
Abstract
Platelet activation is a frontline response to injury, not only essential for clot formation but also important for tissue repair. Indeed, the reparative influence of platelets has long been exploited therapeutically where application of platelet concentrates expedites wound recovery. Despite this, the mechanisms of platelet-triggered cytoprotection are poorly understood. Here, we show that activated platelets accumulate in the brain to exceptionally high levels following injury and release factors that potently protect neurons from apoptosis. Kinomic microarray and subsequent kinase inhibitor studies showed that platelet-based neuroprotection relies upon paracrine activation of the epidermal growth factor receptor (EGFR) and downstream DNA-dependent protein kinase (DNA-PK). This same anti-apoptotic cascade stimulated by activated platelets also provided chemo-resistance to several cancer cell types. Surprisingly, deep proteomic profiling of the platelet releasate failed to identify any known EGFR ligand, indicating that activated platelets release an atypical activator of the EGFR. This study is the first to formally associate platelet activation to EGFR/DNA-PK – an endogenous cytoprotective cascade.
Similar content being viewed by others


Main
Platelets are small discoid anuclear cell fragments that are key players in haemostasis – a frontline physiological response to acute tissue injury. Under basal conditions, platelets circulate at 150–400 × 109 per litre and sequester a diverse array of bioactive molecules within their intracellular granules.1 Upon tissue injury, however, vascular disruption triggers localised platelet deposition, activation and release of their granular contents. These platelet-released molecules (PRMs) further recruit and activate platelets, resulting in a multi-cellular aggregate that restricts blood loss. In conjunction, the coagulation cascade becomes activated, leading to thrombin generation which consolidates the growing thrombus by promoting further platelet activation and by catalysing fibrin formation.
In addition to this critical role in haemostasis, platelets also participate in numerous non-haemostatic processes, including inflammation,2 tissue repair,3 angiogenesis and lymphatic development.4 Platelets also promote tumour cell proliferation and metastasis via peri-cellular adhesion and signalling.5, 6, 7 Such pleiotropy is attributed to the array of molecules that are released from activated platelets.
The reparative influence of platelets also has important clinical utility. For decades, ‘platelet concentrates’ have been applied to sites of injury to expedite the recovery of organs such as bone,8 skin9 and tendon.10 The beneficial effects of platelet concentrates are thought to be due to the many growth factors that are released from activated platelets.3 This reasoning, however, may be considered speculative given the lack of direct experimental evidence. Furthermore, little-to-no information exists about the platelet-derived peri-cellular signals that facilitate tissue repair.
Platelets are normally restricted to the intravascular compartment. However, extravascular platelet accumulation can also occur under certain pathological conditions. One notable example of this is neurotrauma. Here, we find that exceptionally high levels of platelet activation occur in the brain after neurotrauma, thereby providing in-principle support that platelet products may influence neuronal survival. We therefore examined the influence of PRMs on injured primary cortical neurons. As neurons are post-mitotic, this approach allowed the elucidation of effects that were unrelated to proliferation. We find that PRMs selectively and potently reduce neuronal apoptosis via paracrine activation of the epidermal growth factor receptor (EGFR) and downstream activation of DNA-dependent protein kinase (DNA-PK) – a ubiquitous DNA repair enzyme. Strikingly, the same platelet-dependent mechanism also protects several non-neuronal human cancer cell types from chemotherapy-induced apoptosis. Thus, activated platelets trigger a potent and broad-acting paracrine signal that attenuates apoptosis.
Our findings highlight a beneficial action of platelets that likely operates within the injured brain where existing knowledge about platelets has been limited to its haemostatic role. Interestingly, we also find that this underappreciated cytoprotective action of platelets may have implications in cancer where the association between higher platelet count and poorer patient prognosis has been well established.11, 12, 13, 14 In addition, this newly identified anti-apoptotic mechanism should encourage rationally designed improvements to the clinical use of platelet concentrates.
Results
Understanding about platelets during acquired brain injuries largely pertains to their role in haemostasis. Given their reparative influence on other tissues, however, platelets also likely exert a non-haemostatic beneficial effect on the injured brain. We first sought to determine the extent to which PRMs were present in the brain after neurotrauma, an acquired brain injury where extreme platelet activation likely occurs. To this end, we measured soluble GPVI (sGPVI; a platelet-specific marker that is shed from activated platelets15), in the cerebrospinal fluid (CSF) of neurotrauma patients (Figure 1a). Whereas control patients had little-to-no sGPVI in their CSF (mean=0.45 ng/ml), sGPVI levels in the CSF of neurotrauma patients were significantly elevated (mean=35.42 ng/ml). To our knowledge, this is the first quantification of any PRM in the brain after neurotrauma. Remarkably, the level of sGPVI in the CSF of neurotrauma patients was comparable to that in plasma during disseminated intravascular coagulation, which lie between 39.9 and 72.8 ng/ml.15 Thus, profound intracranial release of platelet-derived molecules occurs after neurotrauma.

Platelets accumulate and activate in the brain after neurotrauma. (a) ELISA measuring soluble GPVI in the CSF of non-neurotrauma patients (n=9, black opened circles) and neurotrauma patients (n=31, blue closed circles). The mean±95% confidence intervals are indicated. ***P<0.001 by two-tailed Mann–Whitney test. (b) (i) Representative immunofluorescence for the platelet marker integrin αIIbβ3 shows marked platelet deposition in the injured ipsilateral cortex 30 min after neurotrauma compared with the corresponding contralateral cortex. Diameter of impactor is indicated by the red discontinuous line. The depth of the impact is indicated by the continuous red line. (ii) Magnified image of an area of the ipsilateral cortex (indicated by the yellow box in i). Immunosignal of integrin αIIbβ3 is depicted in white and the Hoechst 33342 signal is in blue. Closed arrowheads show intravascular platelet deposits. Opened arrowheads show large extravascular platelet deposits. (c) Representative immunoblot of mouse brain lysates taken from the injured ipsilateral (ipsi) and uninjured contralateral (contra) hemispheres. Integrin αIIbβ3 was used as a platelet marker. The cleaved fragment of GPIb (GPIbα-tail) was used as a platelet activation marker. Albumin was used as an indicator of blood–brain barrier breakdown. GAPDH was used as a loading control. Scale bar: (b) (i) 500 μm. (ii) 50 μm
To establish the spatiotemporal characteristics of platelet activation/deposition after neurotrauma, we used a mouse model of neurotrauma; where a single unilateral 2 mm blunt-force impact was delivered to the cerebral cortex. After injury, mice were transcardially perfused to remove free-flowing blood and their brains subjected to immunofluorescence for the platelet-specific marker, integrin αIIbβ3. As shown in Figure 1b, considerable platelet accumulation was evident within the injured ipsilateral hemisphere, but not within the uninjured contralateral hemisphere 30 min after neurotrauma. Notably, platelets were deposited not only within the initial impact site (red-coloured axes; Figure 1bi) but also at distal secondary lesion sites within the ipsilateral hemisphere where apoptotic cell death predominates. Platelet deposition within the trauma lesion was not confined to blood vessels, with numerous extravascular platelet-rich aggregates occurring within the injured brain parenchyma (Figure 1bii). Similar results were observed when immunofluorescence for the platelet-specific marker, GPIbα, was performed (data not shown). These studies confirm that widespread platelet activation occurs throughout the ipsilateral hemisphere after focal neurotrauma.
To establish a time course of platelet activation after injury, mice were transcardially perfused 5–60 min after neurotrauma and lysates of the ipsilateral and contralateral cortex were prepared. These lysates were then subjected to immunoblot analysis. As shown in Figure 1c, a greater extent of platelet deposition (presence of the platelet-specific integrin, αIIbβ3) and platelet activation (presence of the platelet activation-specific marker, the ∼17-kDa GPIbα fragment) was observed in the injured ipsilateral cortex relative to the uninjured contralateral cortex. Importantly, higher levels of serum albumin were also observed in the ipsilateral cortex across all time points relative to the corresponding contralateral cortex (Figure 1c), indicating that blood–brain barrier breakdown had occurred. Hence, shortly after neurotrauma (30–60 min), pronounced platelet deposition/activation and blood–brain barrier disruption coincide within the ipsilateral cortex.
Altogether, these experiments show that overlapping blood–brain barrier breakdown and platelet deposition/activation allow PRMs to enter the injured brain parenchyma. The spatial deposition of platelets is vast and spans well beyond the initial contusion site. In addition, the magnitude of platelet deposition/activation is such that the concentration of platelet-derived sGPVI in the CSF after neurotrauma approximates that found in plasma during disseminated intravascular coagulation. Importantly, this comparison provided us with a pathophysiologically relevant concentration for using PRMs in vitro (PRMs were used in vitro at less than half the concentration to that found in the CSF of neurotrauma patients).
PRMs attenuate neuronal apoptosis
We next investigated the influence of PRMs on neuronal injury in vitro. Primary mouse cortical neurons were exposed to a variety of stressors (including etoposide, L-glutamate, linsidomine and oxygen-glucose deprivation) in the presence or absence of PRMs and cellular injury assessed 24 h later. The most striking observation from these studies was that PRMs from activated platelets, but not from resting platelets, markedly reduced the magnitude of etoposide-induced injury as indicated by a PRM-mediated preservation of membrane integrity (Figures 2a and b) and cellular metabolism (Figure 2c). As etoposide is a topoisomerase-II inhibitor that induces DNA damage and subsequent apoptosis,16 we monitored the effect of PRMs on caspase activation. PRMs attenuated etoposide-induced activation of the upstream initiator caspases -8 and -9, reduced downstream activation of the effector caspase-3/7 (Figure 2d) and prevented caspase-dependent cleavage of PARP (Figure 2e). Hence, PRMs block an early stage apoptosis before the activation of major intrinsic/extrinsic caspases.

PRMs attenuate etoposide-induced toxicity in neurons. Primary mouse cortical neuronal cultures were treated with vehicle, 2 mg/l etoposide, etoposide+supernatant from unactivated platelets (PRMs low) or etoposide+supernatant from thrombin-activated platelets (PRMs). The degree of injury was assessed 24 h later by measuring plasma membrane disruption (via lactate dehydrogenase release (LDH); a; n=4), measuring cellular metabolism (via MTS conversion; c; n=5) and measuring caspase-8, -9, -3/7 activation (d; n=4). The mean with S.E.M. is indicated. *P<0.05; **P<0.01; ***P<0.001 and ‘ns’ non-significant by one-way ANOVA with Newman–Keuls correction. (b) Representative images of neuronal cultures treated for 24 h with etoposide±PRMs, then incubated with Hoechst 33342 (coloured in blue) and 7AAD (coloured in pink). The differential interference contrast (DIC) image of the cultures is depicted in the upper row of micrographs. The epifluorescence image of the cultures is depicted in the bottom row of micrographs, with healthy cells in blue and dead cells in pink. (e) Representative immunoblot of lysates from neuronal cultures treated with vehicle, PRMs and etoposide±PRMs for 24 h. Immunoblots show that PRMs reduce etoposide-induced PARP cleavage. Tubulin was used as a loading control. Scale bar: (b) 100 μm
PRMs also protected neurons from linsidomine (Supplementary Figure S2a) – a potent nitrosative stressor. Thus, PRMs protect neurons from mechanistically distinct injuries. In contrast, PRMs failed to rescue neurons from cell death induced by L-glutamate (Supplementary Figure S2b) or oxygen-glucose deprivation (Supplementary Figure S2c). Interestingly, whereas treatment of neurons with etoposide or linsidomine triggered caspase-3 activation, L-glutamate exposure and oxygen-glucose deprivation failed to elicit any discernible caspase-3 activation (Supplementary Figures S2d and e). These findings suggest that PRMs do not protect from caspase-independent forms of cell death, but rather that PRMs specifically attenuate neuronal injury by dampening events upstream of pro-apoptotic caspase activation.
To gain further insight into the cytoprotective actions of PRMs, we assessed its influence on proteasome stress. Despite the fact that inhibition of the proteasome can activate the apoptotic cascade in both neurons and non-neuronal cells,17, 18 PRMs did not attenuate injury caused by MG132 treatment (Supplementary Figure S2f) or bortezomib treatment (Supplementary Figure S2g). Cellular injury caused by proteasome inhibition differs from both etoposide and linsidomine injury in that it does not principally involve DNA damage. These results therefore suggest that not only do PRMs preferentially attenuate caspase-mediated injury, but also PRMs effectively protect against apoptosis caused by DNA damage.
Characterisation of PRM-mediated cytoprotection
As neuroprotection had not previously been directly linked to platelet activation, we further characterised the capacity of platelets to attenuate apoptosis. First, we assessed whether neuroprotection could still be elicited by in situ activated platelets (rather than by purified PRMs), because our protocol for preparing PRMs removes molecules <3 kDa and allows the degradation of labile platelet-derived agents (e.g., thromboxane A2). These omitted molecules, however, do not principally alter the cytoprotective effect of PRMs, as in situ activation of washed platelets still reduced etoposide-induced injury in neuronal cultures (*P<0.05; n=3; data not shown). This finding shows that the net effect of platelet activation is indeed neuroprotection. We also examined the potency of PRM-mediated neuroprotection. As shown in Figures 3a and b, PRMs still reduced neuronal injury caused by a 5-fold higher concentration of etoposide. In addition, the cytoprotective influence of PRMs was observed even after 48 h of etoposide exposure (Figures 3c and d). Hence, PRMs transmit a potent and long-lasting anti-apoptotic signal to neurons. Notably, the beneficial influence of PRMs was unrelated to cellular proliferation as no bromodeoxyuridine incorporation was observed in neuronal cultures after 24 h of PRMs treatment (data not shown). Next, to assess the therapeutic potential of PRMs, we added PRMs to neuronal cultures 1 h after the commencement of etoposide injury. Despite the fact that apoptosis had already been initiated, post-injury application of PRMs significantly attenuated etoposide-induced neuronal injury (Figures 3e and f). Hence, the capacity of PRMs to rescue neurons following the onset of apoptosis suggests that the active agent within PRMs has therapeutic potential.

PRMs protect against high-dose etoposide and pre-initiated etoposide injury. (a and b) Primary neuronal cultures were stimulated with vehicle, 10 mg/l etoposide±PRMs. (c and d) Primary neuronal cultures were stimulated with vehicle, 2 mg/l etoposide±PRMs. (e and f) Primary neuronal cultures were stimulated with vehicle or 2 mg/l etoposide for 1 h before the addition of PRMs. The degree of injury was assessed 24 h (a, b, e and f) or 48 h (c and d) later by measuring plasma membrane disruption (via lactate dehydrogenase (LDH) release; a, n=4; c, n=4; e, n=3) and measuring cellular metabolism (via MTS conversion; b, n=5; d, n=4; f, n=7). Mean with S.E.M. is indicated. *P<0.05; **P<0.01; ****P<0.0001 and ‘ns’ non-significant by one-way ANOVA with Newman–Keuls correction
Finally, we investigated whether PRMs could also attenuate apoptosis in other cell types. Given the well-recognised contribution of platelets to cancer progression,6 we assessed the ability of PRMs to inhibit etoposide-induced apoptosis in a range of human cancer cell lines. PRMs reduced the extent of etoposide-induced cell death in monocytic lymphoma cells (U937, Figure 4a), myelocytic plasma cells (JIM-1, Figure 4b) and myelocytic lymphocytes (KMS26, Figure 4c). These findings broaden the importance of platelet-mediated cytoprotection and are significant as etoposide is a chemotherapeutic used to treat malignancies, including small cell lung carcinoma, germ-line cancers, sarcoma, lymphoma and leukaemia.19

PRMs attenuate apoptosis in non-neuronal cells. The cell lines (indicated on the left) were treated with vehicle, etoposide±PRMs. The degree of injury was assessed 24 h by staining with the nuclear dye 7AAD (FL-3) and CaspACE FITC-VAD-FMK (FL-1). The three left-hand columns depict the raw data (as a scatter plot of FL-1 versus FL-3) of a single representative experiment, with the live cell gate superimposed. The right-hand column depicts the portion of live cells collated across multiple experiments showing that etoposide toxicity was reduced by PRMs in (a) monocytic cells U937 (n=8), (b) plasma cells JIM-1 (n=3) and (c) lymphocyte-like cells KMS26 (n=3). Mean with S.E.M. is indicated. *P<0.05, **P<0.01 and ‘ns’ is non-significant by one-way ANOVA with Newman–Keuls correction
Identifying signalling pathways that underlie PRM-mediated cytoprotection
We next wanted to identify the paracrine signalling pathway responsible for PRM-mediated cytoprotection. We first tested for kinase dependency using staurosporine (a pan-kinase inhibitor that triggers DNA damage and neuronal apoptosis), and found that PRMs failed to alter the extent of staurosporine-induced neuronal injury (Supplementary Figure S3). This finding suggests that the neuroprotective action of PRMs relies upon kinase activity. To identify which kinases are activated by PRMs, we performed a kinomic screen whereby neuronal cultures were injured with etoposide in the presence/absence of PRMs. Lysates were harvested after 30 and 180 min of treatment and then subjected to Kinexus antibody microarray (which utilises ∼500 pan- and ∼340 phospho-specific antibodies). The complete microarray data sets are presented in Supplementary Table 1. Pair-wise comparison and collation of the microarray data across both time points produced a high-confidence list of signalling proteins that were differentially regulated by PRMs (Figure 5a). Importantly, the kinomic screen showed that PRMs caused a 44% reduction in caspase-3 function, verifying the integrity of the initial cell culture experiment (Supplementary Table 1).

Kinomic screening and prediction of the signalling events that underlie PRM-mediated cytoprotection. (a) Neuronal cultures 30 min and 3 h after etoposide±PRMs stimulation were lysed and subject to kinomic microarray analysis yielding a short list of signalling proteins significantly altered (red indicates a >1.2 × upregulated event and green indicates a >0.55 × downregulated event) by PRMs following etoposide injury. (b) The short-listed signalling proteins (Supplementary Table 1) were subjected to Ingenuity Pathway Analysis (IPA) to yield six canonical signalling pathways (data not shown) for PRM-mediated cytoprotection. Signalling proteins that showed interplay with caspase or other apoptotic processes were manually merged (across the predicted canonical signalling pathways) to generate a single predicted signalling pathway that underlies PRM-mediated cytoprotection. Red symbols represent upregulated events. Green symbols represent downregulated events. Subsequent kinase inhibitor studies based on this putative signalling pathway highlighted DNA-PK as a key kinase effector of PRM-mediated cytoprotection
To offer system-wide insight, the kinomic data sets were subjected to Ingenuity Pathway Analysis (Redwood City, CA, USA) where the analyst (from the Australian Proteome Analysis Facility) was blinded to both the experimental design and the overall project hypothesis. Five of the top six canonical pathways identified by Ingenuity Pathway Analysis indicated a critical PRM-mediated impact on apoptosis and predicted a reduction in caspase function (data not shown). These top five pathways were then manually collated into a single signalling pathway that putatively underlies PRM-mediated cytoprotection (Figure 5b).
PRMs inhibit apoptosis via the preservation of DNA-PK activity
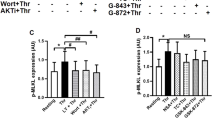
On the basis of the putative signalling pathway outlined in Figure 5b, selective pharmacological inhibitors of p38α, JNK1, JAK1, DAPK1 (data not shown) and DNA-PK were sequentially tested for their capacity to abrogate PRM-mediated cytoprotection. Of the tested inhibitors, only the DNA-PK-selective compound, NU7441, attenuated PRM-mediated cytoprotection in both mouse cortical neurons and human U937 monocytes (Figures 6a and b, respectively). Hence, paracrine activation of DNA-PK mediates the cytoprotective influence of PRMs.

PRM-mediated cytoprotection is via EGFR and downstream DNA-PK activation. Neuronal cultures (a; n=5) or U937 cells (b; n=4) were stimulated with vehicle or 1–2 mg/l etoposide±PRMs±10 nM NU7441 (a DNA-PK-specific inhibitor). The degree of injury was assessed 24 h later by measuring caspase-3/7 activation (a) or cell viability (b; % negatively staining for 7AAD and CaspACE FITC-VAD-FMK). (c) Representative images of neuronal cultures stimulated with vehicle, 2 mg/l etoposide or 2 mg/l etoposide+PRMs. Cultures were fixed after 1 h and subjected to immunofluorescence for the DNA-PK catalytic subunit (DNA-PKcs). The top micrographs depict the Hoechst 33342 nuclear counterstain and the bottom micrographs depict the corresponding DNA-PKcs immunosignal. (d) Representative immunoblot of neuronal cultures stimulated with 2 mg/l etoposide±PRMs. Cultures were lysed 30 min post stimulation and subjected to immunoblot analyses for total and phosphorylated EGFR (pEGFR). Subtext is the densitometry analysis comparing the average ratio of pEGFR/EGFR (mean±S.D.; n=4; #P<0.05 between groups with/without PRMs using unpaired t-test with Welsh correction). Cultures treated in the presence of PRMs have significantly elevated pEGFR/EGFR levels compared with cultures treated in the absence of PRMs. Neuronal cultures (e) or U937 cells (f) were stimulated with vehicle or 1–2 mg/ml etoposide±PRMs±Gefitinib (an EGFR-specific inhibitor; 0.4 μM for neuronal cultures; 10 μM for U937s). The degree of injury was assessed 24 h later by measuring cellular metabolism (via MTS conversion; e; n=4) or cell viability (f; n=4; % negatively stained for 7AAD and CaspACE FITC-VAD-FMK). *P<0.05, **P<0.01, ***P<0.001 and ‘ns’ is non-significant by one-way ANOVA with Newman–Keuls correction. #P<0.05 by unpaired t-test with Welsh correction. Scale bar: (c) 10 μm
DNA-PK is a ubiquitous enzyme responsible for the repair of double-stranded DNA breaks.20 Interestingly, our kinomic screen indicated that DNA-PK levels were 317% higher in neuronal cultures following etoposide+PRM treatment, relative to cultures treated with etoposide alone (Figure 5a; Supplementary Table 1). In support of these findings, immunofluorescence studies showed that 1 h of etoposide treatment dramatically reduced DNA-PK levels, whereas co-treatment with PRMs fully restored DNA-PK to basal levels (Figure 6c). Hence, our data suggest that an immediate-early degradation of DNA repair machinery contributes to etoposide toxicity and that PRMs protect by preserving DNA-PK-mediated DNA repair.
Platelets inhibit apoptosis via atypical activation of the EGFR
While DNA-PK is a recognised cytoprotectant,21, 22 the receptors that trigger its activation are poorly understood. The only well-established receptor that promotes DNA-PK activation is the EGFR.23, 24 Notably, our kinomic screen showed that PRMs, in addition to increasing DNA-PK levels, also triggered a concomitant 192% increase in phospho-EGFR levels (Figure 5a; Supplementary Table 1). We therefore hypothesised that PRMs protect via activation of the ‘EGFR→DNA-PK’ cascade. To test this hypothesis, we first confirmed that PRMs did indeed increase activation of EGFR (Figure 6d). Next, we blocked the EGFR using the anti-cancer compound, Gefitinib – a small-molecule inhibitor of the EGFR.25, 26 As shown in Figures 6e and f, Gefitinib significantly diminished PRM-mediated cytoprotection against etoposide exposure in both mouse neurons and human U937 monocytes. These findings suggest that the cytoprotective signal transduced by PRMs is mediated by EGFR activation.
The EGFR has a well-defined repertoire of extracellular ligands, namely: EGF, Transforming Growth Factor-α, heparin-binding-EGF, Amphiregulin, Betacellulin, Epigen, Epiregulin and Neuregulins 1–6.25 We performed deep proteomic profiling to determine whether any EGFR ligands were present in our preparations of PRMs. In all, 394 proteins were identified in PRMs, of which 266 were confidently identified by two-or-more peptides (Supplementary Table 2). However, no EGFR ligand was confidently identified in our preparations of PRMs. Similarly, no other prior characterisation of the platelet releasome has detected the presence of a recognised EGFR ligand (Wijten et al1 and references therein). While no recognised EGFR ligand was detected in PRMs, proteins containing an ‘EGF-like domain’ represented the most abundant class of proteins in PRMs (Supplementary Table 2). Hence, activated platelets release a novel ligand of the EGFR that, in turn, promotes cell survival during genotoxic insult via the maintenance of DNA-PK-dependent DNA repair.
Discussion
Platelet activation is a primary response to tissue injury. While thrombus formation is the best characterised consequence of platelet activation, proper initiation of inflammation, angiogenesis, proliferation and tissue repair also depends upon platelet activation. Recent research suggests that peri-cellular protection may be another consequence of platelet activation.7, 27, 28 Our study uniquely shows that activated platelets release an atypical agonist of the EGFR, which in turn, transduces a paracrine signal that potently attenuates apoptosis via the preservation of DNA-PK-dependent DNA repair. Figure 7 presents our working model of platelet-based cytoprotection, whereby activated platelets release an EGF-like protein that triggers peri-cellular activation and nuclear translocation of the EGFR, leading to EGFR:DNA-PK complex formation, the preservation of DNA-PK levels and the promotion of DNA repair. In support of this mechanism, prior studies have shown that EGFR:DNA-PK complex maintains DNA repair23 presumably via blockade of caspase-mediated degradation of DNA-PK during genotoxic stress.29 Hence, a ‘therapeutic window’ likely exists for DNA-PK-mediated DNA repair during genotoxic stress. We theorise that injury-induced platelet activation aligns with this ‘therapeutic window’ and helps to prevent the irreversible activation of intrinsic and extrinsic apoptosis (Figure 7).

Working model of PRM-mediated cytoprotection against etoposide injury. (a) Etoposide injury in the absence of platelet activation allows etoposide to injure cells by inhibiting DNA-topoisomerase II and inducing double-stranded DNA damage. Initial caspase activation following DNA damage leads to DNA-PK degradation, thereby preventing DNA repair and promoting irreversible initiation of the caspase-8 (extrinsic) and caspase-9 (intrinsic) arms of apoptosis. (b) Etoposide injury in the presence of platelet activation does not eventuate in apoptosis. This is because activated platelets release an EGF-like protein (blue) that binds to and promotes phospho-activation of the EGFR. The phospho-activated EGFR translocates to the nucleus and forms a complex with DNA-PK. As the EGFR:DNA-PK complex is resistant to caspase-mediated degradation, any etoposide-induced DNA damage is amenable to repair and thus leads to cell survival
Although our data indicate that EGFR-DNA-PK signalling is a major mediator of platelet-triggered cytoprotection, they do not exclude the contribution of other pro-survival signalling cascades. Indeed, the predicted canonical pathway outlined in Figure 5b stipulates that other kinases including p38α, JNK1, JAK1, 4E-BP1 and DAPK1 are activated by PRMs. Studies should now address whether PRM-mediated activation of these additional kinases subserves other non-haemostatic influences of platelets.
The anti-apoptotic factor(s) released from activated platelets has several favourable characteristics. First, PRMs offer protection to a range of cell types (Figure 4) and, importantly, can be used to rescue injured cells after apoptosis has been initiated (Figures 3e and f). The potency of platelet-based anti-apoptosis is also noteworthy, with PRMs often completely protecting neurons from injury (Figures 2a and 6e; Supplementary Figure S2a). This potency may be due to the fact that PRMs interfere with a very early step in apoptotic induction; with PRMs diminishing activation of initiator caspases (Figure 2d), preventing an immediate-early drop in DNA-PK levels after etoposide treatment (Figure 6c), and attenuating etoposide-induced phosphatidylserine exposure in U937 cells (data not shown). It is also notable that PRMs maintained their protective action after heating (>50 oC; data not shown) and that the protective agent lacked pro-thrombotic activity when purified from other PRM constituents (data not shown). Thus, the anti-apoptotic factor in PRMs harbours desirable traits that encourage its future identification and development as a potential therapeutic.
Several points pertaining to the preparation and use of PRMs warrant mention. First, PRMs were prepared by pooling the supernatants from washed activated human platelets across ≥4 independent donors. Our preparations of PRMs were also of high purity, significantly overlapping with the other published in-depth characterisation of the platelet releasome (Wijten et al.1 and references therein). In addition, numerous independent batches of PRMs were used throughout this study, with each batch offering potent cytoprotection. Hence, our study denotes that a potent anti-apoptotic agent is constitutively expressed and reproducibly released from activated human platelets.
Platelets, EGFR and DNA-PK in the context of brain injury
Only recently have studies begun to define the non-haemostatic roles of platelets in the brain. For example, recent studies show that platelets drive neuroinflammation during experimental stroke and multiple sclerosis.30, 31 However, discrepant findings exist with respect to the capacity of platelets to influence neurotoxicity. One research group has shown that activated platelets are toxic to the central nervous system,32, 33 whereas another study shows that intracranial administration of platelet lysates (rather than PRMs) reduces post-stroke lesion volume and neurological deficit in spontaneously hypertensive rats.34 Hayon et al.34 attributed the beneficial effect of platelet lysates to an increase in angiogenesis and proliferation and also concluded that a platelet-based promotion of cell survival likely contributed to the dramatic improvement in post-stroke injury. Our observations that platelets offer potent neuroprotection complement and provide an underlying molecular mechanism for the findings of Hayon et al. 34 Following stroke and neurotrauma, for example, such a platelet-based mechanism might delay apoptosis, affording time for the restoration of tissue homeostasis. Indeed, a platelet-based neuroprotective mechanism capitalises on the fact that platelets are frontline responders to injury capable of releasing anti-apoptotic effectors in a spatially and temporally appropriate manner.
In comparison with the emerging neural roles of platelets, it is well established that EGFR signalling can protect the brain from injury.35, 36, 37, 38, 39 Similarly, DNA-PK is a recognised neuroprotectant, with studies showing that DNA-PK−/− neurons are hypersensitive to injury.22, 40 Surprisingly, no prior study has established whether EGFR-mediated activation of DNA-PK is a protective pathway that operates during brain injury. Thus, a PRM that activates the EGFR-DNA-PK cascade (our present study) warrants further investigation.
Platelets, EGFR and DNA-PK in the context of cancer
It is well accepted that activated platelets deleteriously contributes to cancer aetiology.6, 41 It is thought that activated platelets ensheath circulating cancer cells, allowing them to evade immune detection, protecting them from vascular shear forces and helping vessel wall attachment and metastasis.41, 42, 43 In addition, activated platelets release paracrine factors that promote cancer cell proliferation,44 chemo-resistance7 and angiogenesis.45, 46 Hence, platelet count is commonly used as a prognostic factor whereby patients with high platelet counts have reduced survival rates in a range of tumour types including cervical47 and lung cancer.48 Our data extend these observations and show that activated platelets increase the survival of cancer cells exposed to etoposide – a widely used chemotherapeutic. Therefore, we predict that blockade of platelet-based cytoprotection, in conjunction with radio/chemotherapy may enhance the efficacy of cancer treatments.
Interestingly, ectopic activation of the EGFR-DNA-PK cascade has also been closely linked to tumourigenesis and radio/chemo-resistance.49, 50, 51, 52, 53 Indeed, as a high proportion of malignancies feature hyperactivation/mutation of EGFR, the EGFR is a mainstay of anti-cancer research.25, 54, 55, 56 Similarly, DNA-PK-selective antagonists, including NU7441, are promising new anti-cancer drugs.51, 57 In this context, discovery of platelets as a potent, atypical and injury-specific activator of EGFR-DNA-PK signalling is of explicit relevance to the field of cancer. Clinical trial records should now be stratified to determine whether the efficacy of EGFR-inhibitory regimes correlates with platelet counts in cancer patients.
Concluding remarks
The widespread medical use of platelet concentrates to facilitate tissue repair led us to investigate whether pro-survival factors are released by activated platelets. We find that molecules released from platelets that are yet to be identified, potently protect a variety of cell types from apoptosis by transactivation of the EGFR and stimulation of downstream DNA-PK-mediated DNA repair. Our study is the first to connect platelets, frontline responders to tissue injury, to the highly studied EGFR-DNA-PK cascade. While this connection uncovers new avenues for neuroprotection, it also provides a plausible mechanistic basis for the correlative actions of platelets, EGFR and DNA-PK during cancer. Future research should now address platelet-based cytoprotection during neurotrauma, cancer, the clinical use of platelet concentrates, and indeed, any situation where large-scale platelet activation and apoptosis coincide.
Materials and Methods
Materials
All reagents were from Life Technologies (Carlsbad, CA, USA) unless otherwise indicated. Etoposide, L-glutamate, staurosporine, MG132 and linsidomine (3-morpholinosydnonimine hydrochloride; SIN-1) were form Sigma (St. Louis, MO, USA). NU7441 was from Tocris Bioscience (Bristol, UK). Gefitinib was from Cayman Chemical (Ann Arbor, MI, USA).
Acquisition of human CSF
CSF collection was performed in accordance with the Australian National Health and Medical Research Council’s (NHMRC) National Statement on Ethical Conduct in Research Involving Humans and was approved by the Alfred Hospital Human Ethics Committee. Thirty-one patients (7 female and 24 males; aged between 15 and 73 years old; mean age of 32.1 years) were recruited via the Trauma Service of the Alfred Hospital, Melbourne, with delayed informed consent obtained from the next of kin. Patients who suffered from severe neurotrauma determined by a post-resuscitation and pre-intubation Glasgow Coma Scale Score (GCS) ≤9 and the requirement of an extraventricular drain for monitoring intracranial pressure (ICP) were included in the study. CSF was drained from patients when the ICP was greater than 20 mm Hg and collected in bags in cooled 4 °C containers over 24 h. CSF samples collected within 4 days of admission were centrifuged at 2000 r.c.f. for 15 min at 4 °C, and stored at −80 °C until needed. Exclusion criteria included pregnancy, known history of neurodegenerative diseases, HIV and other chronic infection or inflammatory diseases, history of previous neurotrauma and age <15 years. Patients with focal brain injury (classified as the presence of either an evacuated lesion or a non-evacuated high- or mixed-density mass lesion greater than 25 ml in volume) or/and diffuse brain injury (defined by the presence of Grade I to IV diffuse injury based on the degree of compression of cisterns and the occurrence of a midline shift) were included in the study.
Animals
All animal procedures adhered to the NHMRC Code of Practice for the Care and Use of Animals for Experimental Purposes in Australia and were approved by the institutional Animal Ethics Committee. All experiments used C57Black/6 mice. Experimental neurotrauma was performed on 8- to 12-week-old male C57Black/6 mice. Neuronal cultures were prepared from the embryos of 4- to 12 week-old pregnant mice.
Measuring sGPVI in CSF
The measurement of sGPVI was performed as in Al-Tamimi et al.58 with minor modifications. CSF samples were analysed in duplicate. CSF from neurotrauma patients was diluted 1:5 in artificial CSF (0.3 M NaCl, 6 mM KCl, 2.8 mM CaCl2.2 H2O, 1.6 mM MgCl2.6 H2O, 1.6 mM Na2HPO4.7 H2O, 0.4 mM NaH2PO4.7 H2O) to a final volume of 100 μl. Each ELISA plate contained standards of recombinant GPVI ectodomain (0–100 ng/ml final concentration) serially diluted in artificial CSF.
Experimental neurotrauma
Neurotrauma on mice was induced using the controlled cortical impact model as in Samson et al.59 In brief, a burr hole was drilled over the left parietal cortex of anaesthetised mice and a single blunt impact (5 m/s velocity; 2 mm impact depth; 150 ms dwell time) was delivered by a controlled impactor device. Mice that were designated as ‘sham’ mice were subjected only to the craniotomy procedure after anaesthesia.
Extraction and homogenisation brain tissue
Mice were transcardially perfused at fixed time points after neurotrauma with 30 ml PBS (pH 7.4) containing 10 mM EDTA. Brains were removed and 2 mm coronal sections cut using a Mouse Brain Blocker (Kopf, Tujunga, CA, USA). The cortex was then dissected from these sections, weighed and homogenised to a final concentration of 150 mg of tissue/ml in PBS+1% Triton X-100, containing 10 μM GM6001 as well as 1 × phosphatase and protease inhibitor cocktail tablets (Roche, Mannheim, Germany).
Immunohistochemistry
PBS/EDTA-perfused brains were removed and formaldehyde fixed (4 °C overnight). Brains were then dehydrated overnight and paraffin embedded using an automatic tissue processor. In all, 6 μm tissue sections were cut and processed for immunohistochemistry. Sections were stained with 1:100 rat anti-mouse integrin αIIbβ3 (BD Biosciences, San Jose, CA, USA) or 1:100 rat anti-mouse GPIbα (Emfret Analytics, Eibelstadt, Germany) overnight at 4 °C. Sections were washed and incubated with a fluorescent-conjugated secondary antibody and for 2 h at room temperature, followed by 5 mg/l Hoechst 33342 for 5 min. Slides were washed and cover-slipped in fluorescence mounting media (DAKO, Glostrup, Denmark). Images were captured on the Zeiss Cell Observer system (microscope: Axio Observer.Z1, Jena, Germany; objective#1 (EC Plan-Neofluar, × 40 magnification, 0.75 numerical aperture, air immersion) or objective #2 (Plan-Apochromat 63 × magnification, 1.40 numerical aperture, oil immersion); sequential excitation and emission filters for 4’,6-diamidino-2-phenylindole, fluorescein, and Texas Red fluorophores; camera: AxioCamMR3; acquisition software: AxioVision 4.8, Jena, Germany). Images were processed using the ImageJ v1.47q software (National Institutes of Health, Bethesda, MD, USA).
Immunoblot analysis
Samples were boiled in SDS-loading buffer with dithiothreitol (DTT), subjected to sodium dodecyl sulphate-polyacrylamide gel electrophoresis and transferred onto polyvinylidene fluoride membranes. Primary antibodies were rabbit anti-human GPIbα-tail,58 mouse anti-human GPIbα (WM23, DAKO), rat anti-mouse integrin αIIbβ3 (BD Biosciences), rabbit anti-human GPVI (12A5;Al-Tamimi et al.58), rabbit anti-polyclonal cyclophilin A (Abcam, Cambridge, UK), rabbit anti-polyclonal ADP-ribose polymerase (PARP, Santa Cruz Biotechnology, Dallas, TX, USA), goat anti-human albumin (Bethyl Laboratories Inc., Montgomery, TX, USA), mouse anti-GAPDH (Merck Millipore, Billerica, MA, USA), mouse anti-β tubulin (Sigma), rabbit anti-EGFR (D38B1, Cell Signaling, Danvers, MA, USA), rabbit anti-phospho-EGFR Tyr1173 (53A5, Cell Signaling). Appropriate HRP-conjugated secondary antibodies were used and signals revealed by chemiluminescence (Thermo Scientific, Bremen, Germany).
Preparation of platelet-rich plasma and washed platelets
All human blood collection was in accordance with the ethical standards of the Monash University Standing Committee for Ethics involving Research into Humans and with the Helsinki Declaration of 1975. Human plasma from healthy donors was collected and platelets were washed as in Kulkarni et al.60 In brief, human whole blood was collected into acid citrate dextrose at a ratio of 6:1 (blood:acid). In all, 20 U/ml Clexane was added and incubated for 15 min at 37 °C. All subsequent procedures were performed at 37 °C. Separation of platelet-rich plasma and red blood cells was achieved by centrifugation (300 r.c.f., 16 min). The platelet-rich plasma was collected and left to rest for 10 min before centrifugation (1700 r.c.f., 7 min). The platelet pellet was then resuspended in platelet wash buffer (4.3 mM K2HPO4, 4.3 mM Na2HPO4, 24.3 mM NaH2PO4, 113 mM NaCl, 5.5 mM D-glucose, and 10 mM theophylline, pH 6.5). Platelets were allowed to rest for 30 min before use.
Preparation of PRMs
Two different preparations of PRMs were used for this study: (1) directly from whole blood as described above or (2) from expired platelet bags obtained through the Alfred Hospital Blood Services. The cytoprotective effect of PRMs was evident irrespective of the preparation methodology. Platelets (from either procedure) were centrifuged (800 r.c.f., 7 min, 37 °C) and resuspended in modified Tyrode’s buffer (10 mM HEPES, 12 mM NaHCO3, 13.7 mM NaCl, 2.7 mM KCl, and 5.5 mM D-glucose, pH 7.3) containing 1 mM CaCl2. Platelets were allowed to rest for 30 min before activated via the addition of 1 U/ml bovine thrombin (Merck Millipore). The activated platelet suspension was placed on a rocker (10 min, 37 °C) to ensure maximal platelet activation. As an ‘unactivated PRMs’ control, supernatants were collected from platelets resuspended in Tyrode’s buffer with 1 mM CaCl2, 20 mM theophylline, 20 U/L apyrase and 5 mM EGTA. The vehicle control for PRMs was modified Tyrode’s buffer with 1 mM CaCl2, 1 U/ml thrombin and DMSO (equivalent in volume and concentration to the addition of corresponding compounds). Flow cytometry for P-selectin and fibrinogen exposure was performed to confirm the activated/resting status of platelets (FACs Calibur; Becton Dickinson, Franklin Lakes, NJ, USA; Supplementary Figure S1a). The platelet suspensions were then centrifuged (800 r.c.f., 7 min) and supernatants collected. Supernatants were pooled across 4–15 donors and concentrated 15-fold using a 3-kDa molecular weight cutoff Amicon ultrafiltration device (Merck Millipore). The ultrafiltration of PRMs was validated by immunoblot (Supplementary Figure S1B).
Primary neuronal cultures
Primary cortical neuronal cultures were prepared as in Samson et al.59 Cultures were seeded into 48-well plates at a concentration of 112 500 cells per well. All experiments were conducted on DIV7. Before experimentation, media was replaced with phenol-red free neurobasal media (NBM) containing 2% antioxidant-free B27, 0.5 mM L-glutamine, 50 U/ml penicillin/streptomycin (P/S). Cultures were challenged with 2–20 mg/l etoposide, 10 μM L-glutamate, 60 μM 3-morpholinosydnonimine hydrochloride (linsidomine, SIN-1), 500 nM MG132, 200 nM bortezomib, 100 nM staurosporine or oxygen-glucose deprivation (OGD). For OGD, media was replaced with glucose-free NBM containing antioxidant-free B27, 0.5 mM L-glutamine, 50 U/ml P/S and cultures sealed in a modular incubator (Billups-Rothenburg Inc., Del Mar, CA, USA) filled with 95% nitrogen and 5% carbon dioxide for 4 h at 37 °C. Media was then replaced with NBM and cultures maintained under humidified 5% CO2, 8% O2 conditions for 20 h. PRMs were added at a dose equivalent to that released by 6 × 108 platelets/ml. All measurements were taken 24 h after the commencement of injury unless otherwise indicated in the corresponding legend.
Cancer cell lines
Cancer cell lines (U937, JIM-1 and KMS26) were maintained in RPMI supplemented with 10% (v/v) heat-inactivated fetal calf serum, 2 mM L-glutamine, 50 U/ml P/S under humidified 5% CO2. Before experimentation, cells were seeded at a density of 3.5–5.0 × 105 cells/ml. Cultures were treated with etoposide (1 mg/l for U937; 2 mg/l for JIM-1 and KMS26)±PRMs (to a dose equivalent to that released by 12 × 108 platelets/ml). All measurements were taken 24 h after the commencement of injury, whereby cells were centrifuged, washed and resuspended in 100 μL PBS containing 2.5–5 μM CaspACE FITC-VAD-FMK (Promega, Madison, WI, USA) and 5–10 mg/l 7AAD. Cells were incubated for 20 min, washed and resuspended in 150 μl of PBS before analysis via by flow cytometry (BD FACS Calibur; 25 000 events per sample). Data were analysed using the Flowjo v7.6.5 software (Tree Star Inc., Ashland, OR, USA).
CytoTox96 non-radioactive cytotoxicity (LDH) assay, CellTitre 96 aqueous non-radioactive cell proliferation (MTS) assay, caspase 8-, caspase 9- and caspase 3/7-Glo assays
All assays were from Promega and performed according to the manufacturer’s instructions.
Cytochemistry
Primary neuronal cultures were seeded at 112 500 cells/well in μ-well plates (Ibidi, Munich, Germany), then maintained and stimulated as described above. After 24 h, cultures were PBS washed and incubated with 300 μl PBS containing 5 mg/l Hoechst 33342 and 10 mg/l 7AAD dye for 30 min. Cells were imaged using the Zeiss Cell Observer system as described above. Images were processed using the ImageJ v1.47q software.
Immunofluorescence of DNA-PK
Immunofluorescence staining was performed as in Samson et al.59 with the minor modification that cells were blocked with goat serum, stained with a 1:100 dilution of rabbit-anti-human DNA-PK antibody (Abcam) and counterstained with 10 mg/l Hoechst 33342. Cells were imaged on a Nikon A1r-si resonant scanning confocal system (microscope: Nikon Ti, Tokyo, Japan; objective: Apo LWD, × 40 magnification, 1.15 numerical aperture, water immersion; sequential excitation: 405 nm and 546 nm laser lines; emission filters: 450/50 nm and 595/50 nm; acquisition software, NIS Elements Advanced Research, Tokyo, Japan). Images were processed using the ImageJ v1.47q software.
Cell homogenisation
Cell homogenisation was performed as in Samson et al.59
Phospho-protein microarray
Neuronal cultures were treated with etoposide±PRMs. At t=30 and t=180 min post treatment, homogenates were prepared and sent to Kinexus Bioinformatics (Vancouver, BC, Canada) for blinded kinomic analysis using the KAM-1.2 chip equipped with ∼500 pan-specific and ∼300 phospho-site specific antibodies.
Mass spectrometry of PRMs
In all, 30 μg of purified PRMs was diluted in 1.7 g/l ammonium bicarbonate (Sigma) and 1.7 g/l Tris(2-carboxyethyl)phosphine hydrochloride (Sigma) and incubated for 5 min at 95 oC. The sample was cooled to room temperature and alkylated with 1.8 g/l iodoacetamide (Sigma) for 20 min in the dark. The samples were then digested with trypsin (Promega, Sequencing Grade) at 1:100 w/w for 3 h at 37 °C. In all, 2 μl of formic acid (Sigma) was added to stop proteolysis and the resulting peptides analysed by LC-MS/MS using the Q Exactive mass spectrometer (Thermo Scientific) coupled online with an UltiMate 3000 Rapid Separation LC (RSLC) nano HPLC system (Thermo Scientific). Samples were loaded on a 100 μm, 2 cm nanoviper pepmap100 trap column (Thermo Scientific) in 2% acetonitrile, 0.1% formic acid at a flow rate of 15 μl/min. Peptides were eluted and separated at a flow rate of 300 μl/min on Thermo RSLC nanocolumn 75 μm × 50 cm, pepmap100 C18, 3 μm 100 Å pore size, with a linear acetonitrile gradient from 10 to 26% in 0.1% formic acid for 55 min followed by a linear increase to 34% acetonitrile in 0.1% formic acid over 5 min and additional increase up to 80% acetonitrile in 0.1% formic acid over 5 min. This was followed by reduction in acetonitrile back to 2% and re-equilibration. The eluent was nebulised and ionised using the Thermo nano electrospray source with a distal coated fused silica emitter (New Objective, Woburn, MA, USA) with a capillary voltage of 1.8–2.2 kV. The Q Exactive instrument was operated in the data-dependent mode to automatically switch between full scan MS and MS/MS acquisition. Survey full scan MS spectra (m/z 375–1800) were acquired in the Orbitrap with 70 000 resolution (at m/z 200) after accumulation of ions to a 3 × 106 target value with maximum injection time of 120 ms. Dynamic exclusion was set to 30 s. The 12 most intense multiply charged ions (z≥2) were sequentially isolated and fragmented in the octopole collision cell by higher-energy collisional dissociation (HCD) with a fixed injection time of 120 ms 17 500 resolution and AGC target of 1 × 105 counts. A 2.0-Da isolation width was chosen. Under fill ratio was at 1% dynamic exclusion was set to 15 s. Mass spectrometric conditions were spray voltage, 2 kV; no sheath and auxiliary gas flow; heated capillary temperature, 275 °C; normalised HCD collision energy 27%. The mass spectrometry data were analysed online using X! tandem (release 2013.09.01; Craig and Beavis61) at the Global Proteome Machine site (www.thegpm.org). Raw data files were converted to peak lists in MGF format using MSconvert (ProteoWizard version 3.0.4922; Kessner et al.62) and were searched against Human proteins sequences (Ensemble GRC) appended with known contaminates (http://www.thegpm.org/cRAP/index.html) and mixed with reversed sequences. Search parameters include: trypsin cleavage specificity with two missed cleavage, cysteine carbamidomethyl as fixed modification, methionine oxidation and protein N-terminal acetylation as variable modifications. Peptide tolerance and MS/MS tolerance were set at 20 p.p.m. The remaining search parameters were default settings. The search results from the Global Proteome Machine site are provided in Worksheet 1, Supplementary Table 2.
Statistical analysis
Statistical analysis was performed using GraphPad Prism v6 (GraphPad Software, Inc., La Jolla, CA, USA). Unless stated otherwise, experiments were performed on a minimum of three independent occasions. The statistical test applied to each data set is stipulated in the corresponding legend. A P-value of <0.05 was considered as statistically significant.
Abbreviations
- CSF:
-
cerebrospinal fluid
- DIC:
-
differential interference contrast
- DNA-PK:
-
DNA-dependent protein kinase
- EGFR:
-
epidermal growth factor receptor
- pEGFR:
-
phosphorylated activate EGFR
- GP:
-
glycoprotein
- PARP:
-
poly ADP ribose polymerase
- PRMs:
-
platelet-released molecules
- STS:
-
staurosporine
- veh:
-
vehicle
- SIN-1:
-
linsidomine
- OGD:
-
oxygen-glucose deprivation
References
Wijten P, van Holten T, Woo LL, Bleijerveld OB, Roest M, Heck AJ et al. High precision platelet releasate definition by quantitative reversed protein profiling–brief report. Arterioscler Thromb Vasc Biol 2013; 33: 1635–1638.
Liu Y, Gao XM, Fang L, Jennings NL, Su Y, Q X et al. Novel role of platelets in mediating inflammatory responses and ventricular rupture or remodeling following myocardial infarction. Arterioscler Thromb Vasc Biol 2011; 31: 834–841.
Nurden AT . Platelets, inflammation and tissue regeneration. Thromb Haemost 2011; 105 (Suppl 1): S13–S33.
Bertozzi CC, Schmaier AA, Mericko P, Hess PR, Zou Z, Chen M et al. Platelets regulate lymphatic vascular development through CLEC-2-SLP-76 signaling. Blood 2010; 116: 661–670.
Erpenbeck L, Schon MP . Deadly allies: the fatal interplay between platelets and metastasizing cancer cells. Blood 2010; 115: 3427–3436.
Bambace NM, Holmes CE . The platelet contribution to cancer progression. J Thromb Haemost 2011; 9: 237–249.
Radziwon-Balicka A, Medina C, O'Driscoll L, Treumann A, Bazou D, Inkielewicz-Stepniak I et al. Platelets increase survival of adenocarcinoma cells challenged with anticancer drugs: mechanisms and implications for chemoresistance. Br J Pharmacol 2012; 167: 787–804.
Gentile P, Bottini DJ, Spallone D, Curcio BC, Cervelli V . Application of platelet-rich plasma in maxillofacial surgery: clinical evaluation. J Craniofac Surg 2010; 21: 900–904.
Rozman P, Bolta Z . Use of platelet growth factors in treating wounds and soft-tissue injuries. Acta Dermatovenerol Alp Panonica Adriat 2007; 16: 156–165.
Cervelli V, Lucarini L, Spallone D, Brinci L, de Angelis B . Use of platelet rich plasma and hyaluronic acid on exposed tendons of the foot and ankle 2010 J Wound Care 19: 186, 188–190.
Hernandez E, Lavine M, Dunton CJ, Gracely E, Parker J . Poor prognosis associated with thrombocytosis in patients with cervical cancer. Cancer 1992; 69: 2975–2977.
Shimada H, Oohira G, Okazumi S, Matsubara H, Nabeya Y, Hayashi H et al. Thrombocytosis associated with poor prognosis in patients with esophageal carcinoma. J Am Coll Surg 2004; 198: 737–741.
Ikeda M, Furukawa H, Imamura H, Shimizu J, Ishida H, Masutani S et al. Poor prognosis associated with thrombocytosis in patients with gastric cancer. Ann Surg Oncol 2002; 9: 287–291.
Taucher S, Salat A, Gnant M, Kwasny W, Mlineritsch B, Menzel RC et al. Impact of pretreatment thrombocytosis on survival in primary breast cancer. Thromb Haemost 2003; 89: 1098–1106.
Al-Tamimi M, Grigoriadis G, Tran H, Paul E, Servadei P, Berndt MC et al. Coagulation-induced shedding of platelet glycoprotein VI mediated by factor Xa. Blood 2011; 117: 3912–3920.
Montecucco A, Biamonti G . Cellular response to etoposide treatment. Cancer Lett 2007; 252: 9–18.
Suh J, Lee YA, Gwag BJ . Induction and attenuation of neuronal apoptosis by proteasome inhibitors in murine cortical cell cultures. J Neurochem 2005; 95: 684–694.
Concannon CG, Koehler BF, Reimertz C, Murphy BM, Bonner C, Thurow N et al. Apoptosis induced by proteasome inhibition in cancer cells: predominant role of the p53/PUMA pathway. Oncogene 2007; 26: 1681–1692.
Baldwin EL, Osheroff N . Etoposide, topoisomerase II and cancer. Curr Med Chem Anticancer Agents 2005; 5: 363–372.
Polo SE, Jackson SP . Dynamics of DNA damage response proteins at DNA breaks: a focus on protein modifications. Genes Dev 2011; 25: 409–433.
Kong X, Shen Y, Jiang N, Fei X, Mi J . Emerging roles of DNA-PK besides DNA repair. Cell Signal 2011; 23: 1273–1280.
Culmsee C, Bondada S, Mattson MP . Hippocampal neurons of mice deficient in DNA-dependent protein kinase exhibit increased vulnerability to DNA damage, oxidative stress and excitotoxicity. Brain Res Mol Brain Res 2001; 87: 257–262.
Liccardi G, Hartley JA, Hochhauser D . EGFR nuclear translocation modulates DNA repair following cisplatin and ionizing radiation treatment. Cancer Res 2011; 71: 1103–1114.
Bandyopadhyay D, Mandal M, Adam L, Mendelsohn J, Kumar R . Physical interaction between epidermal growth factor receptor and DNA-dependent protein kinase in mammalian cells. J Biol Chem 1998; 273: 1568–1573.
Eccles SA . The epidermal growth factor receptor/Erb-B/HER family in normal and malignant breast biology. Int J Dev Biol 2011; 55: 685–696.
D'Incecco A, Cappuzzo F . Gefitinib for non-small-cell lung cancer treatment. Expert Opin Drug Saf 2011; 10: 987–996.
Muto T, Kokubu T, Mifune Y, Sakata R, Nagura I, Nishimoto H et al. Platelet-rich plasma protects rotator cuff-derived cells from the deleterious effects of triamcinolone acetonide. J Orthop Res 2013; 31: 976–982.
Fukaya Y, Kuroda M, Aoyagi Y, Asada S, Kubota Y, Okamoto Y et al. Platelet-rich plasma inhibits the apoptosis of highly adipogenic homogeneous preadipocytes in an in vitro culture system. Exp Mol Med 2012; 44: 330–339.
Song Q, Lees-Miller SP, Kumar S, Zhang Z, Chan DW, Smith GC et al. DNA-dependent protein kinase catalytic subunit: a target for an ICE-like protease in apoptosis. EMBO J 1996; 15: 3238–3246.
Sotnikov I, Veremeyko T, Starossom SC, Barteneva N, Weiner HL, Ponomarev ED . Platelets recognize brain-specific glycolipid structures, respond to neurovascular damage and promote neuroinflammation. PLoS One 2013; 8: e58979.
Thornton P, McColl BW, Greenhalgh A, Denes A, Allan SM, Rothwell NJ . Platelet interleukin-1alpha drives cerebrovascular inflammation. Blood 2010; 115: 3632–3639.
Joseph R, Tsering C, Grunfeld S, Welch KM . Platelet secretory products may contribute to neuronal injury. Stroke 1991; 22: 1448–1451.
Joseph R, Saroff DM, Delfs JR . Platelet secretory products have a damaging effect on neurons. Neurosci Lett 1992; 135: 153–158.
Hayon Y, Dashevsky O, Shai E, Varon D, Leker RR . Platelet lysates stimulate angiogenesis, neurogenesis and neuroprotection after stroke. Thromb Haemost 2013; 110: 323–330.
Jin K, Sun Y, Xie L, Childs J, Mao XO, Greenberg DA . Post-ischemic administration of heparin-binding epidermal growth factor-like growth factor (HB-EGF) reduces infarct size and modifies neurogenesis after focal cerebral ischemia in the rat. J Cereb Blood Flow Metab 2004; 24: 399–408.
Justicia C, Planas AM . Transforming growth factor-alpha acting at the epidermal growth factor receptor reduces infarct volume after permanent middle cerebral artery occlusion in rats. J Cereb Blood Flow Metab 1999; 19: 128–132.
Opanashuk LA, Mark RJ, Porter J, Damm D, Mattson MP, Seroogy KB . Heparin-binding epidermal growth factor-like growth factor in hippocampus: modulation of expression by seizures and anti-excitotoxic action. J Neurosci 1999; 19: 133–146.
Scafidi J, Hammond TR, Scafidi S, Ritter J, Jablonska B, Roncal M et al. Intranasal epidermal growth factor treatment rescues neonatal brain injury. Nature 2013; 506: 230–234.
Sun D, Bullock MR, Altememi N, Zhou Z, Hagood S, Rolfe A et al. The effect of epidermal growth factor in the injured brain after trauma in rats. J Neurotrauma 2010; 27: 923–938.
Neema M, Navarro-Quiroga I, Chechlacz M, Gilliams-Francis K, Liu J, Lamonica K et al. DNA damage and nonhomologous end joining in excitotoxicity: neuroprotective role of DNA-PKcs in kainic acid-induced seizures. Hippocampus 2005; 15: 1057–1071.
Nieswandt B, Hafner M, Echtenacher B, Mannel DN . Lysis of tumor cells by natural killer cells in mice is impeded by platelets. Cancer Res 1999; 59: 1295–1300.
Kunita A, Kashima TG, Ohazama A, Grigoriadis AE, Fukayama M . Podoplanin is regulated by AP-1 and promotes platelet aggregation and cell migration in osteosarcoma. Am J Pathol 2011; 179: 1041–1049.
Alonso-Escolano D, Medina C, Cieslik K, Radomski A, Jurasz P, Santos-Martinez MJ et al. Protein kinase C delta mediates platelet-induced breast cancer cell invasion. J Pharmacol Exp Ther 2006; 318: 373–380.
Boucharaba A, Serre CM, Gres S, Saulnier-Blache JS, Bordet JC, Guglielmi J et al. Platelet-derived lysophosphatidic acid supports the progression of osteolytic bone metastases in breast cancer. J Clin Invest 2004; 114: 1714–1725.
Pinedo HM, Verheul HM, D'Amato RJ, Folkman J . Involvement of platelets in tumour angiogenesis? Lancet 1998; 352: 1775–1777.
Feng W, Madajka M, Kerr BA, Mahabeleshwar GH, Whiteheart SW, Byzova TV . A novel role for platelet secretion in angiogenesis: mediating bone marrow-derived cell mobilization and homing. Blood 2011; 117: 3893–3902.
Rodriguez GC, Clarke-Pearson DL, Soper JT, Berchuck A, Synan I, Dodge RK . The negative prognostic implications of thrombocytosis in women with stage IB cervical cancer. Obstet Gynecol 1994; 83: 445–448.
Gonzalez Barcala FJ, Garcia Prim JM, Moldes Rodriguez M, Alvarez Fernandez J, Rey Rey MJ, Pose Reino A et al. Platelet count: association with prognosis in lung cancer. Med Oncol 2010; 27: 357–362.
Mukherjee B, McEllin B, Camacho CV, Tomimatsu N, Sirasanagandala S, Nannepaga S et al. EGFRvIII and DNA double-strand break repair: a molecular mechanism for radioresistance in glioblastoma. Cancer Res 2009; 69: 4252–4259.
Dittmann K, Mayer C, Rodemann HP . Inhibition of radiation-induced EGFR nuclear import by C225 (Cetuximab) suppresses DNA-PK activity. Radiother Oncol 2005; 76: 157–161.
Ciszewski WM, Tavecchio M, Dastych J, Curtin NJ . DNA-PK inhibition by NU7441 sensitizes breast cancer cells to ionizing radiation and doxorubicin. Breast Cancer Res Treat 2014; 143: 47–55.
Javvadi P, Makino H, Das AK, Lin YF, Chen DJ, Chen BP et al. Threonine 2609 phosphorylation of the DNA-dependent protein kinase is a critical prerequisite for epidermal growth factor receptor-mediated radiation resistance. Mol Cancer Res 2012; 10: 1359–1368.
Kang KB, Zhu C, Wong YL, Gao Q, Ty A, Wong MC . Gefitinib radiosensitizes stem-like glioma cells: inhibition of epidermal growth factor receptor-Akt-DNA-PK signaling, accompanied by inhibition of DNA double-strand break repair. Int J Radiat Oncol Biol Phys 2012; 83: e43–e52.
Dhomen NS, Mariadason J, Tebbutt N, Scott AM . Therapeutic targeting of the epidermal growth factor receptor in human cancer. Crit Rev Oncog 2012; 17: 31–50.
Pao W, Chmielecki J . Rational, biologically based treatment of EGFR-mutant non-small-cell lung cancer. Nat Rev Cancer 2010; 10: 760–774.
Troiani T, Martinelli E, Capasso A, Morgillo F, Orditura M, De Vita F et al. Targeting EGFR in pancreatic cancer treatment. Curr Drug Targets 2012; 13: 802–810.
Salles B, Calsou P, Frit P, Muller C . The DNA repair complex DNA-PK, a pharmacological target in cancer chemotherapy and radiotherapy. Pathol Biol 2006; 54: 185–193.
Al-Tamimi M, Mu FT, Moroi M, Gardiner EE, Berndt MC, Andrews RK . Measuring soluble platelet glycoprotein VI in human plasma by ELISA. Platelets 2009; 20: 143–149.
Samson AL, Knaupp AS, Sashindranath M, Borg RJ, Au AE, Cops EJ et al. Nucleocytoplasmic coagulation: an injury-induced aggregation event that disulfide crosslinks proteins and facilitates their removal by plasmin. Cell Rep 2012; 2: 889–901.
Kulkarni S, Woollard KJ, Thomas S, Oxley D, Jackson SP . Conversion of platelets from a proaggregatory to a proinflammatory adhesive phenotype: role of PAF in spatially regulating neutrophil adhesion and spreading. Blood 2007; 110: 1879–1886.
Craig R, Beavis RC . TANDEM: matching proteins with tandem mass spectra. Bioinformatics 2004; 20: 1466–1467.
Kessner D, Chambers M, Burke R, Agus D, Mallick P . ProteoWizard: open source software for rapid proteomics tools development. Bioinformatics 2008; 24: 2534–2536.
Acknowledgements
We thank Mr Andrew Webb (Alfred Hospital Blood Services) for the provision of expired platelet bags. We thank Mr Steve Cody (Monash Microimaging, Alfred Hospital) for advice and support relating to the microscopic experiments. We thank the Burnet Institute for use of their Zeiss Cell Observer system. We thank the Myeloma Research Group (Australian Centre for Blood Diseases, Monash University) for donating the JIM-1 and KMS26 cell lines as well as provision of bortezomib. We thank the Australian Proteome Analysis Facility (Macquarie University, Australia) for performing the Ingenuity Pathway Analysis. We thank Dr Simone M Schoenwaelder and Dr Sue L Cranmer (Australian Centre for Blood Diseases, Monash University) for advice, support and reagents pertaining to the washing and activation of human platelets. We thank the Signalling Network Laboratory (Department of Biochemistry, Monash University) for providing the EGFR and phospho-EGFR antibodies. This work was generously supported by two grants from the Brain Foundation awarded to RLM, AE-LA and ALS. AE-LA was supported by an Australian Postgraduate Award.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by H-U Simon
Supplementary Information accompanies this paper on Cell Death and Disease website
Rights and permissions
Cell Death and Disease is an open-access journal published by Nature Publishing Group. This work is licensed under a Creative Commons Attribution 3.0 Unported License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/3.0/
About this article

Cite this article
Au, AL., Sashindranath, M., Borg, R. et al. Activated platelets rescue apoptotic cells via paracrine activation of EGFR and DNA-dependent protein kinase. Cell Death Dis 5, e1410 (2014). https://doi.org/10.1038/cddis.2014.373
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cddis.2014.373
This article is cited by
-
Platelet-derived chemokines promote skeletal muscle regeneration by guiding neutrophil recruitment to injured muscles
Nature Communications (2023)
-
Protective effects of platelet-rich plasma against lidocaine cytotoxicity on canine articular chondrocytes
Acta Veterinaria Scandinavica (2018)
-
Clinical baseline and prognostic difference of platelet lymphocyte ratio (PLR) in right-sided and let-sided colon cancers
BMC Cancer (2017)
-
Altered B-lymphopoiesis in mice with deregulated thrombopoietin signaling
Scientific Reports (2017)

{kind=link}
{kind=link}
{kind=link}