
Abstract
Dihydroartemisinin (DHA) is a widely used antimalarial drug isolated from the plant Artemisia annua. Recent studies suggested that DHA has antitumor effects utilizing its reactive oxygen species (ROS) yielding mechanism. Here, we reported that DHA is inhibitory on lipopolysaccharide (LPS)-induced osteoclast (OC) differentiation, fusion and bone-resorption activity in vitro. Intracellular ROS detection revealed that DHA could remarkably increase ROS accumulation during LPS-induced osteoclastogenesis. Moreover, cell apoptosis was also increased by DHA treatment. We found that DHA-activated caspase-3 increased Bax/Bcl-2 ratio during LPS-induced osteoclastogenesis. Meanwhile, the translocation of apoptotic inducing factor (AIF) and the release of cytochrome c from the mitochondria into the cytosol were observed, indicating that ROS-mediated mitochondrial dysfunction is crucial in DHA-induced apoptosis during LPS-induced osteoclastogenesis. In vivo study showed that DHA treatment decreased OC number, prevents bone loss, rescues bone microarchitecture and restores bone strength in LPS-induced bone-loss mouse model. Together, our findings indicate that DHA is protective against LPS-induced bone loss through apoptosis induction of osteoclasts via ROS accumulation and the mitochondria-dependent apoptosis pathway. Therefore, DHA may be considered as a new therapeutic candidate for treating inflammatory bone loss.
Similar content being viewed by others

Main
Bone is a dynamic organ that undergoes continuous remodeling throughout life. Bone homeostasis is maintained by a balanced bone-resorbing and bone-forming process. In this process, hematopoietic stem cells or monocytes/macrophage progenitor cell-derived osteoclasts (OCs) are mainly responsible for bone resorption.1 Abnormal OC function is associated with numerous diseases, and most of them are due to excessive osteoclastic activity. These diseases include osteoporosis, rheumatoid arthritis and periodontitis.2, 3 Two of the most important regulating factors during OC differentiation are receptor activator of nuclear factor κB ligand (RANKL) and macrophage-colony-stimulating factor (M-CSF).4, 5 Binding of RANKL to RANK results in the initiation of the TNF receptor-associated factor 6 signaling, which activates nuclear factor-κB, Akt and MAP kinase (ERk, JNK and p-38), and eventually leads to the proliferation, differentiation and maturation of OCs.6, 7
Lipopolysaccharide (LPS) is an important component of the outer membrane of Gram-negative bacteria. In LPS-induced bone loss, many factors are involved including local host response, prostanoids and cytokine production, inflammatory cell recruitment and OC activation.8, 9, 10 Experimental evidence have shown that LPS-mediated inflammation is highly dependent on reactive oxygen species (ROS) and the associated downstream MAPK signaling pathways including ERK, JNK and p-38.11, 12 ROS has been shown having an important role in the process of OC differentiation, survival, activation and bone resorption.13, 14, 15, 16 It has also been proved that ROS production in OC and intracellular hydrogen peroxide accumulation is critical for osteoclastogenesis and skeletal homeostasis.17 Recently, a study reported that LPS induces OC formation via the ROS-mediated JNK and STAT3 pathway, which could be blocked by peroxiredoxin II.18
Dihydroartemisinin (DHA) is the main active metabolite isolated from the plant Artemisia annua. DHA has been widely used as first-line therapeutics against falciparum malaria.19 Recent evidence suggested that DHA has antitumor effects because of its unique cytotoxicity mechanism.20 In particular, studies reported that DHA is pro-apoptotic in tumor cell lines regarding breast and prostate cancer.21, 22 Although the detailed mechanism of DHA cytotoxicity and pro-apoptotic effects is not fully understood, DHA-mediated ROS production has a central role.23, 24 However, the effect of DHA on bone health has not been studied.
In the present study, we reported that DHA could attenuate LPS-induced OC differentiation, fusion and bone-resorption activity in vitro. Our data showed that DHA-induced cell apoptosis during LPS-induced osteoclastogenesis via intracellular ROS generation and mitochondria-mediated pathways. DHA administration in LPS-induced mouse models decreased OC number and reversed bone loss in vivo.
Results
DHA inhibits LPS-induced OC differentiation
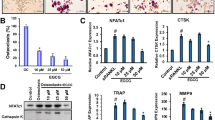
We first evaluated the toxicity of LPS and found that LPS can decrease cell viability (100 ng/ml), increase cell apoptosis rate (1000 ng/ml) and induce cell cycle arrest (10 ng/ml) in bone marrow macrophages (BMMs; Supplementary Figure S1). However, LPS at the dose of 1000 ng/ml could stimulate the highest OC number in BMMs (Supplementary Figure S4). On the basis of this, we use LPS of 1000 ng/ml in the following studies. The chemical formula of DHA is shown (Figure 1a). The cytotoxicity of DHA was evaluated by CCK-8 assay and cell apoptosis assay, suggesting that DHA dose at 100 μg/ml had no cytotoxicity in BMMs (Supplementary Fig. S5). Tartrate-resistant acid phosphatase (TRAP) stain was then performed to test the effects of DHA on LPS-induced osteoclastogenesis (Figure 1b). TRAP-positive (red) cells with more than three nuclei were counted as osteoclasts. Quantification analysis revealed that DHA significantly decreased LPS-induced OC number (P<0.01; Figure 1c). In accordance, qPCR results also showed that mRNA expression of OC-specific marker including TRAP, Ctsk, MMP-9 and DC-STMAP were significantly suppressed by DHA treatment (P <0.01; Figure 1d).

DHA inhibits LPS-induced osteoclast differentiation. (a) Chemical formula of DHA. (b) Representative images of preosteoclasts stained for TRAP (red) treated with LPS (1μg/ml), DHA (100 μg/ml) or both for 3 days. Scale bar represents 200 μm. (c) Quantification of TRAP (+) cells with more than three nuclei in each well (96-well plate). The data in the figures represent the averages±S.D. (d) Relative mRNA expression levels of TRAP, Ctsk, MMP-9 and DC-STAMP of BMMs in different groups on day 3. The data in the figures represent the averages±S.D. Significant differences between the treatment and control groups are indicated as *P<0.05 or **P<0.01
DHA inhibits LPS-induced OC fusion and actin ring formation
To further study the effects of DHA on LPS-induced OC formation, actin cytoskeleton and focal adhesion stain was performed for better visualization of the actin ring. From the stain results we figured out that actin ring formation was hindered by DHA treatments (Figure 2a). Consistent with TRAP stain results, OC number was decreased by DHA treatment (P<0.01; Figure 2b). In addition, average nucleus number of OCs was also significantly decreased by DHA treatments (P<0.01), suggesting that OC fusion was also inhibited (Figure 2c).

DHA inhibits LPS-induced osteoclast fusion and actin ring formation. (a) Representative images of focal and adhesion staining of preosteoclasts treated with LPS (1 μg/ml), DHA (100 μg/ml) or both for 3 days. Scale bar represents 100 μm. (b) Quantification of osteoclasts (nuclei⩾3) in each well (96-well plate). The data in the figures represent the averages±SD. (c) Quantification of average nucleus number in each group. The data in the figures represent the averages±S.D. Significant differences between the treatment and control groups are indicated as *P<0.05 or **P<0.01
DHA inhibits LPS-induced OC bone-resorption activity
To further test the effects of DHA on LPS-induced OC function, pit formation assay was performed on bone slices and osteo surface (Figures 3a and c). BMMs were seeded on bone slices and osteo surface according to group settings. After 5 days’ incubation, cells were removed and resorption pits were quantified. Consistent with previous results, DHA showed strong inhibitory effects on OC resorption activity on both bone slices and osteo surface (P <0.01; Figures 3b and d).

DHA inhibits LPS-induced osteoclast bone-resorption activity. (a) Representative images of preosteoclasts cultured on bovine bone slices treated with LPS (1 μg/ml), DHA (100 μg/ml) or both for 5 days. Scale bar represents 400 μm. (b) Quantification of bone-resorption area on the bone slices. The data in the figures represent the averages±S.D. (c) Representative images of Osteoassay surface 96-well plate after removal of osteoclasts. Scale bar represents 400 μm. (d) Quantification of bone-resorption area on the osteo surface. The data in the figures represent the averages±S.D. Significant differences between the treatment and control groups are indicated as *P<0.05 or **P<0.01
DHA increases intracellular ROS generation and cell apoptosis rate during LPS-induced osteoclastogenesis
To explain the inhibitory effects of DHA on osteoclastogenesis, intracellular ROS was detected using 2′, 7′-dichlorofluorescein diacetate (DCFH)-DA (Figure 4a). BMMs treated with DHA alone showed higher ROS intensity compared with the control group (P<0.01). Cells treated with LPS alone also exhibited upregulated ROS intensity; however, BMMs treated with both LPS and DHA showed significantly higher ROS intensity compared with other groups (Figure 4b). Annexin-V/PI staining was then used to test the effects of DHA on cell apoptosis (Figure 4c). Quantification analysis showed that LPS treatment alone showed the higher early and late cell apoptosis rate compared with control groups (Figures 4d and e). The apoptosis rate induced by LPS was further increased with DHA treatments (P<0.01; Figures 4d and e). The results suggested that DHA could increase intracellular ROS intensity and apoptosis rate in the presence of LPS.

DHA increases intracellular ROS generation and cell apoptosis rate during LPS-induced osteoclastogenesis. (a) Representative images of ROS-positive preosteoclasts treated with LPS (1 μg/ml), DHA (100 μg/ml) or both for 3 days. Scale bar represents 100 μm. (b) Quantification of ROS green fluorescence intensity in each well (96-well plate). The data in the figures represent the averages±S.D. (c) FCM analysis of the cell apoptosis rate of preosteoclasts treated with LPS (1 μg/ml), DHA (100 μg/ml) or both for 3 days. (d) Quantification analysis of the early-stage cell apoptosis rate. The data in the figures represent the averages±S.D. (e) Quantification analysis of the late-stage cell apoptosis rate. The data in the figures represent the averages±S.D. Significant differences between the treatment and control groups are indicated as *P<0.05 or **P<0.01
DHA induces apoptosis in LPS-induced osteoclastogenesis through the mitochondria-dependent pathway
To investigate the underlying mechanisms of DHA-induced apoptosis in LPS-induced osteoclastogenesis, western blot analysis was performed to detect the expression of apoptosis-related proteins in preosteoclasts of different groups after 24-h culture (Figure 5a). As shown in Figure 5b, the protein expression of Bax and activated caspase-3 were remarkably increased, whereas the expression of Bcl-2 was significantly decreased in groups treated with both LPS and DHA. As the accumulation of ROS is crucial for mitochondrial apoptosis,25 we further detected pro-apoptotic proteins including cytochrome c and apoptotic inducing factor (AIF) released from mitochondria to the cytosol (Figure 5c). Notably, when preosteoclasts were treated with LPS and DHA together, the expression level of AIF and cytochrome c decreased in the mitochondria and increased in the cytosol (Figures 5 and d). The results indicated that DHA-induced apoptosis during LPS-induced osteoclastogenesis was associated with mitochondrial dysfunction. It is well recognized that cytochrome c activates apoptosis effector caspase-3, which is consistent with results showed in Figure 5a.26

DHA induces apoptosis in LPS-induced osteoclasts through the mitochondria-dependent pathway. (a) Representative western blot images of cleaved caspase-3, Bax, Bcl-2 and β-actin from preosteoclasts treated with LPS (1 μg/ml), DHA (100 μg/ml) or both for 3 days. (b) Quantification of normalized protein expression intensity of caspase-3, Bax and Bcl-2. The data in the figures represent the averages±S.D. (c) Representative western blot images of AIF, cytochrome c and β-actin in the cytosol; AIF, Cytochrome c and COX-IV in mitochondria. (d) Quantification of normalized protein expression intensity of AIF in the cytosol and mitochondria. The data in the figures represent the averages±S.D. (e) Quantification of normalized protein expression intensity of cytochrome c in the cytosol and mitochondria. The data in the figures represent the averages±S.D. Significant differences between the treatment and control groups are indicated as *P<0.05 or **P< 0.01
DHA reduces LPS-induced osteoclastogenesis and bone loss in vivo
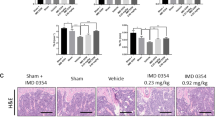
To investigate whether the in vitro effects of DHA still function in vivo, we used LPS-induced bone-loss mouse model. Mice were injected with LPS intraperitoneally and treated with DHA or vehicle. Mice were killed after 4 weeks of treatment. μCT analysis was performed using dissected femurs. For trabecular bone analysis of the distal femur, an upper 3-mm region beginning 0.8 mm proximal to the most proximal central epiphysis of the femur was contoured showed with red dashed box (Figure 6a). Quantification analysis showed that DHA administration in LPS-induced bone-loss mouse models could significantly increase bone mineral density, trabecular bone volume fraction (BV/TV) and trabecular number (Tb. N; P<0.01; Figure 6b). TRAP stain of the distal femur showed that OC surface/bone surface ratio was significantly higher in LPS-treated mice compared with the control group (Figure 6c). DHA administration remarkably decreased the OC surface ratio (Figure 6d). The in vivo results indicated that DHA administration could attenuate LPS-induced osteoclastogenesis and bone loss.

DHA reduces LPS-induced osteoclastogenesis and bone loss in vivo. (a) Representative μCT longitudinal section images of the femurs, cross-sectional view of the distal femurs and reconstructed trabecular structure of the ROI (red dashed box). Color scale bar represents the bone mineral density level. (b) Quantitative μCT analysis of distal femoral volumetric bone mineral density, BV/TV and trabecular number (Tb. N) in each group. The data in the figures represent the averages±S.D. (c) Representative images of histological slides of TRAP stain focusing on the metaphyseal region of the distal femur from mice of different groups. Scale bar represents 800 μm. (d) Quantitative analysis of OC surface/bone surface ratio. The data in the figures represent the averages±S.D. Significant differences between the treatment and control groups are indicated as *P<0.05 or **P<0.01
Discussion
DHA is famous for its efficient therapeutic effects in treating malaria. In recent years, accumulating evidence showed that DHA inhibits cell growth, arrests cell cycle at the G0/G1 phase and induces apoptotic cell death in different kinds of tumor cells.27, 28, 29 It has been proved that the cytotoxicity of DHA is based on ROS and carbon-centered radical generation.30 The role of ROS during RANKL-induced osteoclastogenesis is essential. MAPK including JNK and p-38 are crucial for OC differentiation, and both of these two genes can be activated by ROS.15, 31 Numerous reports were made on the inhibitory effects in osteoclastogenesis of antioxidant compounds.32, 33 Current evidence suggest that most antioxidants could inhibit osteoclastogenesis via scavenging ROS. Although the reports are few, excessive intracellular ROS generation in OCs could also cause oxidative damage and eventually lead to DNA damage and cell apoptosis.34 In this regard, we were curious about the effects of DHA in osteoclastogenesis.
Many kinds of inflammatory bone loss are due to LPS.10 LPS produces a significant amount of ROS during the inflammatory process, which contributes to accelerated OC differentiation and activation.35 The in vitro research of our study showed that LPS could significantly increase the intracellular ROS level during osteoclastogenesis (Figure 4a). When DHA was introduced into the LPS-mediated osteoclastogenesis process, the ROS level was further elevated (Figure 4a). Accordingly, the apoptosis rate increased along with the ROS accumulation (Figures 4d and e). ROS have many sources, including the mitochondrial electron transport chain, xanthine oxidase, cytochrome P-450 enzymes, uncoupled NO synthases and NADPH oxidases.18, 36 As mitochondria are susceptible to oxidative damage, which lead to enhanced mitochondrial ROS generation,37 we further examined the mitochondrial apoptotic factors including cytochrome c and AIF from both inner mitochondria and the cytosol. Cytochrome c in the cytosol activates caspase-8 and caspase-9, which further activate executioner caspase-3 to induce cell apoptosis.38 In our study, DHA treatment together with LPS significantly increased cytochrome c and AIF release from mitochondria to the cytosol and activated caspase-3. In addition, pro-apoptotic protein Bax expression was increased and anti-apoptotic protein Bcl-2 was decreased, leading to an increased ratio of Bax/Bcl-2 (Figures 5a and b). Taken together, these results strongly indicated that DHA induces OC apoptosis in LPS-induced osteoclastogenesis through accumulation of ROS and the mitochondrial apoptotic pathway.
It is interesting to notice that DHA alone could not induce cell apoptosis in BMMs (Supplementary Figure S6); it was only pro-apoptotic in the presence of LPS. To explain this, one should notice that the redox balance in the culture condition regulates in vitro LPS-induced OC formation.39 An antioxidant enhances LPS-induced OC formation, whereas a pro-oxidant reduces it. DHA is known for its induction of iron-dependent oxidative stress and has been proved potential to serve as a redox chemotherapeutic that selectively induces cell apoptosis.40 As ROS generation is necessary during OC differentiation, combined with our results, we concluded that OCs are sensitive to DHA-induced apoptosis because of upregulation of cellular oxidative stress. In contrast, BMMs have low cellular oxidative stress before their differentiation toward OCs, which make them less sensitive to DHA-induced apoptosis. Very recently, two studies reported that DHA could also inhibit RANKL-induced osteoclastogenesis through inhibiting RANKL-induced signaling pathways.41, 42 It is also worthy to test whether the redox balance works in the inhibitory effects of DHA in RANKL-induced osteoclastogenesis.
In summary, the present study demonstrated that DHA treatment during LPS-induced osteoclastogenesis increased cell apoptotic death via accumulation of ROS, increased release of cytochrome c and AIF from mitochondria into the cytosol, activation of caspase-3 and increase in the Bax/Bcl-2 ratio. Moreover, in vivo studies suggested that DHA administration effectively prevented bone loss, rescued bone microarchitecture and restored bone strength in LPS-induced mouse models. Although more research is needed to explore the underlying mechanism, our data imply the protective role of DHA in LPS-induced bone loss, which could be useful in the development of new treatments for patients with inflammatory bone loss.
Materials and Methods
Reagents
Recombinant Mouse RANKL and Recombinant Mouse M-CSF were purchased from R&D Systems (Minneapolis, MN, USA). Antibodies against AIF, cytochrome c, cleaved caspase-3, Bax, Bcl-2 and β-actin were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Osteo Assay Surface for Bone Resorption was purchased from Corning (Corning, NY, USA). Bovine cortical bone slices were obtained from Boineslices.com (Jelling, Denmark). Cell Counting Kit-8 was obtained from Dojindo Molecular Technologies (Dojindo, Japan). The TRAP stain kit was obtained from Sigma-Aldrich (St. Louis, MO, USA). The Actin Cytoskeleton and Focal Adhesion Staining Kit was purchased from Millipore (Darmstadt, Germany). The DCFDA cellular ROS detection assay kit was obtained from ABcam (Cambridge, UK). Alpha minimal essential Medium (α-MEM) and fetal bovine serum (FBS) were purchased from Gibco (Life Technologies, Carlsbad, CA, USA). Penicillin–streptomycin solution was obtained from Hyclone (Thermo Scientific, Waltham, MA, USA). DHA was purchased from Sigma-Aldrich.
Mice
Eight-week-old female C57BL/6 mice were provided by the animal center of Third Military Medical University. All experimental procedures were approved by Third Military Medical University and were performed according to the guidelines of laboratory animal care and use. All efforts were made to reduce the number of animals tested and their suffering. Mice were divided into four groups: phosphate-buffered saline (PBS)-treated (LPS−), DHA (100 mg/kg) only-treated (DHA), LPS (5 mg/kg) only-treated (LPS+) and DHA (100 mg/kg)-treated LPS (5 mg/kg) groups (L+DHA). LPS and DHA dissolved in PBS was injected intraperitoneally three times a week for 4 weeks. Mice were weighed daily, and concentration was calculated for DHA administration. All treated mice were killed by cervical dislocation 1 day after last administration.
In vitro assays for OC differentiation, fusion and function
Bone marrow cells were separated and cultured with M-CSF (50 ng/ml) for 24 h to obtain BMMs. BMMs were cultured in MEM containing 10% FBS and 1% penicillin–streptomycin solution. BMMs were then treated with M-CSF (30 ng/ml) and RNAKL (100 ng/ml) for 36 h to generate preosteoclasts.
For the TRAP stain, preosteoclasts were cultured in a 96-well plate at a density of 5 × 103 cells/well with different treatments for 3 days. Cells were fixed in 4% paraformaldehyde for 20 min and then stained with TRAP staining solution according to the manufacturers’ instructions. Relative TRAP activity was measured by colorimetric analysis.
For actin cytoskeleton and focal adhesion stain, preosteoclasts were cultured on glass sheet in a 12-well plate at a density of 4 × 104 cells/well with different treatments for 3 days. Procedures were described in previous study.43 In brief, on day 4, cells were washed and fixed for permeabilization. After blocking, cells were incubated in primary antibody solution for 1 h at room temperature. After wash, cells were incubated with secondary antibody and TRITC-conjugated Phalloidin for 1 h at room temperature. Nucleus counterstaining was performed by DAPI for 5 min, followed by fluorescence microscopy observation.
For pit formation assay, preosteoclasts were incubated in 96-well plates (Corning Osteo Assay Surface) of 2 × 103 cells/well and in 48-well plates covered with bovine bone slices of 1 × 104 cells/well. Cells were induced with different treatments for 5 days. Methylene blue stain was performed to evaluate the resorption area on bone slices. Bleach solution was added to remove cells. Detailed analysis of the pit formation area was described previously.44
Intracellular ROS detection
Intracellular ROS detection was performed using the DCFDA cellular ROS detection assay kit (ABcam). Preosteoclasts (5 × 103 cells/well in 96-well plates) were treated with LPS (1 μg/ml), DHA (100 μg/ml) or both for 72 h. The intracellular ROS level was measured by 2′, 7′-dichlorofluorescein diacetate (DCFH), which can be oxidized into fluorescent DCF. Cells were washed in 1 × PBS and then incubated in the dark for 30 min with 10 μM DCFH-DA. Images were taken using the fluorescence microscopy (Olympus, Tokyo, Japan).
Annexin-V/PI staining
Cell apoptosis was determined with Annexin-V/PI staining. Preosteoclasts were cultured for 72 h with treatments of LPS, DHA or both. Cells were washed twice with cold PBS and then resuspended in 500 μl of binding buffer (10 mM HEPES/NaOH (pH 7.4), 140 mM NaCl, 2.5 mM CaCl2) at a concentration of 1 × 106 cells/ml. Cells were then stained with 5 μl of annexin V-FITC (Life Technologies) and 10 μl of 20 μg/ml PI. Apoptosis was analyzed using a FAC Star flow cytometer (BD, Triangle, NC, USA).
μCT and histological analyses
For μCT analysis,the Bruker MicroCT Skyscan 1272 system (Kontich, Belgium) with an isotropic voxel size of 10.0 μm was used to image the whole femur. Scans were conducted in 4% paraformaldehyde and used an X-ray tube potential of 60 kV, an X-ray intensity of 166 μA and an exposure time of 1700 ms. Trabecular bones were thresholded at 86–255 (8-bit gray scale bitmap). μCT scans of the whole body of mice (except skull) were performed using isotropic voxel sizes of 148 μm. Reconstruction was accomplished by Nrecon (Ver. 1.6.10, Kontich, Belgium). 3D images were obtained from contoured 2D images by methods based on distance transformation of the gray scale original images (CTvox, Kontich, Belgium, Ver. 3.0.0). 3D and 2D analyses were performed using software CT Analyser (Kontich, Belgium, Ver. 1.15.4.0). All images presented are representative of the respective groups.
For the bone histological analysis, femurs were dissected and fixed in 4% paraformaldehyde in PBS for 48 h. Femurs were then decalcified by daily change of 15% tetrasodium EDTA for 2 weeks. Tissues were dehydrated by passage through an ethanol series, cleared twice in xylene, embedded in paraffin and sectioned at 8 μm thickness along the coronal plate from anterior to posterior. Decalcified femoral sections were stained with TRAP.
RT-qPCR
Total RNA was isolated using Trizol reagent (Life Technologies). Single-stranded cDNA was prepared from 1 μg of total RNA using reverse transcriptase with oligo-dT primer according to the manufacturer’s instructions (Promega, Fitchburg, WI, USA). Two microlitres of each cDNA were subjected to PCR amplification using specific primers as follows: TRAP (F) 5′-AGACGAGGTTACGCTGTGC-3′, (R) 5′-TCGGGGACAATTCGGTAAACT-3′; Ctsk (F) 5′-GCGGCATTACCAACAT-3′, (R) 5′-CTGGAAGCACCAACGA-3′; MMP-9 (F) 5′-ACCCGAAGCGGACATT-3′, (R) 5′-GGCATCTCCCTGAACG-3′; DC-STAMP (F) 5′-TTATGTGTTTCCACGAAGCCCTA-3′, (R) 5′-ACAGAAGAGAGCAGGGCAACG-3′.
Immunoblotting
Cells were lysed in a lysis buffer containing 10 mM Tris, pH 7.2, 150 M NaCl, 5 mM EDTA, 0.1% SDS, 1% Triton X-100 and 1% deoxycholic acid. For western blots, 30 μg of protein samples were subjected to SDS-PAGE, followed by transfer onto PVDF membranes. After blocking in 5% skim milk, membranes were incubated with rabbit antibodies against CaMKKβ, Pyk2, c-FOS, NFATc1, COX-IV and β-actin overnight at 4 °C, followed by 1-h incubation with secondary antibody (1:2000). COX-IV and β-actin were detected in parallel as a loading control for mitochondrial and cytoplasmic proteins, respectively.
Statistical analysis
All data are representative of at least three experiments of similar results performed in triplicate, unless otherwise indicated. Data are expressed as mean±S.D. One-way ANOVA, followed by Student–Newman–Keuls post hoc tests, was used to determine the significance of difference between results, with *P<0.05, **P<0.01 being regarded as significant.
Abbreviations
- DHA:
-
dihydroartemisinin
- OC:
-
osteoclast
- RANKL:
-
receptor activator of nuclear factor κB ligand
- M-CSF:
-
macrophage-colony-stimulating factor
- LPS:
-
lipopolysaccharide
- ROS:
-
reactive oxygen species
- AIF:
-
apoptotic inducing factor
- BMMs:
-
bone marrow macrophages
- TRAP:
-
tartrate-resistant acid phosphatase
- BV/TV:
-
bone volume fraction
- Tb. N:
-
trabecular number
References
Boyle WJ, Simonet WS, Lacey DL . Osteoclast differentiation and activation. Nature 2003; 423: 337–342.
Hadjidakis DJ, Androulakis II . Bone remodeling. Ann N Y Acad Sci 2006; 1092: 385–396.
Rodan GA, Martin TJ . Therapeutic approaches to bone diseases. Science 2000; 289: 1508–1514.
Lacey DL, Timms E, Tan HL, Kelley MJ, Dunstan CR, Burgess T et al. Osteoprotegerin ligand is a cytokine that regulates osteoclast differentiation and activation. Cell 1998; 93: 165–176.
Arai F, Miyamoto T, Ohneda O, Inada T, Sudo T, Brasel K et al. Commitment and differentiation of osteoclast precursor cells by the sequential expression of c-Fms and receptor activator of nuclear factor kappaB (RANK) receptors. J Exp Med 1999; 190: 1741–1754.
Roodman GD . Advances in bone biology: the osteoclast. Endocr Rev 1996; 17: 308–332.
Teitelbaum SL . Bone resorption by osteoclasts. Science 2000; 289: 1504–1508.
Henderson B, Nair SP . Hard labour: bacterial infection of the skeleton. Trends Microbiol 2003; 11: 570–577.
Nagasawa T, Kiji M, Yashiro R, Hormdee D, Lu H, Kunze M et al. Roles of receptor activator of nuclear factor-kappaB ligand (RANKL) and osteoprotegerin in periodontal health and disease. Periodontol 2000 2007; 43: 65–84.
Nair SP, Meghji S, Wilson M, Reddi K, White P, Henderson B . Bacterially induced bone destruction: mechanisms and misconceptions. Infect Immunity 1996; 64: 2371–2380.
Pawate S, Shen Q, Fan F, Bhat NR . Redox regulation of glial inflammatory response to lipopolysaccharide and interferongamma. J Neurosci Res 2004; 77: 540–551.
Sun J, Ramnath RD, Zhi L, Tamizhselvi R, Bhatia M . Substance P enhances NF-kappaB transactivation and chemokine response in murine macrophages via ERK1/2 and p38 MAPK signaling pathways. Am J Physiol Cell Physiol 2008; 294: C1586–C1596.
Garrett IR, Boyce BF, Oreffo RO, Bonewald L, Poser J, Mundy GR . Oxygen-derived free radicals stimulate osteoclastic bone resorption in rodent bone in vitro and in vivo. J Clin Invest 1990; 85: 632–639.
Ha H, Kwak HB, Lee SW, Jin HM, Kim HM, Kim HH et al. Reactive oxygen species mediate RANK signaling in osteoclasts. Exp Cell Res 2004; 301: 119–127.
Lee NK, Choi YG, Baik JY, Han SY, Jeong DW, Bae YS et al. A crucial role for reactive oxygen species in RANKL-induced osteoclast differentiation. Blood 2005; 106: 852–859.
Bhatt NY, Kelley TW, Khramtsov VV, Wang Y, Lam GK, Clanton TL et al. Macrophage-colony-stimulating factor-induced activation of extracellular-regulated kinase involves phosphatidylinositol 3-kinase and reactive oxygen species in human monocytes. J Immunol 2002; 169: 6427–6434.
Bartell SM, Kim HN, Ambrogini E, Han L, Iyer S, Serra Ucer S et al. FoxO proteins restrain osteoclastogenesis and bone resorption by attenuating H2O2 accumulation. Nat Commun 2014; 5: 3773.
Park H, Noh AL, Kang JH, Sim JS, Lee DS, Yim M . Peroxiredoxin II negatively regulates lipopolysaccharide-induced osteoclast formation and bone loss via JNK and STAT3. Antioxidants Redox Signal 2015; 22: 63–77.
Li Y . Qinghaosu (artemisinin): chemistry and pharmacology. Acta Pharmacol Sin 2012; 33: 1141–1146.
Krishna S, Bustamante L, Haynes RK, Staines HM . Artemisinins: their growing importance in medicine. Trends Pharmacol Sci 2008; 29: 520–527.
Mao H, Gu H, Qu X, Sun J, Song B, Gao W et al. Involvement of the mitochondrial pathway and Bim/Bcl-2 balance in dihydroartemisinin-induced apoptosis in human breast cancer in vitro. Int J Mol Med 2013; 31: 213–218.
He Q, Shi J, Shen XL, An J, Sun H, Wang L et al. Dihydroartemisinin upregulates death receptor 5 expression and cooperates with TRAIL to induce apoptosis in human prostate cancer cells. Cancer Biol Ther 2010; 9: 819–824.
Efferth T, Benakis A, Romero MR, Tomicic M, Rauh R, Steinbach D et al. Enhancement of cytotoxicity of artemisinins toward cancer cells by ferrous iron. Free Radical Biol Med 2004; 37: 998–1009.
Efferth T . Molecular pharmacology and pharmacogenomics of artemisinin and its derivatives in cancer cells. Curr Drug Targets 2006; 7: 407–421.
Roue G, Bitton N, Yuste VJ, Montange T, Rubio M, Dessauge F et al. Mitochondrial dysfunction in CD47-mediated caspase-independent cell death: ROS production in the absence of cytochrome c and AIF release. Biochimie 2003; 85: 741–746.
Garrido C, Galluzzi L, Brunet M, Puig PE, Didelot C, Kroemer G . Mechanisms of cytochrome c release from mitochondria. Cell Death Differ 2006; 13: 1423–1433.
Lu JJ, Chen SM, Zhang XW, Ding J, Meng LH . The anti-cancer activity of dihydroartemisinin is associated with induction of iron-dependent endoplasmic reticulum stress in colorectal carcinoma HCT116 cells. Invest New Drugs 2011; 29: 1276–1283.
Chen H, Sun B, Pan S, Jiang H, Sun X . Dihydroartemisinin inhibits growth of pancreatic cancer cells in vitro and in vivo. Anticancer Drugs 2009; 20: 131–140.
Chen T, Li M, Zhang R, Wang H . Dihydroartemisinin induces apoptosis and sensitizes human ovarian cancer cells to carboplatin therapy. J Cell Mol Med 2009; 13: 1358–1370.
O'Neill PM, Barton VE, Ward SA . The molecular mechanism of action of artemisinin—the debate continues. Molecules 2010; 15: 1705–1721.
Lander HM . An essential role for free radicals and derived species in signal transduction. FASEB J 1997; 11: 118–124.
He X, Andersson G, Lindgren U, Li Y . Resveratrol prevents RANKL-induced osteoclast differentiation of murine osteoclast progenitor RAW 264.7 cells through inhibition of ROS production. Biochem Biophys Res Commun 2010; 401: 356–362.
Moon HJ, Ko WK, Han SW, Kim DS, Hwang YS, Park HK et al. Antioxidants, like coenzyme Q10, selenite, and curcumin, inhibited osteoclast differentiation by suppressing reactive oxygen species generation. Biochem Biophys Res Commun 2012; 418: 247–253.
Sharma R, Callaway D, Vanegas D, Bendele M, Lopez-Cruzan M, Horn D et al. Caspase-2 maintains bone homeostasis by inducing apoptosis of oxidatively-damaged osteoclasts. PLoS One 2014; 9: e93696.
Suda K, Woo JT, Takami M, Sexton PM, Nagai K . Lipopolysaccharide supports survival and fusion of preosteoclasts independent of TNF-alpha, IL-1, and RANKL. J Cell Physiol 2002; 190: 101–108.
Schroder K . NADPH oxidases in bone homeostasis and osteoporosis. Cell Mol Life Sci 2015; 72: 25–38.
Balaban RS, Nemoto S, Finkel T . Mitochondria, oxidants, and aging. Cell 2005; 120: 483–495.
Wang X . The expanding role of mitochondria in apoptosis. Genes Dev 2001; 15: 2922–2933.
Odkhuu E, Koide N, Tsolmongyn B, Jambalganiin U, Naiki Y, Komatsu T et al. Involvement of redox balance in in vitro osteoclast formation of RAW 264.7 macrophage cells in response to LPS. Innate Immun 2015; 21: 194–202.
Cabello CM, Lamore SD, Bair WB 3rd, Qiao S, Azimian S, Lesson JL et al. The redox antimalarial dihydroartemisinin targets human metastatic melanoma cells but not primary melanocytes with induction of NOXA-dependent apoptosis. Invest New Drugs 2012; 30: 1289–1301.
Zhou L, Liu Q, Yang M, Wang T, Yao J, Cheng J et al. Dihydroartemisinin, an anti-malaria drug, suppresses estrogen deficiency-induced osteoporosis, osteoclast formation and RANKL induced signalling pathways. J Bone Miner Res 2015 (e-pub ahead of print; doi:10.1002/jbmr.2771).
Feng MX, Hong JX, Wang Q, Fan YY, Yuan CT, Lei XH et al. Dihydroartemisinin prevents breast cancer-induced osteolysis via inhibiting both breast caner cells and osteoclasts. Sci Rep 2016; 6: 19074.
Dou C, Zhang C, Kang F, Yang X, Jiang H, Bai Y et al. MiR-7b directly targets DC-STAMP causing suppression of NFATc1 and c-Fos signaling during osteoclast fusion and differentiation. Biochim Biophys Acta 2014; 1839: 1084–1096.
Dou C, Li J, Kang F, Cao Z, Yang X, Jiang H et al. Dual effect of cyanidin on RANKL-induced differentiation and fusion of osteoclasts. J Cell Physiol 2016; 231: 558–567.
Acknowledgements
This work was funded by grants from the Nature Science Foundation of China (81572164), the National Key Technology Research and Development Program of China (2012BAI42G01), the National High-tech R&D Program of China (863 Program, 2015AA020315) and Key project of Logistics Research Plan of PLA (BWS13C014).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by G Melino
Supplementary Information accompanies this paper on Cell Death and Disease website
Supplementary information
Rights and permissions
Cell Death and Disease is an open-access journal published by Nature Publishing Group. This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Dou, C., Ding, N., Xing, J. et al. Dihydroartemisinin attenuates lipopolysaccharide-induced osteoclastogenesis and bone loss via the mitochondria-dependent apoptosis pathway. Cell Death Dis 7, e2162 (2016). https://doi.org/10.1038/cddis.2016.69
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cddis.2016.69
This article is cited by
-
Circulating Exosomes from Mice with LPS-Induced Bone Loss Inhibit Osteoblast Differentiation
Calcified Tissue International (2022)
-
Phytochemicals in Breast Cancer-Induced Osteoclastogenesis and Bone Resorption: Mechanism and Future Perspective
Current Pharmacology Reports (2022)
-
lncRNA RMRP Prevents Mitochondrial Dysfunction and Cardiomyocyte Apoptosis via the miR-1-5p/hsp70 Axis in LPS-Induced Sepsis Mice
Inflammation (2020)
-
RETRACTED ARTICLE: Matrine derivate MASM uncovers a novel function for ribosomal protein S5 in osteoclastogenesis and postmenopausal osteoporosis
Cell Death & Disease (2017)
