individual with a germline c.38G>A (p.(Gly13Asp)) variant in NRAS and a clinical diagnosis of JMML.32
Herein we report 19 novel patients with disease-causing germline NRAS variants from 13 unrelated families, further expanding the RASopathy-associated spectrum of germline NRAS alterations and providing a more complete picture of their associated clinical spectrum.
Materials and methods
Subjects and phenotyping
Patients with a clinical diagnosis of a RASopathy who were found to carry a disease-causing variant in the NRAS gene and who have not been reported previously were eligible for this study. We included a total 19 affected individuals, thereof nine sporadic cases (patients 1, 2, 6, 7, 8, 9, 11, 12, and 13; see Table 1) and four familial cases (cases 3, 4, 5, and 10; see Table 1) with in total 10 affected individuals. Patients were clinically assessed by experienced clinical geneticists. All patients described here had molecular confirmation in a diagnostic setting, except for the clinically affected family member 5–3, who declined genetic testing. The standardized patient data were collected via the NSEuroNet database (www.nseuronet.com) and entered by the referring clinician. This database uses a comprehensive online questionnaire to facilitate the collection and improve the standardization of clinical information. Furthermore, all variants reported in this study were entered into the Leiden Open Variation Database (www.LOVD.nl/NRAS; patient IDs 00100620, 00100623-00100639).
The clinical data and samples for genetic testing were obtained from all individuals with informed consent of the patients’ parents/legal guardians or the patients themselves, and all studies were performed in accordance with the Declaration of Helsinki and the national legal regulations. Specific written permission was obtained for the use clinical photographs for publication in this report.
Molecular analysis
NRAS (LRG_92, identical to NM_002524.3) variants were identified by routine genetic testing of the known RASopathy genes using Sanger sequencing or targeted resequencing as well as whole-exome sequencing (WES) (Patients 10-1, 10-2, 11, and 12) of DNA extracted from venous blood samples, except for case 2, which was diagnosed prenatally on DNA extracted from CVS. Where available, DNA samples from parents and additional affected family members were investigated for the variants discovered in the index case in order to demonstrate the de novo occurrence of the variant or co-segregation with the RASopathy phenotype (see Table 1). In selected cases, DNA samples from various tissue sources were examined in addition to leukocyte DNA to prove that the variant was not a clonal event in hematopoietic cells. These sources included buccal cells and finger nails in patient 1, urine and saliva in patient 8, finger nails in patient 12, and fibroblasts in patient 13.
Functional characterization of the NS-causing NRASThr58Ile variant
The missense changes resulting in the p.Thr58Ile (c.173C>T) and p.Gly12Val (c.35G>T) amino acid substitutions were introduced by site-directed mutagenesis in an N-terminal FLAG-tagged human NRAS cDNA cloned in pFLAG-CMV vector.
HEK293T cells were cultured in Dulbecco’s modified Eagle’s medium (EuroClone, Milan, Italy) supplemented with 10% heat-inactivated FBS (EuroClone). Cells were transfected at 70–80% confluence with each NRAS construct, using Fugene6 transfection reagent (Promega, Madison, WI, USA). Twelve hours after transfection, cells were serum-starved for 18 h, and stimulated with EGF (30 ng/ml, Invitrogen, Carlsbad, CA, USA) for 5 min or left unstimulated.
ERK and AKT activation status was assessed by immunoblotting with anti-phospho-p44/42 ERK (p.Thr202/p.Tyr204) and anti p-AKT (p.Ser473) antibodies (Cell Signaling, Danvers, MA, USA). Membranes were then stripped and reprobed with anti-p44/42 (ERK) and anti-AKT antibodies (Cell Signaling) for protein normalization. To evaluate FLAG-NRAS and β-tubulin protein levels, 10 ng of total lysates were immunoblotted with anti-FLAG and anti-β-tubulin (Sigma Aldrich, St Louis, MO, USA) antibodies.
For GTP-bound Ras pull-down assays, HEK293T cells were transfected with each NRAS expression plasmids or left untransfected. 12 h after transfection, cells were serum-starved for 18 h and collected in ice-cold Mg2+ lysis/wash buffer (Millipore, Temecula, CA, USA), supplemented with protease inhibitor cocktail (Sigma Aldrich). Lysates were centrifuged at 4 °C, and each supernatant containing 500 μg of proteins was incubated with 10 μl of RAF1 Ras-binding domain-conjugated agarose beads (Millipore), rotated at 4 °C for 30 min, washed three times with lysis/wash buffer, boiled for 5 min in Laemmli buffer under reducing conditions, and separated by SDS/PAGE. To evaluate the activation level of NRAS proteins the membrane was probed with an anti-FLAG antibody. The same antibody was used on aliquots of corresponding cell lysates to normalize experiments.
All images and tables presented here were prepared using Adobe Illustrator CS6, Adobe Photoshop CS6 and Microsoft Office 2010.
Results
Spectrum of NRAS variants
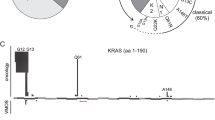
We identified a total of 19 affected individuals from 13 unrelated families with disease-causing variants in NRAS. In nine sporadic cases, de novo occurrence of the variant was proven by testing of parental DNA, if available. In four instances of familial NS, the respective NRAS substitution was demonstrated to co-segregate with the phenotype. In total, nine different heterozygous NRAS variants were identified. All changes affected highly conserved residues (Figure 1). Of note, the variants segregating in the four families, c.71T>A (p.(Ile24Asn)), c.149C>T (p.(Thr50Ile)) and c.179G>A (p.(Gly60Glu)), had all been documented in NS, previously.22, 23 Six changes, c.34G>C (p.(Gly12Arg)), c.35G>A (p.(Gly12Asp)), c.34G>A (p.(Gly12Ser)), c.35G>T (p.(Gly12Val)), c.112-1_113dupGGA (p.(Glu37dup)), and c.173C>T (p.(Thr58Ile)), had not been described as RASopathy-associated germline variants in NS previously and occurred exclusively in sporadic cases. De novo occurrence was demonstrated for the missense changes c.34G>C (p.(Gly12Arg)), c.35G>A (p.(Gly12Asp)), c.34G>A (p.(Gly12Ser)), and c.35G>T (p.(Gly12Val)). The germline nature of the variants c.34G>C (p.(Gly12Arg)), c.35G>A (p.(Gly12Ser)), c.34G>A (p.(Gly12Asp)), and c.173C>T (p.(Thr58Ile) was further supported by their occurrence in a heterozygous pattern in non-hematopoietic tissues. Regarding the c.112-1_113dupGGA (p.(Glu37dup)) variant, the father was unavailable for testing, and in the case of c.173C>T (p.(Thr58Ile)) the father was found to have a mosaicism for the variant in his peripheral blood leukocytes (Supplementary Figure S1).

NRAS domain structure and spectrum of germline variants. Upper panel: The sequences of amino acids 1–70 of three RAS proteins are aligned. RASopathy-associated and germline variants in NRAS are represented above the sequence alignment. Length of bars and numerals at their basis indicate the numbers of unrelated observations for each disease-causing variant. Bold font indicates disease-causing variants described previously, which were also identified in our cohort. Novel changes identified in this study are written in italics. Variants previously reported to cause NS but not found in our cohort are written in normal font. *Notably, the reported case with the c.38G>A (p.(Gly13Asp)) variant had a primary diagnosis of JMML and mild Noonan-like features were only noted retrospectively. Bars and numerals below the alignment represent the numbers of somatic variants associated with cancer at the respective position (according to COSMIC, Sept 2016). Lower panel: Domain structure of entire NRAS protein. Numbers represent amino acids, black boxes: coding exons, dark grey boxes: functional domains, HVR, hypervariable region; P-L, P-Loop; SW I, switch I region; SW II, switch II region.
Functional characterization of the NS-causing NRASThr58Ile variant
The c.173C>T (p.(Thr58Ile)) variant was inherited from the apparently unaffected father, who showed mosaicism for the substitution. While the same amino acid substitution in KRAS has been demonstrated to perturb protein function and upregulate signal flow through the MAPK cascade,33, 34 no sign suggestive of RASopathy was documented in the proband’s father. Consequently, the impact of this variant on NRAS function and ERK and AKT signalling was assessed in transiently transfected HEK293T cells to further validate the causative role of the variant. As expected, overexpression of the wild-type protein resulted in enhanced ERK and AKT phosphorylation following EGF stimulation (Figure 2a). By contrast, ectopic expression of either NRASThr58Ile or NRASGly12Val promoted enhanced activation of ERK and AKT, even in absence of stimulation (Figure 2a). Consistent with these data, higher level of NRASThr58Ile in its GTP-bound, active form was observed, which was however lower compared to the NRASGly12Val protein (Figure 2b). Overall, these data support the pathogenetic role of the c.173C>T (p.(Thr58Ile)) substitution.

Functional characterization of the NS-causing NRASThr58Ile variant: (a) ERK1/2 and AKT phosphorylation assays. HEK293T cells were transfected with the indicated FLAG-tagged NRAS construct. Following starvation (18 h) and EGF stimulation (30 ng/ml, 5 min), ERK (anti p-ERK1/2) and AKT (anti p-AKT) phosphorylation levels were evaluated. Normalization for transfection efficiency and total protein amount was determined by the use of anti-β-tubulin and anti-FLAG antibodies, respectively. Representative blots of three performed experiments are shown. (b) Determination of basal GTP-bound NRAS level in 293 T cells transiently expressing wild-type or mutant FLAG-tagged NRAS. Normalization was determined by using an anti-FLAG antibody on aliquots of the corresponding cell lysates. Assays were performed in serum-free condition. A representative blot of three performed experiments is shown.
Clinical presentation of patients with NRAS alterations
Our cohort of individuals with heterozygous RASopathy-associated NRAS variants consisted of nine males and ten females with a median age of 7.1 years (range: 3 months to 50 years). The majority (15 patients) had a clinical diagnosis of NS. However, in two patients the proposed diagnosis based on clinical assessment was CFCS, and in one patient aged 1 year CS was initially suspected (see patient 12 below). One case was a prenatal diagnosis where genetic testing for a RASopathy was performed because of a large hygroma colli. The clinical data are summarized in Table 1.
All assessed subjects had craniofacial features that were classified as typical or suggestive for a RASopathy. Short and broad or webbed neck was found in 94%, and ocular ptosis in 82% of the cases. Ten out of 17 patients (59%) had cardiac anomalies with hypertrophic cardiomyopathy (HCM) being present in 35% of the cases. Of note, septal defects and pulmonary stenosis (PST) were reported only in a minority of our cohort (12 and 6%, respectively). Similarly, short stature was present in only 4 out of 15 cases (27%). Motor delay and intellectual or learning disabilities were recorded in 38 and 42% of patients, respectively. Learning disabilities, if present, were of mild degree except for patient 7 who had severe global developmental delay (discussed below). In addition, the majority of patients had prenatal abnormalities (69%) with polyhydramnios being reported in 46% of the cases. Nuchal oedema and fetal chylothorax/hydrops as prenatal abnormalities were seen in 15 and 23%, respectively. Cryptorchidism occurred in 63% of affected males. Bleeding diathesis was reported in 3 out of 15 individuals, but only one patient had a confirmed coagulopathy (von Willebrand disease, patient 8). Neoplasias were observed in two individuals: one patient had a JMML-like myeloproliferative disorder (patient 13) and another patient had a brain tumour of unclear aetiology (patient 12). Selected patients are presented in more detail in the following.
Patient 1—(c.34G>A, p.(Gly12Ser)) presented with typical physical signs of NS: facial anomalies (Figure 3), left ventricular hypertrophy, atrial and ventricular septal defects (corrected by surgery), a persistent ductus arteriosus, and short stature. She exhibited marked muscular hypotonia in infancy. Her development was mildly delayed; she was able to walk freely with 2.3 years and spoke 2–3 word sentences at the age of 2.5 years. Her language skills at last evaluation at the age of 5.5 years were classified as normal for age. Intellectual development was also tested normal for age, but deficits were noted in spatial performance and attention. She had regular follow-up in the paediatric haemato-oncology during infancy, and had no signs of myeloproliferative disorder. The heterozygous NRAS variant was found in leukocyte DNA, confirmed in DNA from buccal mucosal epithelial cells and finger nail keratinocytes, and was absent in the parents (Supplementary Figure S1).

Clinical photographs documenting the craniofacial phenotype of patients with heterozygous NRAS variants. The patients’ ID numbers (according to Table 1) are given in the upper left corner of each photograph. Written consent was obtained from the patient or his/her legal guardian for publication of the images. P1: p.(Gly12Ser), 4 years old; P3–1, P3–2, P3-3: p.(Ile24Asn), familial observation with affected family members shown at age 10, 13, and 36 years, respectively; P5-1: p.(Gly60Glu), 17 years old; P7: p.(Glu37dup), 6 month old; P10-1: p.(Gly60Glu), 18 years old; P11: p.(Thr50Ile), 11 years old; P13: p.(Gly12Asp), 1 year old.
Patient 2—(c.35G>T, p.(Gly12Val)) was a fetus with a suspected RASopathy based on the finding of a very large cystic nuchal hygroma (14.3 mm) at 12 weeks of gestation. Progressive hydrops foetalis developed, eventually leading to intrauterine death at 22 weeks of gestation. No autopsy data were available but hydropic appearance of the fetus was reported. No heart defect could be detected. Fetal karyotyping was performed on a CVS sample and resulted in a normal male karyotype. The heterozygous NRAS variant was demonstrated in DNA from cultured chorionic villus fibroblasts. Parents were negative for the variant.
Patient 5–2—(c.179G>A, p.(Gly60Glu)) is an affected child of a three generation family with obvious features of NS in her father and grandfather. She died on the first day of life from severe hydrops. Post-mortem MRI revealed bilateral pleural effusions, a small pericardial effusion and left ventricular hypertrophy.
Patient 7—(c.112-1_113dupGGA, p.(Glu37dup)) came to medical attention because of developmental issues and an atrial septal defect. She had a prenatal history of fetal nuchal oedema, polyhydramnios, an intracerebral arachnoid cyst, and bilateral hydronephrosis detected by antenatal ultrasound. Subcutaneous oedema was noted at birth. She required gavage feeding due to severe failure to thrive. In addition, she developed epilepsy and a sleep disorder. She had duplex collecting system and recurrent urinary tract infections due to ureteroceles requiring surgical treatment. An MRI in the newborn period revealed thinning of the cortex with white matter changes, consistent with hypoxic ischemic encephalopathy. At the age of 2 years and 5 months, her stature was below third centile (−2.69 SD) and she had a short webbed neck, shield chest, wide-spaced nipples, while facial anomalies were less typical (Figure 3). She was suspected to have CFCS because of her severe developmental delay, hypotonia and ectodermal abnormalities. She continued to demonstrate head lag at age 4 years old and unsupported sitting was not possible. Other features included microcephaly (43 cm at 2y5 m (−3.27 SD)), ocular abnormalities, including nystagmus and strabismus, hearing deficits and haemangioma. The NRAS variant was detected in leukocyte DNA. Additionally, she was identified to harbour a 1.24 Mb gain from chromosome 22q11.23 detected by array CGH analysis (arr[GRCh38] 22q11.23(23408742_24595702)x3). This duplication was considered to have an impact on phenotypic complexity and modification of the RASopathy phenotype, specifically the severity of neurodevelopmental issues. Neither genetic abnormality was detected in the mother, but the father was unavailable for testing. Because of the complex phenotype and its probable modification by the 22q11.23 duplication and hypoxic ischemic encephalopathy this patient was excluded from the aggregate clinical tables and statistics.
Patient 8—(c.173C>T, p.(Thr58Ile)) presented with suggestive facial features of NS, ocular ptosis, strabismus, and pectus excavatum, but without cardiac anomalies and short stature. She displayed muscular hypotonia and mild motor delay. The germline origin of the NRAS variant was supported by its identification in DNA from urine, saliva, and peripheral blood. The patient's father was found to have a mosaicism for the variant in his peripheral blood (Supplementary Figure S1).
Patient 11—(c.149C>T, p.(Thr50Ile)) presented with typical facial features of a RASopathy, and mitral valve prolapse. At age 11, her height was in the low normal range (−1.47 SD). The suspected clinical diagnosis was CFCS because of typical facial features with substantial supraorbital hypoplasia and prominent ectodermal findings (curly hair, sparse eyebrows, keratosis pilaris, facial keratosis and multiple nevi), while her motor and intellectual development was only mildly delayed (IQ 65). Her perinatal history was characterized by multiple complications that may have contributed to her developmental problems. She had perinatal asphyxia (Apgar scores 2/5/7) and required mechanical ventilation for 17 days. Also, she developed sepsis and portal vein thrombosis after umbilical vein catheter. She was discharged from the hospital at 45 days of life. Brain MRI at 4 months of age revealed non-obstructive hydrocephalus requiring ventriculoperitoneal shunting at 6 months. The NRAS variant was demonstrated in leukocyte DNA.
Patient 12—(c.34G>C, p.(Gly12Arg)) had a RASopathy-typical facial phenotype, short webbed neck, macrocephaly, and HCM. She was born at 36 weeks of gestation after a pregnancy complicated by polyhydramnios. Birth weight was increased at 4,145 g. The postnatal period was complicated by prolonged hyperinsulinemic hypoglycemia, severe feeding difficulties, including oral aversion and respiratory difficulties due to upper airway collapse. She was treated with a percutaneous gastrostomy and a tracheostomy with continuous positive airway pressure ventilation. A brain MRI at 8 months of age revealed an expansive lesion of 0.8x0.7x0.6 cm in the hypothalamus proposed to represent a hamartoma or lipoma (Supplementary Figure S2). Her EEG was normal, and she has been followed by a clinical neurologist. Her early motor development was delayed (unsupported sitting at age 10 months). She had sparse, thin hair, full lips, deep palmar and plantar creases, several small nevi and one café-au-lait spot. On the basis of the clinical findings, CS was suggested, but no HRAS change was found. The heterozygous NRAS variant was detected by WES. It was confirmed in DNA obtained from finger nail keratinocytes by Sanger sequencing, and was not found in DNA samples from both parents. On follow-up by the age of 1 year and 9 months her initial problems improved. The tracheostomy could be removed at 1 year and 8 months. She was able to make up for her motor delay and started walking at 1 year and 5 months. Neither surgery nor biopsy have been performed so far for her hypothalamic tumour because of the absence of any endocrinological or specific neurological deficits attributable to this lesion.
Patient 13—(c.35G>A, p.(Gly12Asp)) is a male born after an uneventful pregnancy except for mild fetal pyelocalyceal dilatation (7 mm) of the left kidney detected on ultrasound scans. Birth weight at term was 4060 g, body length 51 cm, and head circumference 37.5 cm. He had no feeding problems and no lymphatic anomalies. A few café-au-lait spots were noted on neonatal examination, and he was therefore followed up because of a suspected diagnosis of neurofibromatosis. At the age of 3 months, a blood cell count showed leucocytosis with 3% blast cells on a peripheral blood smear, mild anemia and thrombocytopenia. On examination, he showed four café-au-lait spots over 0.5 cm diameter and three melanocytic nevi on lower limbs and buttocks, macrocephaly with suggestive facial features of NS (Figure 3), short neck but no thorax deformities, hepato-splenomegaly (4 and 3 cm, respectively), normal male genitalia with descended testes, and normal hands and feet. Bone marrow biopsy showed findings consistent with a myeloproliferative disorder. A brain MRI showed subdural fluid accumulations but no brain structural anomalies. Cardiological examination and echocardiography were normal. The heterozygous NRAS variant was identified in leukocyte DNA and later confirmed in DNA extracted from skin fibroblasts, but was absent in both parents. Although he met clinical and analytical/cytological criteria for the diagnosis of JMML, in view of the stability of the blood cell counts and absence of complications it was decided not to start treatment and follow him up closely. Until the age of 11 months, this child has remained stable haematologically, without proliferative phenomena or infectious complications. He was not short in stature, and his motor development was still within the normal range. A detailed developmental assessment was not possible yet due to his young age.
Discussion
We describe 19 new patients with a RASopathy due to germline NRAS variants. The size of this cohort exceeds the total number of previously reported patients harbouring disease-causing germline variants in NRAS.22, 23, 24, 25, 26, 27 We report de novo as well as familial occurrence of the already known changes c.71T>A (p.(Ile24Asn)), c.149C>T (p.(Thr50Ile)), and c.179G>A (p.(Gly60Glu)), confirming these codons to be hotspots for RASopathy-associated germline changes in NRAS. In addition, we identified six novel alterations, affecting residues Gly12, Glu37, and Thr58 to cause a RASopathy phenotype.
The assumption that the novel sequence variants, c.34G>C, 35G>A, 34G>A, c.35G>T (p.(Gly12Arg/Asp/Ser/Val) respectively), c.112-1_113dupGGA (p.(Glu37dup)), and c.173C>T (p.(Thr58Ile)), indeed represent disease-causing changes is supported by several lines of evidence: All amino acid substitutions affect highly conserved residues of NRAS, and they are located at or near known hotspots for somatic or disease-causing germline NRAS variants, respectively (Figure 1). Additionally, all novel NRAS changes described here have previously been reported as disease-causing variants in the other RAS genes KRAS and/or HRAS, and the activating effects have been shown in functional studies (present data and ref 22, 33, 34, 35, 36, 37, 38, 39).
Missense changes affecting p.Gly12 in NRAS or other RAS proteins are among the most common somatic changes in cancer, and result in impaired GTPase activity and GAP resistance, leading to constitutive activation.33, 34, 39 While for position p.Glu37 in NRAS neither somatic nor germline variants had previously been described, the same duplication p.(Glu37dup) in HRAS has been reported in a case of CS.38 HRASGlu37dup was shown to predominate in the active GTP-bound state due to lower intrinsic GTPase activity and complete resistance to GAP, but its impact on signal flux through the MAPK and PI3K-AKT cascades was found to be milder compared to HRASG12V, because of reduced binding affinities for effector proteins.38 Sequence changes resulting in a p.(Thr58Ile) missense variant have been described eight times in HRAS as a cause of CS and six times in KRAS mainly associated with NS (www.nseuronet.com). Additionally, this particular alteration has occasionally been observed in RAS isoforms as a somatic variant in various types of malignancy (COSMIC, Forbes, et al.40). Previous functional characterization of the KRASThr58Ile variant provided evidence for a mild overactivated behavior of the GTPase and enhanced activation of downstream effectors.33, 34 Here we demonstrated that the same amino acid change in NRAS underlies NS and has similar gain-of-function effects, promoting a shift towards the constitutively activated GTP-bound conformation, and enhanced ERK and AKT activation basally, even though with a lesser extent compared to the oncogenic NRASGly12Val variant (Figure 2). The four novel RASopathy-associated NRAS changes affecting Gly12 represent well-known oncogenic changes that occur with decreasing prevalence (p.Gly12Asp>p.Gly12Ser>p.Gly12Val>p.Gly12Arg) as somatic alterations in various tumours, particularly in hematopoietic and lymphoid malignancies (COSMIC database, Forbes, et al.40). The previously reported spectrum of NRAS RASopathy-associated germline variants appeared to spare the classical oncogenic hotspots Gly12, Gly13, and Gln61, suggesting that the occurrence of oncogenic NRAS alterations in the germline might lead to embryonic lethality.22 This is therefore the first report to show that apparent non-mosaic germline changes in NRAS affecting one of these hotspots for oncogenic variants may be compatible with life and lead to a clearly recognizable RASopathy phenotype. An overlap of the spectrum of germline variants with those hotspots for oncogenic changes has very rarely been observed for KRAS variants causing CFCS,10 while it is regularly observed for HRAS variants leading to CS. This has been correlated with the significantly increased tumour risk in this particular type of RASopathy. Consistently, one of the present subjects carrying a missense change at codon 12 had a tumour-like lesion in the hypothalamus, the precise nature of which was not delineated by the time of this report (Supplementary Figure S2), and another one had a JMML-like myeloproliferative disorder. Of note, JMML and JMML-like myeloproliferative disorders (MPDs) are particularly related to NS and to somatic changes in RASopathy genes, including NRAS.30, 31, 41, 42, 43 Also, for the stillbirth with the variant c.35G>T (p.(Gly12Val), patient 2), we do not have sufficient data to exclude a myeloproliferative disease or other kind of neoplasia.
There is one previously published case report of suggestive germline status for the NRAS variant c.38G>A (p.(Gly13Asp)) in a patient with JMML.32 The infant who was affected by JMML was reported to have short stature and dysmorphic features reminiscent of NS. In this case, the disease-causing variant was not only in buccal cells and hair bulbs, but also in fibroblasts, suggesting that the alteration was indeed not restricted to hematopoietic tissue. Nevertheless, a mosaic status for this variant cannot be excluded, in principle. Notably, another patient affected by ALPS and no clinical signs of NS was erroneously reported to have the same NRAS variant in the germline, based on the demonstration of this change in DNA from a buccal swab.44 During follow-up, the change was not retrieved in DNA samples from hair bulbs and a repeat buccal swab, thus disproving the germline status (personal communication JB Oliveira, February 2010). This is pointing to the difficulties in assessing the germline status of a DNA sequence change, which is also a possible limitation in the data presented here. It is known that hematopoietic cells may migrate and reside in non-hematopoietic tissues, including buccal mucosa and, more surprisingly, finger nails.45, 46 However, cultured fibroblasts that could be tested in some of the patients are devoid of contamination with hematopoietic cells.26, 41, 42 Together with the systemic clinical features observed in all our patients, it is likely that NRAS changes affecting Gly12 were indeed of germline origin.
Notably, post-zygotic NRAS variants have also been observed in mosaic RASopathies, including systematized and isolated forms of congenital melanocytic naevi and keratinocytic epidermal naevi. Disease-associated missense variants mainly affect the residues Gly13 and Gln61, but there is at least one observation of the p.Gly12Asp variant in a keratinocytic epidermal naevus.47 Patient 13, who was found to carry the same variant in the germline, did not show a prominent skin phenotype. The reason for such phenotypic variability with the same change in the mosaic status versus germline is still unclear.
Our findings suggest that clinically obvious RASopathy features constitute the major phenotype caused by activating NRAS variants occurring in the germline, whereas hematological abnormalities do not invariably emerge even in the presence of variants at oncogenic hotspots. Comparing the clinical symptoms recorded in our patients to previously reported cases with disease-causing germline NRAS variants, we generally found high congruency regarding the frequency of clinical findings in various organ systems. Taking together, the clinical data from a total of 37 affected individuals allows us to further delineate the RASopathy phenotype associated with NRAS variants (Supplementary Table S1). Cardiac anomalies are present in approximately half of cases, but contrasting to what is generally observed in NS, PST is present in only 14% of patients with NRAS-related disease as compared to 50–60% in the overall NS population.6 HCM is the most prevalent cardiac finding in our NRAS cohort affecting 29% of individuals. This is a slightly higher percentage than reported in the overall NS population (20%).6 Septal defects represent the third most common cardiac feature, occurring in 9% of cases with NRAS-related NS.48 The typical craniofacial appearance is commonly seen and indistinguishable from NS of other aetiologies. Short or webbed neck and ocular ptosis were highly prevalent among patients with NRAS variants (80 and 63%, respectively). Notably, short stature was only present in 42% of patients, which is less frequent than in NS in general. Motor delay is relatively common (39%), while learning disabilities are observed in a similar frequency as in NS of other genetic aetiologies (27%). Cryptorchidism in males and the occurrence of prenatal abnormalities are also quite frequent (63%) and comparable to their general frequency in NS.6
While all previously reported patients had a clinical diagnosis of NS, three patients in this cohort were diagnosed as having CFCS or CS based on their clinical features. For patient 7, a working diagnosis of CFCS was initially made primarily based on her striking craniofacial features, profound hypotonia, significant developmental delay, and feeding issues. In addition to the NRAS variant, she was found to have a 1.24 Mb duplication on chromosome 22q11.23, corresponding to the region between LCR-F and LCR-H, whose de novo occurrence could not be confirmed. Similar 22q11.23 duplications have been found in individuals with variable clinical features, including muscular hypotonia, developmental delay, and seizures, with de novo occurrence but also inherited from apparently healthy parents.49, 50 It is therefore possible that this copy number change might contribute to the severity of the developmental phenotype and some atypical features described in this patient. Similarly, patient 11, who carried the c.149C>T (p.(Thr50Ile)) variant, was clinically classified as having CFCS because of significantly delayed motor development, mild intellectual disability (IQ of 65), and prominent ectodermal abnormalities (keratosis pilaris, multiple nevi, sparse eyebrows and curly, sparse hair). Of note, the same disease-causing variant had previously been reported in patients with a NS phenotype and no or only mild developmental problems.22, 25 Patient 12 (c.34G>C, p.(Gly12Arg)) showed severe perinatal complications typically seen in newborns with CS, including polyhydramnios, neonatal hyperinsulinemic hypoglycemia, severe feeding difficulties, and respiratory problems. Furthermore, she had deep palmar and plantar creases and a tumour-like lesion of unknown aetiology in the hypothalamus. Her motor development was delayed. This constellation prompted the diagnosis of CS, but the patient was very young at last follow-up and the phenotype may still change with age.
Considering the relatively severe expression of NS in patient 1 (c.34G>A, p.(Gly12Ser)) and patient 13 (c.35G>A, p.(Gly12Asp)), the CS-like neonatal presentation in patient 12 (c.34G>C, p.(Gly12Arg)), and intrauterine death of patient 2 (c.35G>T, p.(Gly12Val)), it is tentative to speculate that this may be related to the more severe functional impact of these variants at Gly12. Among these, the p.(Gly12Val) change that was associated with intrauterine death is the most common oncogenic variant at codon 12 in NRAS and demonstrated to represent the strongest effect on downstream signalling, while the others have somewhat weaker activating potential.35, 36 Similarly, gradual differences in the phenotypic severity have also been proposed for different CS-associated HRAS variants affecting Gly12.51 Regarding clinical management, it has to be considered that changes in NRAS corresponding to CS-associated HRAS changes may also share a similar risk of malignancy as seen in CS. However, the number of patients is still limited and the observation period for the patients reported here was too short to draw definite conclusions about possible genotype-phenotype correlations.
In summary, we have presented the largest cohort to date of patients with disease-causing germline NRAS variants and their association with the typical clinical phenotype of RASopathies. We showed that alterations affecting Gly12 might be compatible with life when occurring in the germline. This is comparable to the germline variant spectrum previously documented in HRAS,37 but different to the one in KRAS, where RASopathy-associated germline variants and somatic changes occurring in cancer hardly overlap.52 Our findings emphasize the obvious RASopathy phenotype associated with activating germline NRAS variants, thus challenging the germline status of oncogenic NRAS changes in previous reports with hematological phenotypes only. More data is required to ascertain the risk of malignancy in patients with oncogenic NRAS variants in the germline.
References
Barbacid M : ras genes. Annu Rev Biochem 1987; 56: 779–827.
Karnoub AE, Weinberg RA : Ras oncogenes: split personalities. Nat Reviews Mol Cell Biol 2008; 9: 517–531.
Bos JL : ras oncogenes in human cancer: a review. Cancer Res 1989; 49: 4682–4689.
Tartaglia M, Gelb BD, Zenker M : Noonan syndrome and clinically related disorders. Best Pract Res Clin Endocrinol Metab 2011; 25: 161–179.
Tartaglia M, Gelb BD : Disorders of dysregulated signal traffic through the RAS-MAPK pathway: phenotypic spectrum and molecular mechanisms. Ann N Y Acad Sci 2010; 1214: 99–121.
Roberts AE, Allanson JE, Tartaglia M, Gelb BD : Noonan syndrome. Lancet 2013; 381: 333–342.
Aoki Y, Niihori T, Banjo T et al: Gain-of-function mutations in RIT1 cause Noonan syndrome, a RAS/MAPK pathway syndrome. Am J Hum Genet 2013; 93: 173–180.
Yamamoto GL, Aguena M, Gos M et al: Rare variants in SOS2 and LZTR1 are associated with Noonan syndrome. J Med Genet 2015; 52: 413–421.
Cordeddu V, Yin JC, Gunnarsson C et al: Activating mutations affecting the dbl homology domain of SOS2 cause noonan syndrome. Hum Mutat 2015; 36: 1080–1087.
Nava C, Hanna N, Michot C et al: Cardio-facio-cutaneous and Noonan syndromes due to mutations in the RAS/MAPK signalling pathway: genotype-phenotype relationships and overlap with Costello syndrome. J Med Genet 2007; 44: 763–771.
Sarkozy A, Carta C, Moretti S et al: Germline BRAF mutations in Noonan, LEOPARD, and cardiofaciocutaneous syndromes: molecular diversity and associated phenotypic spectrum. Hum Mutat 2009; 30: 695–702.
Flex E, Jaiswal M, Pantaleoni F et al: Activating mutations in RRAS underlie a phenotype within the RASopathy spectrum and contribute to leukaemogenesis. Hum Mol Genet 2014; 23: 4315–4327.
Cordeddu V, Di Schiavi E, Pennacchio LA et al: Mutation of SHOC2 promotes aberrant protein N-myristoylation and causes Noonan-like syndrome with loose anagen hair. Nat Genet 2009; 41: 1022–1026.
Martinelli S, De Luca A, Stellacci E et al: Heterozygous germline mutations in the CBL tumor-suppressor gene cause a Noonan syndrome-like phenotype. Am J Hum Genet 2010; 87: 250–257.
Perez B, Mechinaud F, Galambrun C et al: Germline mutations of the CBL gene define a new genetic syndrome with predisposition to juvenile myelomonocytic leukaemia. J Med Genet 2010; 47: 686–691.
Gripp KW, Aldinger KA, Bennett JT et al: A novel rasopathy caused by recurrent de novo missense mutations in PPP1CB closely resembles Noonan syndrome with loose anagen hair. Am J Med Genet A 2016; 170: 2237–2247.
Rauen KA : The RASopathies. Annu Rev Genomics Hum Genet 2013; 14: 355–369.
Roberts A, Allanson J, Jadico SK et al: The cardiofaciocutaneous syndrome. J Med Genet 2006; 43: 833–842.
Hennekam RC : Costello syndrome: an overview. Am J Med Genet C Semin Med Genet 2003; 117C: 42–48.
Gripp KW : Tumor predisposition in Costello syndrome. Am J Med Genet C Semin Med Genet 2005; 137C: 72–77.
Zenker M : Clinical manifestations of mutations in RAS and related intracellular signal transduction factors. Curr Opin Pediat 2011; 23: 443–451.
Cirstea IC, Kutsche K, Dvorsky R et al: A restricted spectrum of NRAS mutations causes Noonan syndrome. Nat Genet 2010; 42: 27–29.
Runtuwene V, van Eekelen M, Overvoorde J et al: Noonan syndrome gain-of-function mutations in NRAS cause zebrafish gastrulation defects. Dis Models Mech 2011; 4: 393–399.
Digilio MC, Lepri F, Baban A et al: RASopathies: Clinical Diagnosis in the First Year of Life. Mol Syndromol 2011; 1: 282–289.
Denayer E, Peeters H, Sevenants L, Derbent M, Fryns JP, Legius E : NRAS Mutations in Noonan Syndrome. Mol Syndromol 2012; 3: 34–38.
Kraoua L, Journel H, Bonnet P et al: Constitutional NRAS mutations are rare among patients with Noonan syndrome or juvenile myelomonocytic leukemia. Am J Med Genet A 2012; 158A: 2407–2411.
Ekvall S, Wilbe M, Dahlgren J et al: Mutation in NRAS in familial Noonan syndrome—case report and review of the literature. BMC Med Genet 2015; 16: 95.
Estep AL, Tidyman WE, Teitell MA, Cotter PD, Rauen KA : HRAS mutations in Costello syndrome: detection of constitutional activating mutations in codon 12 and 13 and loss of wild-type allele in malignancy. Am J Med Genet A 2006; 140: 8–16.
Kratz CP, Franke L, Peters H et al: Cancer spectrum and frequency among children with Noonan, Costello, and cardio-facio-cutaneous syndromes. Br J Cancer 2015; 112: 1392–1397.
Strullu M, Caye A, Lachenaud J et al: Juvenile myelomonocytic leukaemia and Noonan syndrome. J Med Genet 2014; 51: 689–697.
Niemeyer CM : RAS diseases in children. Haematologica 2014; 99: 1653–1662.
De Filippi P, Zecca M, Lisini D et al: Germ-line mutation of the NRAS gene may be responsible for the development of juvenile myelomonocytic leukaemia. Br J Haematol 2009; 147: 706–709.
Schubbert S, Zenker M, Rowe SL et al: Germline KRAS mutations cause Noonan syndrome. Nat Genet 2006; 38: 331–336.
Gremer L, Merbitz-Zahradnik T, Dvorsky R et al: Germline KRAS mutations cause aberrant biochemical and physical properties leading to developmental disorders. Hum Mutat 2011; 32: 33–43.
Ahmadian MR, Zor T, Vogt D et al: Guanosine triphosphatase stimulation of oncogenic Ras mutants. Proc Natl Acad Sci USA 1999; 96: 7065–7070.
Fasano O, Aldrich T, Tamanoi F, Taparowsky E, Furth M, Wigler M : Analysis of the transforming potential of the human H-ras gene by random mutagenesis. Proc Natl Acad Sci USA 1984; 81: 4008–4012.
Aoki Y, Niihori T, Kawame H et al: Germline mutations in HRAS proto-oncogene cause Costello syndrome. Nat Genet 2005; 37: 1038–1040.
Gremer L, De Luca A, Merbitz-Zahradnik T et al: Duplication of Glu37 in the switch I region of HRAS impairs effector/GAP binding and underlies Costello syndrome by promoting enhanced growth factor-dependent MAPK and AKT activation. Hum Mol Genet 2010; 19: 790–802.
Wey M, Lee J, Jeong SS, Kim J, Heo J : Kinetic mechanisms of mutation-dependent Harvey Ras activation and their relevance for the development of Costello syndrome. Biochemistry 2013; 52: 8465–8479.
Forbes SA, Beare D, Gunasekaran P et al: COSMIC: exploring the world's knowledge of somatic mutations in human cancer. Nucleic Acids Res 2015; 43: D805–D811.
Caye A, Strullu M, Guidez F et al: Juvenile myelomonocytic leukemia displays mutations in components of the RAS pathway and the PRC2 network. Nat Genet 2015; 47: 1334–1340.
Stieglitz E, Taylor-Weiner AN, Chang TY et al: The genomic landscape of juvenile myelomonocytic leukemia. Nat Genet 2015; 47: 1326–1333.
Kratz CP, Niemeyer CM, Castleberry RP et al: The mutational spectrum of PTPN11 in juvenile myelomonocytic leukemia and Noonan syndrome/myeloproliferative disease. Blood 2005; 106: 2183–2185.
Oliveira JB, Bidere N, Niemela JE et al: NRAS mutation causes a human autoimmune lymphoproliferative syndrome. Proc Natl Acad Sci USA 2007; 104: 8953–8958.
Hiramoto R, Imamura T, Muramatsu H et al: Serial investigation of PTPN11 mutation in nonhematopoietic tissues in a patient with juvenile myelomonocytic leukemia who was treated with unrelated cord blood transplantation. Int J Hematol 2015; 102: 719–722.
Imanishi D, Miyazaki Y, Yamasaki R et al: Donor-derived DNA in fingernails among recipients of allogeneic hematopoietic stem-cell transplants. Blood 2007; 110: 2231–2234.
Hafner C, Toll A, Gantner S et al: Keratinocytic epidermal nevi are associated with mosaic RAS mutations. J Med Genet 2012; 49: 249–253.
Marino B, Digilio MC, Toscano A, Giannotti A, Dallapiccola B : Congenital heart diseases in children with Noonan syndrome: An expanded cardiac spectrum with high prevalence of atrioventricular canal. J Pediatr 1999; 135: 703–706.
Coppinger J, McDonald-McGinn D, Zackai E et al: Identification of familial and de novo microduplications of 22q11.21-q11.23 distal to the 22q11.21 microdeletion syndrome region. Hum Mol Genet 2009; 18: 1377–1383.
Chang J, Zhao L, Chen C et al: Pachygyria, seizures, hypotonia, and growth retardation in a patient with an atypical 1.33 Mb inherited microduplication at 22q11.23. Gene 2015; 569: 46–50.
Lorenz S, Petersen C, Kordass U, Seidel H, Zenker M, Kutsche K : Two cases with severe lethal course of Costello syndrome associated with HRAS p.G12C and p.G12D. Eur J Med Genet 2012; 55: 615–619.
Zenker M, Lehmann K, Schulz AL et al: Expansion of the genotypic and phenotypic spectrum in patients with KRAS germline mutations. J Med Genet 2007; 44: 131–135.
Acknowledgements
This work was supported by E-Rare (NSEuroNet to MRA, HC, MT, and MZ), AIRC (IG17583 to MT), Italian Ministry of Health (RF-2011-02349938 and Ricerca Corrente 2016 to MT), Telethon (13107 to MT), German Federal Ministry of Education and Research (BMBF) NSEuroNet (FKZ 01GM1602A to MZ and MRA); GeNeRARe (FKZ 01GM1519A to MZ and MRA); Deutsche Forschungsgemeinschaft (ZE524/10-1 to MZ); LGS Synaptogenetics, CBBS to IS; Raine Clinician Research Fellowship to GB; Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP) 2011/17299-3; 2013/08028-1 and Conselho Nacional de Pesquisa (CNPq) 304130/2016-8 to DB.
Databases
The databases include COSMIC database:http://cancer.sanger.ac.uk/cosmic.40 NSEuroNet database: https://nseuronet.com/php/. Leiden Open Variation Database: http://databases.lovd.nl/shared/variants/NRAS/unique.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on European Journal of Human Genetics website
Rights and permissions
About this article

Cite this article
Altmüller, F., Lissewski, C., Bertola, D. et al. Genotype and phenotype spectrum of NRAS germline variants. Eur J Hum Genet 25, 823–831 (2017). https://doi.org/10.1038/ejhg.2017.65
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2017.65
This article is cited by
-
Consensus reporting guidelines to address gaps in descriptions of ultra-rare genetic conditions
npj Genomic Medicine (2024)
-
Neurofibromatosis-Noonan syndrome and growth deficiency in an Iranian girl due to a pathogenic variant in NF1 gene
Human Genomics (2023)
-
Assessing reproducibility of inherited variants detected with short-read whole genome sequencing
Genome Biology (2022)
-
Variants of SOS2 are a rare cause of Noonan syndrome with particular predisposition for lymphatic complications
European Journal of Human Genetics (2021)

{kind=link}
{kind=link}