
Abstract
Background:
We evaluated the safety, maximum-tolerated dose (MTD), pharmacokinetics, recommended dose for phase II (P2RD), and preliminary anticancer activity of a combination eribulin and S-1 therapeutic in metastatic breast cancer patients pretreated with anthracycline and taxane.
Method:
Patients aged 20–74 years were recruited. In level 1, patients received S-1 (65 mg m−2) from day 1 to 14, and eribulin (1.1 mg m−2) on day 1 and 8 in a 21-day cycle. In level 2, eribulin was increased to 1.4 mg m−2. In level 3, S-1 was increased to 80 mg m−2.
Results:
Twelve patients were enrolled into three cohorts. Planned dose escalation was completed, with one case exhibiting dose-limiting toxicity (grade 3 hypokalaemia) at level 3, without reaching the MTD. The P2RD was determined to be level 2 (eribulin 1.4 mg m−2 and S-1 65 mg m−2). The most common grade 3 or 4 toxicity was neutropenia (83.3%), followed by febrile neutropenia (25.0%). Five of eleven patients (41.7%) with measurable disease had a partial response. Pharmacokinetics were characterised by dose-dependent elimination and nonlinear exposure.
Conclusion:
Dose level 3 was not tolerated owing to febrile neutropenia development. Thus, intermediate dose level 2 was recommended for further evaluation. Preliminary antitumour activity warrants further investigation in this setting.
Similar content being viewed by others


Main
Breast cancer is a leading cause of death among women worldwide (Jemal et al, 2011). Breast cancer mortality has declined in western countries because of multidisciplinary efforts over the last decade, including improved detection through screening, increased specialisation of care (Kingsmore et al, 2003), and better access to more effective treatments, such as improved surgical techniques, targeted radiotherapy, and adjuvant therapies, including tamoxifen (Autier et al, 2010). Nevertheless, clinical outcomes in metastatic breast cancer (MBC) remain poor, and identification of therapeutics to improve treatment is necessary. As MBC remains an incurable disease, the main treatment objectives are to prolong survival time and provide palliative care. The standard first-line chemotherapy for MBC utilises anthracyclines or taxanes (Mincey and Perez, 2004; Hamilton and Hortobagyi, 2005), which are the mainstays of adjuvant therapy for breast cancer. However, tumour cells often develop resistance to these drugs. Thus, novel treatments that improve overall survival (OS) but minimise toxicity and maintain a good quality of life for women with heavily pretreated MBC are necessary.
Eribulin mesylate (Halaven), an analogue of the marine sponge-derived compound halichondrin B, is a non-taxane microtubule dynamics inhibitor with a distinct mechanism of action from other tubulin-targeted drugs (Jain and Vahdat, 2011). Eribulin inhibits tumour growth in taxane-resistant human ovarian cells harbouring β-tubulin mutations, suggesting the potential to overcoming taxane resistance because of gene alterations (Kuznetsov et al, 2009). The side effects of this drug are reported to be manageable, with notable occurrence of neutropenia and fatigue, and a relatively low incidence of peripheral neuropathy (Jain and Vahdat, 2011). Further, eribulin significantly increased OS (median OS for the eribulin-treated group was 13.1 months vs 10.6 months for the group treated by investigator’s choice) in MBC patients who were refractory to both anthracyclines and taxanes (Cortes et al, 2011). Recently, a large-scale phase III trial comparing eribulin to capecitabine, one of the best MBC therapeutics, revealed that the drugs had comparable efficacies. Furthermore, treatment of triple negative breast cancer (TNBC) with eribulin showed a slightly better outcome than capecitabine (Kaufman et al, 2012). Thus, eribulin has become a standard care for MBC.
S-1, an oral fluoropyrimidine capsule formulation that consists of 1 M tegafur (a prodrug of 5-fluorouracil (5-FU)), 0.4 M 5-chloro-2,4-dihydroxypyridine, and 1 M potassium oxonate, has efficient antitumour activity and low gastrointestinal toxicity (Okamoto and Fukuoka, 2009). It has been widely used in Asian countries, including Japan, and is accepted as a standard care for gastric (Sakuramoto et al, 2007; Koizumi et al, 2008; Boku et al, 2009), colorectal (Yamada et al, 2013), non-small-cell lung (Okamoto et al, 2010), and pancreatic cancer (Ueno et al, 2013). S-1 is also recommended as an option for third-line or later MBC treatment in Japan (JapanBreastCancerSociety, 2013), based on phase II studies that showed a response rate of 40.7 and 42.0% as first- or second-line treatment (Saek et al, 2004), and of 21.8% as a salvage treatment (Saeki et al, 2004).
The combination of eribulin and S-1 has not yet been investigated. We recently found that combination of S-1 and eribulin has a synergistic effect in vitro and in vivo (Terashima et al, 2014), supporting the evaluation of this combination in clinical trials. Thus, we conducted a phase I dose-escalation study using combined eribulin and S-1 to evaluate the safety and pharmacokinetic profiles of each drug. Furthermore, we determined a recommended drug dose for phase II trials (phase II trial recommended dose, P2RD).
Patients and Methods
Patient eligibility
Eligible patients were 20–74 years of age with a confirmed diagnosis of MBC. They were required to have an Eastern Cooperative Oncology Group performance status of 0 or 1 and adequate organ function. Previous treatments, including chemotherapy, radiotherapy, and surgery, were allowed if they had been completed at least 4 weeks before registration. However, previous administration of both anthracycline and taxanes was required. Patients with other serious illnesses or medical conditions, such as uncontrolled infection, other malignancies, or central nervous system metastases that were still symptomatic, were also not eligible for participation. All participants received information about the nature and purpose of the study, and provided written informed consent in accordance with the institutional guidelines. The consent was given before any study procedures. Having measurable disease was not a requirement to participate in this study.
Study design and patient selection
The study was designed as a single-centre, open-label, dose-escalation phase I trial. The primary objectives were to determine the maximum-tolerated dose (MTD) and the P2RD for the combination of eribulin and S-1, and to collect overall safety data. Secondary objectives included the determination of pharmacokinetic variables, as well as a preliminary assessment of antitumour activity in the treatment population. The study was reviewed and approved by the Institutional Review Board of Kinki University, Faculty of Medicine. This study has been registered with the UMIN Clinical Trials Registry (UMIN-CTR, UMIN 000009716). The study was performed according to the International Conference on Harmonisation Good Clinical Practices.
Treatment schedule
S-1 was administered orally with approximately 200 ml water and within 30 min after a meal (ideally after breakfast and evening meal, 12 h apart). S-1 was administered in two doses, 65 mg m−2 (25 mg for body surface area (BSA) less than 1.25 m2, 40 mg for BSA from 1.25 to 1.49 m2, 50 mg for BSA greater than 1.5 m2, twice daily) or 80 mg m−2 (40 mg for BSA less than 1.25 m2, 50 mg for BSA from 1.25 to 1.49 m2, 60 mg for BSA greater than 1.5 m2, twice daily) on days 1–14 every 3 weeks, in combination with two doses of eribulin (1.1 or 1.4 mg m−2), given intravenously on days 1 and 8 every 3 weeks. In addition, for the prevention of nausea and vomiting, patients received dexamethasone 12 mg i.v. 30 min before the start of intravenous chemotherapy. Three patients were recruited at each S-1 and eribulin dose level, and at least three patients received at least one cycle and were observed for toxicity for at least 3 weeks before dose escalation was permitted. If no patients experienced a DLT, the dose was escalated to the next level in subsequent patients. If one of the three patients developed a DLT, then that dose level was expanded to six patients. If an additional patient in the six-patient cohort experienced a DLT, no further dose escalation was allowed, and the previous dose level was identified as the MTD. If none of patients at dose level 3 experienced DLTs, dose level 3 was expanded to six patients. If DLTs were exhibited in less than one of six patients at dose level 3, then the level was not escalated further, as the dosages of both drugs corresponded to those approved in Japan, and the trial would not reach a MTD. The MTD was the highest dose level at which no more than one of six patients treated exhibited DLTs. The occurrence of one or more of the following toxicities during the first cycle of chemotherapy was considered dose limiting: any National Cancer Institute Common Toxicity Criteria (NCI CTC) grade 3 or 4 non-haematological toxicity (excluding grade 3 alopecia, grade 3 nausea or vomiting, or grade 3 stomatitis persisting for <3 days), platelet count<25 000 cells per μl or<50 000 cells per μl accompanied by bleeding requiring blood transfusion, neutropenia (absolute neutrophil count <500 cells per μl for >7 days), or grade 4 febrile neutropenia (absolute neutrophil count <500 cells per μl accompanied by a fever ⩾38.5 °C (single evaluation), or a fever>38 °C for >12 h and unable to have S-1 for more than 6 days in the first cycle due to any toxicity).
The dose intensity was calculated as follows: the sum of the actual given dose (mg m−2)/the actual treatment weeks × patient number. Dose delay was incorporated in actual weeks.
Patient evaluation
The safety and tolerability of the eribulin and S-1 combination were assessed according to the NCI CTC version 4.0. Radiological tumour assessment was performed every two cycles to confirm the response until progression. Objective tumour response was evaluated according to the Response Evaluation Criteria in Solid Tumors (RECIST) version 1.1. Progression-free survival (PFS) was calculated as the time from the first day of treatment to the first day of documented progression or death. OS was calculated from the first day of the combination therapy until death from any cause or the date of last contact. The probability of survival as a function of time was estimated with the Kaplan–Meier method. Analyses were performed with STATA version 13.1 (StataCorp, College Station, TX, USA).
Pharmacokinetics
The plasma pharmacokinetics of the combination treatment were investigated in order to assess the potential interactions between eribulin and S-1 at each dose level. The pharmacokinetics of eribulin were evaluated on day 1 immediately before and 0.1, 0.5, 1, 2, 4, 6, 12, and 168 h after administration during cycle 1. The pharmacokinetics of S-1 were evaluated on day 1 immediately before and 1, 2, 4, 6, and 12 h after administration during cycle 1. The plasma concentrations of eribulin and S-1 were measured by Shin Nippon Biomedical Laboratories, LTD (Tokyo, Japan) and FALCO Biosystems (Kyoto, Japan), respectively. All concentrations were determined using liquid chromatography and tandem mass spectrometry (Matsushima et al, 1997). Differences in pharmacokinetic parameters were evaluated using the Student’s t-test, and a P value <0.05 was considered statistically significant.
Results
Patient characteristics
From October 2012 to December 2013, 12 patients were enrolled in this phase I trial. The date of data cutoff was 20 March 2014. The characteristics of the 12 study patients are summarised in Table 1. The median age was 64 years, with a range of 49–70 years. Ten individuals had recurrent disease, whereas two had stage 4 disease. All patients had previously received two to seven chemotherapies, including anthracycline and taxane.
Dose escalation and determination of MTD and P2RD
The dose-escalation scheme, including the number of cycles, patients, and DLTs by dose level, is shown in Table 2. Given that no DLTs were observed at dose level 1 and 2, the dose of S-1 and eribulin was escalated to level 3. Because no DLTs were observed in the initial three patients at dose level 3, an additional three patients were assigned to dose level 3, according to the protocol. Among these three additional patients at dose level 3, one patient experienced a DLT in cycle 1, exhibiting grade 3 hypokalaemia. Short episodes of febrile neutropenia that responded to treatment with oral antibiotics were observed in two of three additional patients during the assessment period. Although a pre-specified DLT was experienced in only a single patient at level 3, and the MTD of eribulin/S-1 combination therapy was not reached, three of six patients at level 3 exhibited grade 3 febrile neutropenia (two patients in cycle 1 and one in cycle 3). Although this was not defined as a DLT, in view of non-DLT grade 3 febrile neutropenia in three patients at 80 mg m−2, DL2 was chosen as the P2RD.
Treatment administered
Sixty-seven cycles of chemotherapy were administered, with a median of five treatment cycles per patient (range 1–11). The mean relative dose intensities of S-1 and eribulin were 68.7% and 67.3%, respectively. Dose reductions were observed in 13 (19.4% of total cycles) cycles in six patients because of neutropenia, followed by skipping a dose in four patients (33.3%), febrile neutropenia in three patients (25.0%), and prolongation of grade 2 peripheral neuropathy in one patient. Treatment delay occurred in 19 cycles (28.3% of total cycles) in nine patients, primarily due to neutropenia.
Toxicities
The toxicity profiles observed over the entire treatment period are shown in Table 3. All patients who received the combination therapy were assessable for toxicity. The most common grade 3 or 4 toxicity was neutropenia in 10 (83.3%) cases, followed by grade 3 febrile neutropenia in 3 (25.0%) cases, both of which were clinically manageable. In contrast, grade 3 or 4 non-haematological toxicities were not observed throughout the study period, except for grade 3 hypokalaemia in one case (8.3%), which was defined as a DLT. Onset of the grade 3 hypokalaemia occurred in cycle 1 on day 11, and the serum potassium level was normalised within 7 days. The possible occurrence of this event was attributable to grade 2 diarrhoea along with fever, followed by decreased dietary intake. Diarrhoea was deemed an adverse event caused by S-1. The patient was receiving concomitant medications, including thiamazole 5 mg per day and pravastatin 10 mg per day, for comorbidities. The other major grade 1 or 2 non-haematological toxicities were fatigue (n=4, 33.3%), diarrhoea (n=2, 16.6%), peripheral neuropathy (n=2, 16.6%), and oral mucositis (n=2, 16.6%). There were no treatment-related deaths. The most frequent reason for discontinuation of therapy was disease progression (n=5, 41.7%), followed by the patients’ refusal (n=2, 16.6%).
Pharmacokinetics
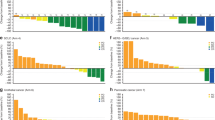
All 12 patients in the dose-escalation phase of the study were evaluated for pharmacokinetic analysis. The plasma concentration vs time curves of 5-FU and eribulin on day 1 of the first treatment cycle are shown in Figure 1A and B, respectively. The plasma concentration of S-1 peaked at 2 h, and declined from the maximum concentration (Cmax) rapidly. In contrast, plasma concentrations of eribulin peaked when the infusion finished, and declined from Cmax rapidly. Both the AUC and Cmax for total 5-FU and eribulin increased proportionally with increasing dose (Table 4).

PK analysis of enrolled patient population: a relationship between plasma concentration vs time profiles for each (A) S-1 and (B) eribulin dose level.
Efficacy
Eleven of the eleven patients were assessable for antitumour response, with a patient at dose level 3 having no measurable lesions. Five patients showed a partial response, yielding an overall response rate of 41.7% (95% confidence interval (CI): 8.9–74.4), and five other patients had stable disease, giving an overall disease control rate of 91.7% (95% CI: 73.3–NA). At data cutoff, seven patients were alive and one patient remained on the combination therapy. The median PFS for all 12 treated patients was 7.6 months (95% CI: 1.3–NA) and the OS was not reached.
Discussion
Breast cancer is not a single disease, but a combination of many diseases. Regardless of disease stage, therapeutic management for patients with breast cancer should be optimised and individualised based on tumour biology, as well as many other factors surrounding them. TNBC is a distinct subset of disease, and its prognosis is worse than other subtypes, mainly due to a lack of effective targeted medicines. Recently, tremendous efforts have been made to elucidate the tumour biology of this disease, and targeted therapeutics are undergoing evaluation for patients with TNBC, including PARP and mTOR inhibitors. Nevertheless, specific targets for TNBC remain unclear. Thus, cytotoxic agents still have an important role in TNBC treatment. Chemotherapeutic combinations may be useful in patients with rapidly progressing cancer or that previously did not respond to chemotherapy. Indeed, MBC treatment guidelines recommend combination chemotherapy in these cases (Cardoso et al, 2014; Partridge et al, 2014). Thus, we sought to identify an efficacious combination with a favourable toxicity profile.
The most common grade 3 or higher toxicity observed in the current study was neutropenia (83.3%). This toxicity was also seen in previous trials evaluating 1.4 mg m−2 of eribulin monotherapy in the same setting (Cortes et al, 2011; Aogi et al, 2012) and was mild and manageable. Further, incidence of febrile neutropenia following monotherapy was ∼10%. However, we observed 25% febrile neutropenia in the current study. Furthermore, after treatment with 1.4 mg m−2 eribulin and 80 mg m−2 S-1 in this study, all six patients experienced grade 3–4 neutropenia, and three patients developed grade 3 febrile neutropenia. These findings suggest that eribulin-induced neutropenia may be enhanced by S-1, although the PKs of eribulin were not influenced by S-1. Given that MBC patients in this setting often have insufficient bone marrow function because of prior treatments, including several lines of chemotherapy and radiation, haematological toxicities can be prolonged and exacerbated. We thus considered level 2 as the P2RD for future studies, although the MTD was not reached according to the pre-specified DLTs and the protocol. In contrast, non-haematologic DLTs were mild (grade 1 or 2), including fatigue (33.3%), diarrhoea (16.7%), and peripheral neuropathy (16.7%). One patient developed moderate hypokalaemia (grade 3), likely as a consequence of grade 2 diarrhoea, suggesting the importance of appropriate management, even for mild toxicities, in this subset of patients.
In the present study, we investigated the plasma pharmacokinetics of combination eribulin and S-1 treatment to assess potential interactions between the two drugs at each dose level. As compared with previous studies using eribulin monotherapy (Goel et al, 2009; Tan et al, 2009; Mukohara et al, 2012), the plasma concentration profile and pharmacokinetic parameters for eribulin did not appear to be affected by S-1 co-administration. This was consistent with previous observations that eribulin and capecitabine, another oral fluoropyrimidine prodrug used in a phase Ib trial, were not significantly different than eribulin alone (Nasim et al, 2012). These findings suggest that oral fluoropyrimidines, such as S-1 and capecitabine, do not interact with eribulin in terms of plasma concentration profile and pharmacokinetic parameters.
A growing amount of evidence suggests that eribulin has some off-target effects in addition to tubulin disruption. Recent preclinical studies have revealed that eribulin has the ability to convert the epithelial–mesenchymal transition (EMT) state to the mesenchymal–epithelial transition state (Yoshida et al, 2014). EMT has been reported to play a role in the invasive and metastatic potential of cancer progression (Hugo et al, 2007; Peinado et al, 2007; Tsuji et al, 2009), and the acquisition of resistance to several anti-cancer agents, including 5-FU (Thomson et al, 2005; Arumugam et al, 2009; Wang et al, 2009; Singh and Settleman, 2010). We recently observed that 5-FU induced EMT in TNBC, leading to 5-FU resistance, whereas eribulin reversed EMT and sensitised cells to 5-FU (Terashima et al, 2014). Indeed, both in vitro and in vivo, the combination of eribulin and S-1 resulted in significantly higher antitumour activity than eribulin or S-1 alone. These finding suggest that the action of eribulin on mesenchymal–epithelial transition improves 5-FU resistance, resulting in a synergistic effect. Although we are unable to reach any firm conclusion regarding the efficacy of this regimen because of the small size of our phase I trial, the promising antitumour activity of this combination in the current study, with an overall response rate of 41.7% and median PFS of 7.6 months, may reflect this mechanism of action. Thus, we plan to conduct a phase II study of this combination to further evaluate its safety and efficacy.
In conclusion, the MTD of the combination therapy was not reached in this study, and P2RD was set as 1.4 mg m−2 eribulin intravenously injected on days 1 and 8 of a 21-day cycle in combination with 65 mg m−2 oral S-1 for 14 days, followed by 1 week of rest. A further clinical study evaluating the safety and efficacy of this combination is warranted.
Change history
03 March 2015
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
Aogi K, Iwata H, Masuda N, Mukai H, Yoshida M, Rai Y, Taguchi K, Sasaki Y, Takashima S (2012) A phase II study of eribulin in Japanese patients with heavily pretreated metastatic breast cancer. Ann Oncol 23 (6): 1441–1448.
Arumugam T, Ramachandran V, Fournier KF, Wang H, Marquis L, Abbruzzese JL, Gallick GE, Logsdon CD, McConkey DJ, Choi W (2009) Epithelial to mesenchymal transition contributes to drug resistance in pancreatic cancer. Cancer Res 69 (14): 5820–5828.
Autier P, Boniol M, La Vecchia C, Vatten L, Gavin A, Hery C, Heanue M (2010) Disparities in breast cancer mortality trends between 30 European countries: retrospective trend analysis of WHO mortality database. BMJ 341: c3620.
Boku N, Yamamoto S, Fukuda H, Shirao K, Doi T, Sawaki A, Koizumi W, Saito H, Yamaguchi K, Takiuchi H, Nasu J, Ohtsu A Gastrointestinal Oncology Study Group of the Japan Clinical Oncology G (2009) Fluorouracil versus combination of irinotecan plus cisplatin versus S-1 in metastatic gastric cancer: a randomised phase 3 study. Lancet Oncol 10 (11): 1063–1069.
Cardoso F, Costa A, Norton L, Senkus E, Aapro M, André F, Barrios CH, Bergh J, Biganzoli L, Blackwell KL, Cardoso MJ, Cufer T, El Saghir N, Fallowfield L, Fenech D, Francis P, Gelmon K, Giordano SH, Gligorov J, Goldhirsch A, Harbeck N, Houssami N, Hudis C, Kaufman B, Krop I, Kyriakides S, Lin UN, Mayer M, Merjaver SD, Nordström EB, Pagani O, Partridge A, Penault-Llorca F, Piccart MJ, Rugo H, Sledge G, Thomssen C, van’t Veer L, Vorobiof D, Vrieling C, West N, Xu B, Winer E (2014) ESO-ESMO 2nd international consensus guidelines for advanced breast cancer (ABC2). Ann Oncol 25 (10): 1871–1888.
Cortes J, O'Shaughnessy J, Loesch D, Blum JL, Vahdat LT, Petrakova K, Chollet P, Manikas A, Dieras V, Delozier T, Vladimirov V, Cardoso F, Koh H, Bougnoux P, Dutcus CE, Seegobin S, Mir D, Meneses N, Wanders J, Twelves C investigators E (2011) Eribulin monotherapy versus treatment of physician's choice in patients with metastatic breast cancer (EMBRACE): a phase 3 open-label randomised study. Lancet 377 (9769): 914–923.
Goel S, Mita AC, Mita M, Rowinsky EK, Chu QS, Wong N, Desjardins C, Fang F, Jansen M, Shuster DE, Mani S, Takimoto CH (2009) A phase I study of eribulin mesylate (E7389), a mechanistically novel inhibitor of microtubule dynamics, in patients with advanced solid malignancies. Clin Cancer Res 15 (12): 4207–4212.
Hamilton A, Hortobagyi G (2005) Chemotherapy: what progress in the last 5 years? J Clin Oncol 23 (8): 1760–1775.
Hugo H, Ackland ML, Blick T, Lawrence MG, Clements JA, Williams ED, Thompson EW (2007) Epithelial—mesenchymal and mesenchymal—epithelial transitions in carcinoma progression. J Cell Physiol 213 (2): 374–383.
Jain S, Vahdat LT (2011) Eribulin mesylate. Clin Cancer Res 17 (21): 6615–6622.
JapanBreastCancerSociety (2013) The Japanese clinical guideline for breast cancer based on scientific evidences 1: medical treatment. (in Japanese).
Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D (2011) Global cancer statistics. CA Cancer J Clin 61 (2): 69–90.
Kaufman PA, Awada A, Twelves C, Yelle L, Perez EA, Wanders J, Olivo MS, He Y, Dutcus CE, Cortes Norris Cotton (2012) A phase III, open-label, randomized, multicenter study of eribulin mesylate versus capecitabine in patients with locally advanced or metastatic breast cancer previously treated with anthracyclines and taxanes. San Antonio Breast Cancer Symp [S6-6] 6 December 2012 San Antonio, TX, USA.
Kingsmore D, Ssemwogerere A, Hole D, Gillis C (2003) Specialisation and breast cancer survival in the screening era. Br J Cancer 88 (11): 1708–1712.
Koizumi W, Narahara H, Hara T, Takagane A, Akiya T, Takagi M, Miyashita K, Nishizaki T, Kobayashi O, Takiyama W, Toh Y, Nagaie T, Takagi S, Yamamura Y, Yanaoka K, Orita H, Takeuchi M (2008) S-1 plus cisplatin versus S-1 alone for first-line treatment of advanced gastric cancer (SPIRITS trial): a phase III trial. Lancet Oncol 9 (3): 215–221.
Kuznetsov G1, Xu Q, Rudolph-Owen L, Tendyke K, Liu J, Towle M, Zhao N, Marsh J, Agoulnik S, Twine N, Parent L, Chen Z, Shie JL, Jiang Y, Zhang H, Du H, Boivin R, Wang Y, Romo D, Littlefield BA (2009) Potent in vitro and in vivo anticancer activities of des-methyl, des-amino pateamine A, a synthetic analogue of marine natural product pateamine A. Mol Cancer Ther 8 (5): 1250–1260.
Matsushima E, Yoshida K, Kitamura R, Yoshida K (1997) Determination of S-1 (combined drug of tegafur, 5-chloro-2,4-dihydroxypyridine and potassium oxonate) and 5-fluorouracil in human plasma and urine using high-performance liquid chromatography and gas chromatography-negative ion chemical ionization mass spectrometry. J Chromatogr B Biomed Sci Appl 691 (1): 95–104.
Mincey BA, Perez EA (2004) Advances in screening, diagnosis, and treatment of breast cancer. Mayo Clin Proc 79 (6): 810–816.
Mukohara T, Nagai S, Mukai H, Namiki M, Minami H (2012) Eribulin mesylate in patients with refractory cancers: a Phase I study. Invest New Drugs 30 (5): 1926–1933.
Nasim MY, Plummer R, Evans TRJ, Morrison R, Anthoney DA, Haney S, Madi A, Savulsky CI, Johnston C, Carter D, Reyderman L, Gopalakrishna P, Wanders J, Twelves C (2012) A phase Ib dose-escalation study of eribulin mesylate in combination with capecitabine in patients with advanced/metastatic cancer. J Clin Oncol 30 (suppl): abstract 2552.
Okamoto I, Fukuoka M (2009) S-1: a new oral fluoropyrimidine in the treatment of patients with advanced non-small-cell lung cancer. Clin Lung Cancer 10 (4): 290–294.
Okamoto I, Yoshioka H, Morita S, Ando M, Takeda K, Seto T, Yamamoto N, Saka H, Asami K, Hirashima T, Kudoh S, Satouchi M, Ikeda N, Iwamoto Y, Sawa T, Miyazaki M, Tamura K, Kurata T, Fukuoka M, Nakagawa K (2010) Phase III trial comparing oral S-1 plus carboplatin with paclitaxel plus carboplatin in chemotherapy-naive patients with advanced non-small-cell lung cancer: results of a west Japan oncology group study. J Clin Oncol 28 (36): 5240–5246.
Partridge AH, Rumble RB, Carey LA, Come SE, Davidson NE, Di Leo A, Gralow J, Hortobagyi GN, Moy B, Yee D, Brundage SB, Danso MA, Wilcox M, Smith IE (2014) Chemotherapy and targeted therapy for women with human epidermal growth factor receptor 2–negative (or unknown) advanced breast cancer: American Society of Clinical Oncology Clinical Practice Guideline. J Clin Oncol 32 (29): 3307–3329.
Peinado H, Olmeda D, Cano A (2007) Snail, Zeb and bHLH factors in tumour progression: an alliance against the epithelial phenotype? Nat Rev Cancer 7 (6): 415–428.
Saek T, Takashima S, Sano M, Horikoshi N, Miura S, Shimizu S, Morimoto K, Kimura M, Aoyama H, Ota J, Noguchi S, Taguchi T (2004) A phase II study of S-1 in patients with metastatic breast cancer—a Japanese trial by the S-1 Cooperative Study Group, Breast Cancer Working Group. Breast Cancer 11 (2): 194–202.
Saeki T, Takashima S, Sano M, Horikoshi N, Miura S, Shimizu S, Morimoto K, Kimura M, Taguchi T (2004) [A late phase II clinical study of S-1 in patients with progressed, refractory breast cancer]. Gan To Kagaku Ryoho 31 (4): 539–547.
Sakuramoto S, Sasako M, Yamaguchi T, Kinoshita T, Fujii M, Nashimoto A, Furukawa H, Nakajima T, Ohashi Y, Imamura H, Higashino M, Yamamura Y, Kurita A, Arai K Group A-G (2007) Adjuvant chemotherapy for gastric cancer with S-1, an oral fluoropyrimidine. N Engl J Med 357 (18): 1810–1820.
Singh A, Settleman J (2010) EMT, cancer stem cells and drug resistance: an emerging axis of evil in the war on cancer. Oncogene 29 (34): 4741–4751.
Tan AR, Rubin EH, Walton DC, Shuster DE, Wong YN, Fang F, Ashworth S, Rosen LS (2009) Phase I study of eribulin mesylate administered once every 21 days in patients with advanced solid tumors. Clin Cancer Res 15 (12): 4213–4219.
Terashima M, Sakai K, Togashi Y, Hayashi H, Velasco MAD, Tsurutani J, Nishio K (2014) Synergistic antitumor effects of S-1 with eribulin in vitro and in vivo for triple-negative breast cancer cell lines. Springer Plus 3: 147.
Thomson S, Buck E, Petti F, Griffin G, Brown E, Ramnarine N, Iwata KK, Gibson N, Haley JD (2005) Epithelial to mesenchymal transition is a determinant of sensitivity of non-small-cell lung carcinoma cell lines and xenografts to epidermal growth factor receptor inhibition. Cancer Res 65 (20): 9455–9462.
Tsuji T, Ibaragi S, Hu GF (2009) Epithelial-mesenchymal transition and cell cooperativity in metastasis. Cancer Res 69 (18): 7135–7139.
Ueno H, Ioka T, Ikeda M, Ohkawa S, Yanagimoto H, Boku N, Fukutomi A, Sugimori K, Baba H, Yamao K, Shimamura T, Sho M, Kitano M, Cheng AL, Mizumoto K, Chen JS, Furuse J, Funakoshi A, Hatori T, Yamaguchi T, Egawa S, Sato A, Ohashi Y, Okusaka T, Tanaka M (2013) Randomized phase III study of gemcitabine plus S-1, S-1 alone, or gemcitabine alone in patients with locally meta and metastatic pancreatic cancer in Japan and Taiwan: GEST study. J Clin Oncol 31 (13): 1640–1648.
Wang Z, Li Y, Kong D, Banerjee S, Ahmad A, Azmi AS, Ali S, Abbruzzese JL, Gallick GE, Sarkar FH (2009) Acquisition of epithelial-mesenchymal transition phenotype of gemcitabine-resistant pancreatic cancer cells is linked with activation of the notch signaling pathway. Cancer Res 69 (6): 2400–2407.
Yamada Y, Takahari D, Matsumoto H, Baba H, Nakamura M, Yoshida K, Yoshida M, Iwamoto S, Shimada K, Komatsu Y, Sasaki Y, Satoh T, Takahashi K, Mishima H, Muro K, Watanabe M, Sakata Y, Morita S, Shimada Y, Sugihara K (2013) Leucovorin, fluorouracil, and oxaliplatin plus bevacizumab versus S-1 and oxaliplatin plus bevacizumab in patients with metastatic colorectal cancer (SOFT): an open-label, non-inferiority, randomised phase 3 trial. Lancet Oncol 14 (13): 1278–1286.
Yoshida T, Ozawa Y, Kimura T, Sato Y, Kuznetsov G, Xu S, Uesugi M, Agoulnik S, Taylor N, Funahashi Y, Matsui J (2014) Eribulin mesilate suppresses experimental metastasis of breast cancer cells by reversing phenotype from epithelial-mesenchymal transition (EMT) to mesenchymal-epithelial transition (MET) states. Br J Cancer 110 (6): 1497–1505.
Acknowledgements
This study was supported by the Osaka Medical Research Foundation for Intractable Disease. We thank Asami Kitano, Yume Shinkai, Haruka Sakamoto, and Tomoka Yamada from the Department of Medical Oncology, Kinki University Faculty of Medicine, Osaka, Japan. We gratefully acknowledge the participating patients, their families, and study investigators for their invaluable contribution. We thank Editage (http://www.editage.jp) for English language editing.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
This work is published under the standard license to publish agreement. After 12 months the work will become freely available and the license terms will switch to a Creative Commons Attribution-NonCommercial-Share Alike 4.0 Unported License.
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 4.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/
About this article

Cite this article
Sakiyama, T., Tsurutani, J., Iwasa, T. et al. A phase I dose-escalation study of eribulin and S-1 for metastatic breast cancer. Br J Cancer 112, 819–824 (2015). https://doi.org/10.1038/bjc.2015.10
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/bjc.2015.10
