
Abstract
Background
The aim of this study was to investigate the antitumour activity, safety, and tolerability of pamiparib plus tislelizumab in patients with previously treated advanced solid tumours.
Methods
In this study, patients were enrolled into eight arms by tumour type. All received pamiparib 40 mg orally twice daily plus tislelizumab 200 mg intravenously every 3 weeks. The primary endpoint was objective response rate (ORR), assessed by the investigator per Response Evaluation Criteria in Solid Tumours v1.1. Secondary endpoints included duration of response (DoR), safety, and tolerability.
Results
Overall, 180 patients were enrolled. In the overall population, the ORR was 20.0% (range: 0–47.4 across study arms), with median DoR of 17.1 months (95% confidence interval [CI]: 6.2, not estimable [NE]). The highest ORR was observed in the triple-negative breast cancer (TNBC) arm (patients with BRCA1/2 mutations and/or homologous recombination deficiency) (ORR: 47.4%; median DoR: 17.1 months [95% CI: 3.0, NE]). Treatment-emergent adverse events (TEAEs) of ≥Grade 3 occurred in 61.7% of patients. Serious TEAEs occurred in 50.0% of patients.
Conclusions
Pamiparib plus tislelizumab showed a variable level of antitumour activity in patients with advanced solid tumours, with the highest ORR in TNBC and was associated with a manageable safety profile.
Clinical trial registration
ClinicalTrial.gov: NCT02660034.
Similar content being viewed by others


Introduction
PARP 1/2 inhibitors interfere with DNA repair mechanisms by binding directly to PARP enzymes, thereby inhibiting their activity, and through trapping PARP-DNA complexes at the site of DNA damage. These effects lead to genomic instability, prevention of DNA transcription/translation, increased DNA damage, and tumour cell death [1,2,3,4]. PARP inhibitors are synthetically lethal in tumours with homologous recombination deficiencies (HRD), in particular in tumours with either a germline or somatic mutation in the breast cancer type 1/2 susceptibility gene (BRCA1/2) [5]. Several PARP inhibitors are approved for the treatment of ovarian, breast, prostate, and pancreatic cancers, including for patients with deleterious/suspected deleterious BRCA mutations (BRCAmut) and/or tumours with HRD [6,7,8,9].
PARP inhibitors may enhance the antitumour effects of immune checkpoint inhibitors, such as programmed cell death protein 1 (PD-1)/programmed death-ligand 1 (PD-L1) inhibitors [3, 4, 10]. PARP inhibitor-induced tumour cell death leads to tumour neoantigen release, facilitating potential immune responses [3, 10]. PARP inhibitors also have other effects that may potentiate antitumour immune responses and create a favourable environment for immune checkpoint blockade, such as promotion of T-cell infiltration and upregulation of interferons [3, 10, 11]. These effects may be mediated through mechanisms such as activation of the cGMP/AMP-synthase-stimulator-of-interferon genes (cGAS-STING) pathway and increased chemokine levels [3, 10]. However, PARP inhibitors also upregulate PD-L1 expression on tumour cells via various mechanisms (e.g., GSK3β, JAK2/STAT3 signalling pathway, or activation of the cGAS-STING pathway) [10,11,12] which could, in turn, lead to suppression of T-cell-mediated immune responses, and therefore represent a mechanism of resistance to PARP inhibitors [10, 12].
Preclinical studies suggest addition of a PD-(L)1 inhibitor can resensitise cells treated with PARP inhibitors to T-cell cytotoxicity, which restores the reduced antitumour immunity caused by upregulation of PD-L1 expression and enhances antitumour activity [10,11,12]. Furthermore, HRD tumours have been reported to exhibit traits that may favour immune checkpoint blockade, such as high neoantigen load, increased PD-L1 expression, and increased levels of tumour-infiltrating lymphocytes [13]. Consequently, several clinical trials have been initiated to investigate the effects of combination PARP inhibitors with PD-(L)1 inhibitors in various solid tumours [3, 10, 14,15,16,17,18,19].
Pamiparib (developed by BeiGene, Ltd.) is a potent, selective, investigational small molecule inhibitor of PARP1 and PARP2 that has demonstrated brain penetration and PARP-DNA complex trapping in preclinical studies [20, 21]. In phase I and II clinical studies, pamiparib demonstrated antitumour activity and induced durable responses in patients with epithelial ovarian cancer (EOC) and HER2-negative (HER2−) breast cancer with germline BRCAmut [22,23,24,25].
Several PD-(L)1 inhibitors are approved as monotherapy and/or in combination with chemotherapy and/or other agents for the treatment of a range of solid tumours, with survival benefits demonstrated versus placebo and various other comparators [26]. Tislelizumab is an anti-PD-1 monoclonal antibody with high affinity and binding specificity for PD-1 [27, 28] and was specifically engineered to minimise Fc-gamma receptor binding on macrophages [28, 29]. Clinical studies have demonstrated durable antitumour efficacy of tislelizumab in various solid tumours [30,31,32,33], and it is approved in China for the treatment of several tumour types.
A phase Ia/b study was initiated to investigate the combination of pamiparib with tislelizumab in patients with advanced solid tumours [34]. The study comprised two phases: dose escalation (part A) and dose expansion (part B). In the dose-escalation phase, the combination was generally well tolerated and demonstrated antitumour activity, supporting its continued investigation in the dose-expansion phase [34]. The objective response rate (ORR) was 20.4%, with responses seen in patients with gynaecological cancers (ovarian, fallopian or peritoneal) and breast cancer [34]. The recommended phase II dose was pamiparib 40 mg orally twice daily plus tislelizumab 200 mg intravenously (IV) every 3 weeks (Q3W) [34]. Here, we report results from the dose-expansion phase, which sought to investigate the antitumour activity, safety, and tolerability of pamiparib combined with tislelizumab at the recommended phase II dose in patients with a variety of advanced solid tumour types (with and without a germline or somatic BRCAmut and HRD), including cohorts of patients with EOC and TNBC.
Methods
Study design
Part B was a multicentre, open-label, multiple-arm, dose-expansion study that evaluated the antitumour activity, safety, and tolerability of pamiparib plus tislelizumab (NCT02660034). The study enrolled patients into eight arms according to tumour type (Supplementary Fig. S1). The study arms included patients with ovarian, TNBC, prostate, small cell lung, gastric or gastroesophageal junction, urothelial, or pancreatic cancers, and an exploratory arm included patients with non-ovarian gynaecological cancers, and patients with tumours that were mismatch repair deficient or HRD who were not eligible for inclusion in other arms. Patients were not randomised and there was no blinding of study treatments. Study endpoints were evaluated independently in each arm to explore the clinical activity, safety, and tolerability of pamiparib plus tislelizumab in each selected tumour type. All patients provided written informed consent before study participation. All relevant Institutional Review Boards/Independent Ethics Committees approved the study, which was carried out in accordance with the International Conference on Harmonization Good Clinical Practice Guideline, the principles of the Declaration of Helsinki, and local laws and regulations.
Participants
Eligible patients were adults (≥18 years of age) with histologically confirmed malignancies that have progressed to the advanced/metastatic stage, with measurable disease per Response Evaluation Criteria in Solid Tumours (RECIST) v1.1, and an Eastern Cooperative Oncology Group (ECOG) performance status of 0 or 1.
As outlined in Supplementary Fig. S1, tumour-specific eligibility criteria per study arm were as follows: relapsed, platinum-sensitive high-grade epithelial, non-mucinous, ovarian cancer, fallopian tube, or primary peritoneal cancer (termed “EOC” hereafter), with a known germline or somatic BRCAmut and/or HRD (Arm 1a), or without mutation (BRCAwt) and with homologous recombination proficiency (HRP) (Arm 1b); TNBC with either a known germline or somatic BRCAmut and/or with documented HRD (Arm 2); metastatic castration-resistant prostate cancer (mCRPC) with either a known germline or somatic BRCAmut and/or with documented HRD (Arm 3); extensive-stage small cell lung cancer (SCLC) (Arm 4); HER2− gastric or gastroesophageal junction cancer (Arm 5); locally advanced or metastatic urothelial cancer (Arm 6); advanced or metastatic pancreatic adenocarcinoma (Arm 7). The final arm of the study (Arm 8) enrolled patients with advanced or metastatic recurrent non-ovarian gynaecological cancers (endometrial cancer or cancer of the cervix), and patients with tumours known to be either mismatch repair deficient or HRD that were not eligible for inclusion in any other arms of the trial but may be expected to benefit from the combination of a PARP inhibitor and a PD-1 inhibitor (termed “exploratory arm” hereafter). For the purposes of determination of study eligibility, patients’ BRCAmut and HRD/HRP status in Arms 1–3 were determined using historical clinical test results or, when results were not available, based on central testing of samples using the Myriad BRCAnalysis CDx or Myriad myChoice® CDx methods (for a germline BRCAmut and HRD/HRP status, respectively). For patients enrolled on the basis of local historical clinical test results, confirmatory central testing was subsequently performed using archival or fresh samples, where possible.
Prior treatment varied by tumour type, but all patients were required to have received standard of care for the treatment of their disease (Supplementary Fig. S1). Patients who had received treatment in the advanced/metastatic setting were also eligible. If received, chemotherapy or investigational therapy must have been completed ≥4 weeks (or ≥5 half-lives, whichever was longer) prior to administration of the study treatment, and palliative radiotherapy must have been completed ≥2 weeks prior.
Key exclusion criteria were platinum-resistant/refractory EOC and prior treatment with therapies targeting PD-1, PD-L1, or PARP. Full eligibility criteria are provided in the Supplementary Appendix.
Interventions
All patients received pamiparib 40 mg orally twice daily plus tislelizumab 200 mg IV Q3W (Supplementary Fig. S1). The dosing regimen was selected based on the previously reported results of the dose-escalation phase of the study [34]. The initial infusion of tislelizumab was administered over 60 min and subsequently reduced to 30 min if well tolerated. Study treatments were administered until disease progression, unacceptable toxicity, loss to follow-up, death, or discontinuation for other reasons. Continued treatment beyond progression was permitted if “pseudo-progression” was suspected by the investigator, provided protocol-specified criteria were met.
If required to manage adverse events (AEs), pamiparib dose reduction to 20 mg was permitted, and dosing could also be withheld for up to 28 days consecutively. No dose reduction was permitted for tislelizumab, but dose delays of less than 12 weeks were permitted. Whether an AE was possibly related to either pamiparib or tislelizumab alone, or both, was assessed by the investigator, and the study drug(s) considered to be responsible were modified accordingly.
Endpoints and assessments
The primary endpoint of this study was ORR, defined as the proportion of patients with a documented complete response (CR) or partial response (PR) per RECIST v1.1, as assessed by investigators. Tumour imaging was performed within 28 days prior to enrolment, every 9 weeks (±1 week) in the first 12 months of enrolment, and every 12 weeks (±1 week) thereafter. Computed tomography (CT) or magnetic resonance imaging techniques were used, with a preference for CT. The same imaging technique was used throughout the study for each individual patient. All known disease was documented at baseline as target or non-target lesions per RECIST v1.1.
Secondary endpoints included: progression-free survival (PFS), duration of response (DoR), disease control rate (DCR), and clinical benefit rate (CBR), all by investigator per RECIST v1.1; and overall survival (OS), safety and tolerability, pharmacokinetic parameters, and the immunogenicity of tislelizumab. Safety and tolerability were assessed by the incidence and nature of AEs. AEs were coded using the Medical Dictionary for Regulatory Activities (MedDRA) version 22.0 and graded per National Cancer Institute’s Common Terminology Criteria for Adverse Events version 4.03. The assignment of immune-mediated AEs was assessed by investigators based on diagnostic test results and clinical judgement, and the exclusion of alternative explanations, in line with criteria defined in the study protocol. Other safety assessments included vital signs, electrocardiograms, laboratory analyses, and physical and ophthalmologic examinations. In addition, as an elevated incidence of hepatic AEs was observed with the combination of pamiparib and tislelizumab in the dose-escalation part of the study [34], the incidence of hepatic AEs is reported. Pharmacokinetic parameters assessed included Cmax, Ctrough and Tmax for pamiparib and Ctrough for tislelizumab. Immunogenic responses to tislelizumab were assessed in terms of the incidence of anti-drug antibodies (ADAs).
Statistical analyses
This dose-expansion study planned to initially enrol 20 patients in each tumour-specific arm. The probability of observing at least one responder was calculated to be approximately 88% in each dose-expansion arm (n = 20) if the underlying ORR was as low as 10%. Twenty additional patients could be enrolled in any arm to further evaluate antitumour activity if evidence of activity was observed. Since a precise estimate of ORR is difficult to predict due to the heterogeneity of patients enrolled within each arm, the Bayesian predictive probability, which evaluates the statistical strength of the pamiparib plus tislelizumab combination regimen versus the standard chemotherapy, will be used to provide guidance to decide whether to enrol an additional twenty patients. For example, for an arm with a historical response rate of 10%, at least two responders of the initial 20 patients should be observed to have a predictive probability of >10% superiority over the 10% historical rate in a total of 50 patients; however, for an arm with higher expected response rate (e.g., >30% ORR in arm 1a), at least six responders are required to have a >10% predictive probability in order to expand the arm beyond the initial 20 patients. Conversely, a decision could be made to stop enrolment in an arm early due to suboptimal clinical antitumour activity. Efficacy and safety analyses were performed in the safety analysis set, which included all patients who received any dose of any study treatment. Pharmacokinetic analyses were performed in all patients with valid pharmacokinetic sampling after treatment with study drug(s) (the pharmacokinetic analysis set). Immunogenicity analyses were performed among patients with evaluable data (ADA evaluable population).
Data for each arm in this study were analysed independently. For PFS and OS, median durations and event-free rates at various timepoints were estimated using the Kaplan–Meier method: for medians and other quartiles, 95% confidence intervals (CIs) were estimated using the Brookmeyer and Crowley method; for event-free rates, 95% CIs were estimated using the Kaplan–Meier method and the Greenwood formula. Patients who remained alive before data cutoff or discontinuation of the study were censored at the last date the subject was known to be alive. DoR analyses only included patients who responded to treatment, with the same censoring rules used for PFS, and Kaplan–Meier curves were used to estimate median DoR and 95% CIs. The incidence of treatment-emergent AEs (TEAEs), incidence of ADAs, laboratory test results, vital signs, pharmacokinetic parameters, and their changes from baseline were summarised using descriptive statistics. All safety analyses were performed by arms and by the total combination of cohorts in the safety analysis set. All calculations and analyses were conducted using SAS version 9.2 or higher. The study was not powered to detect statistical significance.
Results
Patients and treatment
Patients were recruited from 25 sites across five countries (Australia, France, New Zealand, Spain, and the United States) between 21 July 2017 and 9 April 2019, with a median time of 25.8 months from initial diagnosis to study entry. In total, 180 patients were assigned to the eight study arms. Patient demographics and baseline characteristics, including BRCAmut/HRD status, are presented in Table 1 and Supplementary Table S1. All 180 patients were included in the safety analysis set, of whom all except two patients had received at least one prior line of systemic therapy.
At the data cutoff of 25 September 2020, all 180 patients (100.0%) had discontinued treatment with pamiparib and tislelizumab, and had also discontinued from the study (Supplementary Table S2). Progressive disease (PD) was the most common reason for treatment discontinuation for both pamiparib (73.3% of patients) and tislelizumab (71.7% of patients). Following completion or discontinuation of treatment, patients were followed for their survival status until discontinuation from the study due to patient death or other reasons. In total, 68.9% of patients discontinued the study due to death, 3.3% due to withdrawal by patient, 1.7% due to loss to follow-up, 0.6% due to physician decision, 0.6% due to study termination by the sponsor, and 25.0% due to other reasons, including transfer to a long-term extension study of tislelizumab and/or pamiparib (NCT04164199).
The median duration of exposure to tislelizumab was 104.0 days (range: 21–994 days), and the median number of completed cycles was 5 (range: 1–46 cycles). The median duration of exposure to pamiparib was 104.0 days (range: 6–952 days), the median relative dose intensity was 98% (range: 24–100%), and the median number of completed cycles was 5 (range: 1–46 cycles).
Antitumour activity
ORR per RECIST v1.1 in the overall study population was 20.0%, with responses occurring in 36 patients in total (Table 2). ORR ranged from 0% to 47.4% across study arms. Overall, 12 (6.7%) patients achieved a CR and 24 (13.3%) patients achieved a PR, while stable disease was observed in 50 (27.8%) patients (Table 2). The highest ORR was observed in patients with TNBC (Arm 2; 13 out of the 19 patients had centrally confirmed HRD, of whom six also had a centrally confirmed germline BRCAmut). In this arm, the ORR was 47.4% (nine of 19 patients), with three patients achieving a CR (15.8%; two had a centrally confirmed germline BRCAmut and one had centrally confirmed germline BRCAwt). Patients with EOC with BRCAmut and/or HRD had an ORR of 30.4% (seven of 23 patients), with a CR rate of 8.7% (two of 23 patients; both patients had centrally confirmed HRD and germline BRCAwt). The change in target lesion size from baseline in each study arm is shown in Fig. 1.

*BRCAmut by central testing; †BRCAwt by central testing; ‡BRCA unknown by central testing; §HRD by central testing; |HRP by central testing; ¶HRD unknown by central testing. #Patients with non-ovarian gynaecological cancers (endometrial cancer or cancer of the cervix) and patients with tumours known to be mismatch repair deficient or HRD that are not eligible for inclusion in any other arms of the trial but that may be expected to benefit from the PARP/PD-1 inhibitor combination (see Supplementary Table S1 for the full list of cancer types enrolled in this arm). Dotted lines at −30% and 20% indicate the boundaries for disease response and progression, respectively. Data are presented only for patients with an evaluable post-baseline assessment of target lesions. Data cutoff: 25 September 2020. BRCAmut breast cancer type 1/2 susceptibility gene mutation, BRCAwt breast cancer type 1/2 susceptibility gene wildtype, CR complete response, EOC epithelial ovarian cancer, G/GEJ gastric or gastroesophageal junction, HER2− HER2 negative, HRD homologous recombination deficiency, HRP homologous recombination proficiency, mCRPC metastatic castration-resistant prostate cancer, PD-1 programmed cell death protein 1, PD progressive disease, PR partial response, SCLC small cell lung cancer, SD stable disease, TNBC triple-negative breast cancer.
In the overall population, median DoR among responders was 17.1 months (95% CI: 6.2, not estimable [NE]) (Table 2). Where reached, median DoR ranged from 6.2 months in both EOC with BRCAwt and HRP (95% CI: 3.8, NE), and SCLC (95% CI: 4.3, 8.1), to 17.1 months (95% CI: 3.0, NE) in TNBC. Median DoR was 11.2 months (95% CI: 6.2, NE) in EOC with BRCAmut and/or HRD, but was not reached by responders in the remaining arms. In the overall population, DCR was 51.7% and CBR was 35.0%.
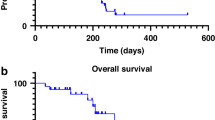
Median PFS and OS were 4.0 months (95% CI: 2.2, 5.2) and 10.4 months (95% CI: 7.7, 14.2), respectively (Table 2). The longest PFS was seen in the arms including patients with mCRPC, TNBC, and EOC with BRCAmut and/or HRD, with observed median PFS of 10.4 (95% CI: 4.3, 16.2), 8.4 (95% CI: 3.9, 19.0), and 8.2 (95% CI: 5.2, 11.8) months, respectively. Median OS in these arms was 21.2 months (95% CI: 10.5, NE) in mCRPC, 20.9 months (95% CI: 13.5, NE) in EOC with BRCAmut and/or HRD, and 15.8 months (95% CI: 10.4, NE) in TNBC. PFS and OS rates at various timepoints are reported in Supplementary Table S3. Kaplan–Meier curves for PFS and OS are presented for all arms in Fig. 2, and are also presented discretely for the TNBC arm in Supplementary Fig. S2.

a Progression-free survival. b Overall survival. *Patients with non-ovarian gynaecological cancers (endometrial cancer or cancer of the cervix) and patients with tumours known to be mismatch repair deficient or HRD that are not eligible for inclusion in any other arms of the trial but that may be expected to benefit from the PARP/PD-1 inhibitor combination (see Supplementary Table S1 for the full list of cancer types enrolled in this arm). Data cutoff: 25 September 2020. BRCAmut breast cancer type 1/2 susceptibility gene mutation, BRCAwt breast cancer type 1/2 susceptibility gene wildtype, EOC epithelial ovarian cancer, G/GEJ gastric or gastroesophageal junction, HER2− HER2 negative, HRD homologous recombination deficiency, HRP homologous recombination proficiency, mCRPC metastatic castration-resistant prostate cancer, OS overall survival, PD-1 programmed cell death protein 1, PFS progression-free survival, SCLC small cell lung cancer, TNBC triple-negative breast cancer.
Safety and tolerability
In the overall study population, all patients except one (99.4%) experienced at least one TEAE (Table 3). TEAEs were defined using MedDRA preferred terms. Nausea was the most commonly reported any grade TEAE (56.1% of patients) (Table 4). TEAEs of ≥Grade 3 were reported in 61.7% of patients. The most common ≥Grade 3 TEAEs were anaemia and alanine aminotransferase (ALT) increased (both 7.2%). Immune-mediated TEAEs occurred in 17.2% of patients. TEAEs related to either pamiparib or tislelizumab were reported in 82.2% of patients, with nausea the most commonly reported TEAE related to either tislelizumab or pamiparib (in 40.6% of patients; Supplementary Table S4).
Half of the study patients experienced at least one serious TEAE (50.0%), and 16.1% of patients experienced a serious TEAE related to either pamiparib or tislelizumab. The most commonly reported serious TEAE related to either pamiparib or tislelizumab was immune-mediated hepatitis (3.3%). Eight (4.4%) patients experienced a TEAE leading to death, none of which were considered related to treatment.
In the overall population, 25.6% of patients experienced at least one hepatic TEAE (Table 3). Hepatic TEAEs were defined using investigator selection of MedDRA preferred terms according to laboratory assessed terms (ALT increased, aspartate aminotransferase [AST] increased, blood bilirubin increased, transaminases increased) or non-laboratory assessed terms (hepatitis, immune-mediated hepatitis, autoimmune hepatitis, hepatic failure).
When assessed based on only the first hepatic TEAE experienced by the patient, the most commonly occurring hepatic TEAEs of any grade were ALT increased (12.2% of patients), hepatitis (3.9%), AST increased (2.8%), blood bilirubin increased (2.2%), and immune-mediated hepatitis (2.2%). In total, 8.3% of patients in the overall population experienced a ≥Grade 3 hepatic TEAE as their first hepatic TEAE, with the majority of these patients (eight of the 15 patients) from the EOC arms. The most common first ≥Grade 3 hepatic TEAEs were ALT increased (2.8%), and hepatitis (2.2%).
When assessed based on total incidence regardless of chronology, the most common hepatic TEAEs (with an incidence of ≥10% in the safety analysis set) were AST increased (16.7%) and ALT increased (16.1%). The most common ≥Grade 3 hepatic TEAEs were ALT increased (7.2% of patients), AST increased (4.4%), hepatitis (3.9%), blood bilirubin increased (2.8%), and immune-mediated hepatitis (2.8%). Among those patients who experienced hepatic TEAEs, the first hepatic TEAEs typically began during Cycle 1 or 2 of treatment (median onset: day 40 of treatment [range: day 6–485]). There were no deaths attributed to hepatic TEAEs.
Of the 46 patients who experienced a hepatic TEAE, five patients had ALT or AST >3 × the upper limit of normal (ULN) with a concomitant total bilirubin that was >2 × ULN within a 3-day period. There were four patients who had concomitant alkaline phosphatase (ALP) >2 × ULN within a 3-day period, whilst the remaining patient showed concomitant ALP <2 × ULN within a 3-day period with a serious AE of immune-mediated hepatitis (meeting the criteria for Hy’s law [ALT or AST >3 × ULN; total bilirubin >2 × ULN; no initial findings of cholestasis [no elevation of ALP to >2 × ULN]; and no other reason for the increase in ALT and bilirubin] [35]). The immune-mediated hepatitis was evaluated as related to tislelizumab but not pamiparib by the investigator, and the event resolved following the discontinuation of study drug and steroid treatment.
Overall, TEAEs led to permanent discontinuation of tislelizumab or pamiparib in 15.0% of patients for each drug, and discontinuation of both drugs in 7.2% of patients (Table 3). The most common (incidence >2.0% of patients) TEAEs leading to discontinuation of pamiparib were in the “hepatobiliary disorders” and “investigations” MedDRA system organ classes (4.4% and 2.8% of patients, respectively), and included hepatitis (2.2%) and ALT increased (2.2%). Similarly, the most common TEAEs leading to discontinuation of tislelizumab were also in the “hepatobiliary disorders” and “investigations” system organ classes (6.7% and 3.9% of patients, respectively), and included immune-mediated hepatitis and ALT increased (each 3.3%). In addition, TEAEs led to dose reduction of pamiparib in 5.6% of patients.
Pharmacokinetics and immunogenicity
The plasma concentrations of pamiparib and tislelizumab indicated that the combination of the two study drugs had no impact on the pharmacokinetic profile of either drug (data not shown). The overall prevalence and incidence of ADAs for tislelizumab were low (3.3% [6/180 patients in the overall population], and 3.2% [5/156 patients in the ADA evaluable population], respectively), and no patients tested positive for neutralising antibodies.
Discussion
The dose-expansion part of this study was conducted in 180 patients with previously treated locally advanced or metastatic solid tumours, including cohorts of patients with either BRCAmut and/or HRD. The combination of pamiparib 40 mg orally twice daily plus tislelizumab 200 mg IV Q3W demonstrated antitumour activity in several tumour types, durable responses in a subset of patients, and a manageable safety profile.
In the overall study population, the ORR was 20.0%, which was consistent with the results previously reported for the dose-escalation part of the study (20.4%) [34], despite variation in tumour type enrolment between the two parts. The highest ORR (47.4%) was seen in the arm enrolling patients with TNBC with BRCAmut and/or HRD. Two phase III trials have reported ORRs of a similar magnitude with PARP inhibitor monotherapy in patients with advanced or metastatic TNBC with a germline BRCAmut, with ORRs of 61.8% for talazoparib (the EMBRACA trial), and 54.7% for olaparib (the OlympiAD trial) [36, 37]. Furthermore, the OlympiAD trial of olaparib in patients with metastatic TNBC with BRCAmut demonstrated a significantly longer PFS compared to standard treatment [37]. However, these trials of PARP inhibitor monotherapy exclusively enrolled patients with BRCAmut, which is known to be a predictive biomarker for PARP inhibitor sensitivity [38]. In contrast, among the patients with TNBC in the present study who underwent central germline BRCA testing, one-third (5/15) had BRCAwt. A BRCAwt population may have been less likely to respond to PARP inhibition, although three of these five patients had centrally confirmed HRD (the remaining two were enrolled on the basis of a historical local assessment of a germline BRCAmut status that was not confirmed during subsequent central assessment). For context, in the Olaparib Expanded trial in patients with metastatic breast cancer (most with oestrogen receptor-positive HER2− subtype breast cancer and a minority with TNBC), the ORR with olaparib monotherapy in the cohort with a germline mutation in a non-BRCA1/2 homologous recombination gene was 33% [39]. In addition, the TNBC patients in the current study were more heavily pre-treated than in the EMBRACA and OlympiAD trials and all had received at least one prior line of systemic therapy in the advanced/metastatic setting. For PD-(L)1 inhibitor monotherapy, low ORRs (typically <10%) have been reported in patients with previously treated metastatic TNBC, albeit with the potential for durable responses in a small subset [40]. The promising response rate for the pamiparib and tislelizumab combination reported herein is of particular interest due to the poor prognosis of patients with metastatic TNBC harbouring BRCAmut and/or HRD who have progressed on prior lines of systemic therapy [41, 42].
Previous studies of combinations of PARP inhibitors with PD-(L)1 inhibitors have demonstrated antitumour activity in a variety of tumour types, including in patients with TNBC with BRCAmut, although there are limited data from head-to-head clinical trials of the combinations versus their component monotherapies [3, 10, 24, 43,44,45]. For example, the phase I/II MEDIOLA trial of olaparib plus durvalumab reported an ORR of 58.8%, a median DoR of 12.9 months, and a median PFS of 4.9 months in patients with metastatic TNBC with BRCAmut [44]. Meanwhile, the single-arm TOPACIO/KEYNOTE-162 phase II study of niraparib combined with pembrolizumab in advanced/metastatic TNBC reported an ORR of 47% among patients with BRCAmut, with a median PFS of 8.3 months [43]. The ORRs in these studies appear comparable to that reported with olaparib alone (54.7%) among patients with metastatic TNBC in the phase III OlympiAD trial [37]; however, cross-trial comparisons should be interpreted cautiously given the potential influence of variations in prior therapy and patient characteristics between studies. In the present study, an ORR of 47.4%, a median DoR of 17.1 months, and a median PFS of 8.4 months were reported with pamiparib plus tislelizumab in patients with TNBC harbouring BRCAmut and/or with HRD, consistent with the promising response rate seen in such patients in the prior trials of PARP inhibitors with PD-(L)1 inhibitors [43, 44]. These results support further investigation of such combinations in this setting.
The second highest ORR (30.4%) was seen in the EOC arm harbouring BRCAmut and/or with HRD. Previously, a phase I/II study of pamiparib monotherapy reported an ORR of 64.6% in previously treated advanced platinum-sensitive ovarian cancer [24], while in the germline BRCA1/2 mutated platinum-sensitive relapsed ovarian cancer cohort of the MEDIOLA trial the ORR was 71.9% with olaparib plus durvalumab [46]. In the recurrent platinum-resistant ovarian cancer cohort (n = 53) of the TOPACIO/KEYNOTE-162 phase I/II trial investigating niraparib and pembrolizumab, regardless of BRCA status, the ORR was 18%, with three (5%) patients experiencing CR and eight (13%) with PR [47]. However, comparisons between these trials should be interpreted cautiously due to differences in patient populations, most notably the presence of germline BRCAmut in all patients in both the pamiparib monotherapy study [24] and in the MEDIOLA trial cohort [46], compared with only 17.4% in the present study. For tislelizumab monotherapy, a phase Ia/b study reported an ORR of 9.8% in previously treated advanced ovarian cancer [48] which, taken together with the results of the present study, suggests that adding pamiparib may enhance antitumour responses. Further investigation into the addition of tislelizumab to pamiparib to improve antitumour activity is needed.
The ORR with pamiparib plus tislelizumab in the urothelial cancer cohort in the present study (28.6%) also appeared encouraging, and was similar to findings reported in the phase III KEYNOTE-045 trial of pembrolizumab as second-line therapy for advanced urothelial cancer (21.1%) [49] and in a phase II trial of tislelizumab in Asian patients with previously treated advanced PD-L1-positive urothelial carcinoma (24.0%) [33]. Although results of the phase II BAYOU trial indicated that addition of olaparib to durvalumab in patients with metastatic urothelial carcinoma did not improve PFS or OS, the combination did appear beneficial in the subgroup of patients with HRD, supporting the continued investigation of PARP inhibitor and PD-(L)1 inhibitor combinations in urothelial cancer [50].
Compared with urothelial cancer, TNBC and EOC harbouring BRCAmut and/or with HRD, ORR was lower in the other tumour-specific treatment arms, including in the patients with EOC without BRCAmut or HRD. These results suggest that the effect of PARP inhibitors combined with PD-(L)1 inhibitors is affected by both tumour type and mutational signature, and emphasise the need for further research into the combination in carefully defined populations.
It is notable that patients with mCRPC in the present study had a median PFS of 10.4 months (95% CI: 4.3, 16.2) and a median OS of 21.2 months (95% CI: 10.5, NE), which were longer than observed in other treatment arms. For comparison, in a recent phase III trial, median PFS and OS of patients with mCRPC receiving olaparib monotherapy were reported to be 5.8 and 17.5 months, respectively [51]. Accepting the limitation that our mCRPC sample was small (n = 20) and should therefore be interpreted cautiously, these findings suggest that pamiparib in combination with a PD-(L)1 inhibitor, such as tislelizumab, may have improved efficacy in patients with mCRPC and should be further investigated. Similar findings were found in a small study (n = 17) of olaparib in combination with the PD-L1 inhibitor durvalumab in mCRPC, which reported a median PFS of 16.1 months among patients with deficiencies in DNA damage repair genes [52].
Overall, the safety profile of the pamiparib plus tislelizumab combination was manageable. The most commonly reported TEAEs were consistent with those reported for the combination in the phase Ia part of the study [34], and more broadly with those reported previously for the individual agents [22, 48, 53]. Although more than half of patients (61.7%) experienced a ≥Grade 3 TEAE, TEAEs led to discontinuation of treatment in a relatively small number of patients (15.0% each for discontinuation of pamiparib and tislelizumab [7.2% discontinued both]). TEAEs leading to dose reduction of pamiparib were also infrequent (5.6% of patients) and lower than reported with other PARP inhibitors (with the caveat of cross-trial comparison) [54,55,56,57].
As seen in the dose-escalation part of the study [34], hepatic TEAEs were reported in some patients in this dose-expansion phase. The most commonly reported any grade and ≥Grade 3 hepatic TEAEs (whether reported as the first or subsequent hepatic TEAE) were AST increased (any grade in 16.7% of patients and ≥Grade 3 in 4.4% of patients) and ALT increased (16.1% and 7.2%, respectively), and there were few cases of ≥Grade 3 hepatitis (3.9% of patients), ≥Grade 3 immune-mediated hepatitis (2.8% of patients), and ≥Grade 3 blood bilirubin increased (2.8% of patients). An increased incidence of hepatic TEAEs (particularly increased transaminase levels) has been previously reported with PD-(L)1 inhibitors, such as nivolumab and atezolizumab, as well as PARP inhibitors, such as rucaparib [58, 59]. However, an increased incidence of hepatic TEAEs has not been reported in prior studies of pamiparib or tislelizumab, particularly in terms of a notable elevation in ≥Grade 3 increased transaminase levels [23, 24, 30,31,32,33, 48, 53]. There was variation in the incidence of hepatic TEAEs between the tumour-specific study arms in the present study, with greatest incidence in the EOC and urothelial cancer arms. Possible explanations for the higher hepatic TEAEs reported in some arms are prior therapies, predisposition to immune-mediated hepatitis, or liver metastases. Nevertheless, the reported hepatic TEAEs do not appear to compromise the safety and tolerability profile of the combination, with only one of the 46 patients who experienced hepatic TEAEs meeting the criteria for Hy’s law.
The strengths of the study included the assessment of the pamiparib and tislelizumab combination in multiple tumour types, including cohorts with EOC and TNBC, with variability in BRCA mutation and HRD status. Potential limitations include the heterogeneous patient population, small sample size, and lack of a head-to-head arms versus PARP inhibitor or PD-1 inhibitor monotherapy, which limits the ability to draw conclusions on the effects of combination therapy versus monotherapy. In addition, the attribution and assignment of the cause of AEs by investigators when patients were on two drugs was subjective and can be challenging. However, the patient numbers and study design are typical of, and appropriate for, a phase I expansion study.
In conclusion, pamiparib in combination with tislelizumab showed evidence of antitumour activity in patients with advanced solid tumours, particularly those with BRCAmut and/or HRD tumours, with a manageable safety profile in keeping with the class of agents. This study supports further investigation of this combination strategy, particularly in patients with TNBC with BRCAmut and/or HRD.
Data availability
On request, and subject to certain criteria, conditions, and exceptions, BeiGene, Ltd., will provide access to individual de-identified participant data from BeiGene-sponsored global interventional clinical studies conducted for medicines (1) for indications that have been approved or (2) in programs that have been terminated. BeiGene will also consider requests for the protocol, data dictionary, and statistical analysis plan. Data requests may be submitted to DataDisclosure@beigene.com.
References
Lupo B, Trusolino L. Inhibition of poly(ADP-ribosyl)ation in cancer: old and new paradigms revisited. Biochim Biophys Acta. 2014;1846:201–15.
Dziadkowiec KN, Gąsiorowska E, Nowak-Markwitz E, Jankowska A. PARP inhibitors: review of mechanisms of action and BRCA1/2 mutation targeting. Prz Menopauzalny. 2016;15:215–9.
Vikas P, Borcherding N, Chennamadhavuni A, Garje R. Therapeutic potential of combining PARP inhibitor and immunotherapy in solid tumors. Front Oncol. 2020;10:570.
Stewart RA, Pilié PG, Yap TA. Development of PARP and immune-checkpoint inhibitor combinations. Cancer Res. 2018;78:6717–25.
Miller RE, Leary A, Scott CL, Serra V, Lord CJ, Bowtell D, et al. ESMO recommendations on predictive biomarker testing for homologous recombination deficiency and PARP inhibitor benefit in ovarian cancer. Ann Oncol. 2020;31:1606–22.
US Food and Drug Administration. RUBRACA® (rucaparib) tablets, for oral use. Prescribing information. Revised 6/2022.
US Food and Drug Administration. LYNPARZA® (olaparib) tablets, for oral use. Prescribing information. Revised 3/2022.
US Food and Drug Administration. ZEJULA (niraparib) capsules, for oral use. Prescribing information. Revised 7/2021.
US Food and Drug Administration. TALZENNA (talazoparib) capsules, for oral use. Prescribing information. Revised 9/2021.
Wu Z, Cui P, Tao H, Zhang S, Ma J, Liu Z, et al. The synergistic effect of PARP inhibitors and immune checkpoint inhibitors. Clin Med Insights Oncol. 2021;15:1179554921996288.
Wang Y, Zheng K, Xiong H, Huang Y, Chen X, Zhou Y, et al. PARP inhibitor upregulates PD-L1 expression and provides a new combination therapy in pancreatic cancer. Front Immunol. 2021;12:762989.
Jiao S, Xia W, Yamaguchi H, Wei Y, Chen MK, Hsu JM, et al. PARP inhibitor upregulates PD-L1 expression and enhances cancer-associated immunosuppression. Clin Cancer Res. 2017;23:3711–20.
Strickland KC, Howitt BE, Shukla SA, Rodig S, Ritterhouse LL, Liu JF, et al. Association and prognostic significance of BRCA1/2-mutation status with neoantigen load, number of tumor-infiltrating lymphocytes and expression of PD-1/PD-L1 in high grade serous ovarian cancer. Oncotarget. 2016;7:13587–98.
Lee JM, Annunziata CM, Houston N, Kohn EC, Lipkowitz S, Minasian L, et al. A phase II study of durvalumab, a PD-L1 inhibitor and olaparib in recurrent ovarian cancer (OvCa). Ann Oncol. 2018;29:viii334.
Lampert EJ, Zimmer A, Padget M, Cimino-Mathews A, Nair JR, Liu Y, et al. Combination of PARP inhibitor alaparib, and PD-L1 inhibitor durvalumab, in recurrent ovarian cancer: a proof-of-concept phase II study. Clin Cancer Res. 2020;26:4268–79.
Meng J, Peng J, Feng J, Maurer J, Li X, Li Y, et al. Niraparib exhibits a synergistic anti-tumor effect with PD-L1 blockade by inducing an immune response in ovarian cancer. J Transl Med. 2021;19:415.
Moore K, Redhead K, Goble S, Bowles O, Lin K, Tripathi A. 312 Aries: a phase 2, open-label study to evaluate rucaparib (PARP inhibitor) in combination with nivolumab (anti-PD-1 antibody) in patients with ovarian or urothelial cancer (UC). Int J Gynecol Cancer. 2019;29:A130.
Konstantinopoulos P, Gockley A, Xiong N, Tayob N, Krasner C, Buss M, et al. LBA35 phase II study of PARP inhibitor talazoparib and PD-L1 inhibitor avelumab in patients (pts) with microsatellite stable (MSS) recurrent/persistent endometrial cancer. Ann Oncol. 2020;31:S1142–215.
ClinicalTrials.gov. NCT04276376. Efficacy and safety of the combination of rucaparib (PARP inhibitor) and atezolizumab (Anti-PD-L1 antibody) in patients with DNA repair-deficient or platinum-sensitive solid tumors (ARIANES) 2020.
Xiong Y, Guo Y, Liu Y, Wang H, Gong W, Liu Y, et al. Pamiparib is a potent and selective PARP inhibitor with unique potential for the treatment of brain tumor. Neoplasia. 2020;22:431–40.
Wang H, Ren B, Liu Y, Jiang B, Guo Y, Wei M, et al. Discovery of pamiparib (BGB-290), a potent and selective poly (ADP-ribose) polymerase (PARP) inhibitor in clinical development. J Med Chem. 2020;63:15541–63.
Lickliter JD, Voskoboynik M, Mileshkin L, Gan HK, Kichenadasse G, Zhang K, et al. Phase 1A/1B dose-escalation and -expansion study to evaluate the safety, pharmacokinetics, food effects and antitumor activity of pamiparib in advanced solid tumours. Br J Cancer. 2022;126:576–85.
Xu B, Yin Y, Dong M, Song Y, Li W, Huang X, et al. Pamiparib dose escalation in Chinese patients with non-mucinous high-grade ovarian cancer or advanced triple-negative breast cancer. Cancer Med. 2021;10:109–18.
Wu X, Zhu J, Wang J, Lin Z, Yin R, Sun W, et al. Pamiparib monotherapy for patients with germline BRCA1/2-mutated ovarian cancer previously treated with at least two lines of chemotherapy: a multicenter, open-label, phase II study. Clin Cancer Res. 2022;28:653–61.
Sun T, Shi Y, Cui J, Yin Y, Ouyang Q, Liu Q, et al. A phase 2 study of pamiparib in the treatment of patients with locally advanced or metastatic HER2-negative breast cancer with germline BRCA mutation. J Clin Oncol. 2021;39:1087.
Chang E, Pelosof L, Lemery S, Gong Y, Goldberg KB, Farrell AT, et al. Systematic review of PD-1/PD-L1 inhibitors in oncology: from personalized medicine to public health. Oncologist. 2021;26:e1786–99.
Hong Y, Feng Y, Sun H, Zhang B, Wu H, Zhu Q, et al. Tislelizumab uniquely binds to the CC′ loop of PD-1 with slow-dissociated rate and complete PD-L1 blockage. FEBS Open Bio. 2021;11:782–92.
Zhang T, Song X, Xu L, Ma J, Zhang Y, Gong W, et al. The binding of an anti-PD-1 antibody to FcγRΙ has a profound impact on its biological functions. Cancer Immunol Immunother. 2018;67:1079–90.
Dahan R, Sega E, Engelhardt J, Selby M, Korman AJ, Ravetch JV. FcγRs modulate the anti-tumor activity of antibodies targeting the PD-1/PD-L1 axis. Cancer Cell. 2015;28:285–95.
Wang J, Lu S, Yu X, Hu Y, Sun Y, Wang Z, et al. Tislelizumab plus chemotherapy vs chemotherapy alone as first-line treatment for advanced squamous non–small-cell lung cancer: a phase 3 randomized clinical trial. JAMA Oncol. 2021;7:709–17.
Lu S, Wang J, Yu Y, Yu X, Hu Y, Ai X, et al. Tislelizumab plus chemotherapy as first-line treatment for locally advanced or metastatic nonsquamous NSCLC (RATIONALE 304): a randomized phase 3 trial. J Thorac Oncol. 2021;16:1512–22.
Xu J, Bai Y, Xu N, Li E, Wang B, Wang J, et al. Tislelizumab plus chemotherapy as first-line treatment for advanced esophageal squamous cell carcinoma and gastric/gastroesophageal junction adenocarcinoma. Clin Cancer Res. 2020;26:4542–50.
Ye D, Liu J, Zhou A, Zou Q, Li H, Fu C, et al. Tislelizumab in Asian patients with previously treated locally advanced or metastatic urothelial carcinoma. Cancer Sci. 2021;112:305–13.
Friedlander M, Meniawy T, Markman B, Mileshkin L, Harnett P, Millward M, et al. Pamiparib in combination with tislelizumab in patients with advanced solid tumours: results from the dose-escalation stage of a multicentre, open-label, phase 1a/b trial. Lancet Oncol. 2019;20:1306–15.
Regev A, Björnsson ES. Drug-induced liver injury: morbidity, mortality, and Hy’s law. Gastroenterology. 2014;147:20–4.
Litton JK, Rugo HS, Ettl J, Hurvitz SA, Gonçalves A, Lee KH, et al. Talazoparib in patients with advanced breast cancer and a germline BRCA mutation. N Engl J Med. 2018;379:753–63.
Robson M, Im SA, Senkus E, Xu B, Domchek SM, Masuda N, et al. Olaparib for metastatic breast cancer in patients with a germline BRCA mutation. N Engl J Med. 2017;377:523–33.
Liu X, Wu K, Zheng D, Luo C, Fan Y, Zhong X, et al. Efficacy and safety of PARP inhibitors in advanced or metastatic triple-negative breast cancer: a systematic review and meta-analysis. Front Oncol. 2021;11:742139.
Tung NM, Robson ME, Ventz S, Santa-Maria CA, Nanda R, Marcom PK, et al. TBCRC 048: phase II study of olaparib for metastatic breast cancer and mutations in homologous recombination-related genes. J Clin Oncol. 2020;38:4274–82.
Thomas R, Al-Khadairi G, Decock J. Immune checkpoint inhibitors in triple negative breast cancer treatment: promising future prospects. Front Oncol. 2020;10:600573.
Pauls M, Chia S, LeVasseur N. Current and new novel combination treatments for metastatic triple-negative breast cancer. Curr Oncol. 2022;29:4748–67.
Wein L, Loi S. Mechanisms of resistance of chemotherapy in early-stage triple negative breast cancer (TNBC). Breast. 2017;34:S27–30.
Vinayak S, Tolaney SM, Schwartzberg L, Mita M, McCann G, Tan AR, et al. Open-label clinical trial of niraparib combined with pembrolizumab for treatment of advanced or metastatic triple-negative breast cancer. JAMA Oncol. 2019;5:1132–40.
Domchek SM, Postel-Vinay S, Im SA, Park YH, Delord JP, Italiano A, et al. Olaparib and durvalumab in patients with germline BRCA-mutated metastatic breast cancer (MEDIOLA): an open-label, multicentre, phase 1/2, basket study. Lancet Oncol. 2020;21:1155–64.
Yap TA, Konstantinopoulos P, Telli ML, Saraykar S, Beck JT, Galsky MD, et al. Abstract P1-19-03: JAVELIN PARP Medley, a phase 1b/2 study of avelumab plus talazoparib: results from advanced breast cancer cohorts. Cancer Res. 2020;80:P1–19-03.
Drew Y, Kaufman B, Banerjee S, Lortholary A, Hong SH, Park YH, et al. 4563 phase II study of olaparib + durvalumab (MEDIOLA): updated results in germline BRCA-mutated platinum-sensitive relapsed (PSR) ovarian cancer (OC). Ann Oncol. 2019;30:v475–532.
Konstantinopoulos PA, Waggoner S, Vidal GA, Mita M, Moroney JW, Holloway R, et al. Single-arm phases 1 and 2 trial of niraparib in combination with pembrolizumab in patients with recurrent platinum-resistant ovarian carcinoma. JAMA Oncol. 2019;5:1141–9.
Desai J, Deva S, Lee JS, Lin CC, Yen CJ, Chao Y, et al. Phase IA/IB study of single-agent tislelizumab, an investigational anti-PD-1 antibody, in solid tumors. J Immunother Cancer. 2020;8:e000453.
Bellmunt J, de Wit R, Vaughn DJ, Fradet Y, Lee JL, Fong L, et al. Pembrolizumab as second-line therapy for advanced urothelial carcinoma. N Engl J Med. 2017;376:1015–26.
Rosenberg JE, Park SH, Kozlov V, Dao TV, Castellano D, Li JR, et al. Durvalumab plus olaparib in previously untreated, platinum-ineligible patients with metastatic urothelial carcinoma: a multicenter, randomized, phase II trial (BAYOU). J Clin Oncol. 2023;41:43–53.
de Bono J, Mateo J, Fizazi K, Saad F, Shore N, Sandhu S, et al. Olaparib for metastatic castration-resistant prostate cancer. N Engl J Med. 2020;382:2091–102.
Karzai F, VanderWeele D, Madan RA, Owens H, Cordes LM, Hankin A, et al. Activity of durvalumab plus olaparib in metastatic castration-resistant prostate cancer in men with and without DNA damage repair mutations. J Immunother Cancer. 2018;6:141.
Shen L, Guo J, Zhang Q, Pan H, Yuan Y, Bai Y, et al. Tislelizumab in Chinese patients with advanced solid tumors: an open-label, non-comparative, phase 1/2 study. J Immunother Cancer. 2020;8:e000437.
Francis KE, Kim SI, Friedlander M, Gebski V, Ray-Coquard I, Clamp A, et al. The impact of olaparib dose reduction and treatment interruption on treatment outcome in the SOLO2/ENGOT-ov21 platinum-sensitive recurrent ovarian cancer. Ann Oncol. 2022;33:593–601.
Kristeleit RS, Oaknin A, Ray-Coquard I, Leary A, Balmaña J, Drew Y, et al. Antitumor activity of the poly(ADP-ribose) polymerase inhibitor rucaparib as monotherapy in patients with platinum-sensitive, relapsed, BRCA-mutated, high-grade ovarian cancer, and an update on safety. Int J Gynecol Cancer. 2019;29:1396–404.
Berek JS, Matulonis UA, Peen U, Ghatage P, Mahner S, Redondo A, et al. Safety and dose modification for patients receiving niraparib. Ann Oncol. 2018;29:1784–92.
Hurvitz SA, Gonçalves A, Rugo HS, Lee KH, Fehrenbacher L, Mina LA, et al. Talazoparib in patients with a germline BRCA-mutated advanced breast cancer: detailed safety analyses from the Phase III EMBRACA trial. Oncologist. 2020;25:e439–50.
Shivaji UN, Jeffery L, Gui X, Smith SCL, Ahmad OF, Akbar A, et al. Immune checkpoint inhibitor-associated gastrointestinal and hepatic adverse events and their management. Ther Adv Gastroenterol. 2019;12:1756284819884196.
O’Cearbhaill RE. Using PARP inhibitors in advanced ovarian cancer. Oncology. 2018;32:339–43.
Acknowledgements
The authors would like to thank the participants of the study and all the study staff for their contributions to the study. Medical writing support, under the direction of the authors, was provided by Helena Crisford, PhD, and Victoria Dagwell, MSc, of Ashfield MedComms, an Inizio company, and was funded by BeiGene, Ltd.
Funding
This study was sponsored by BeiGene, Ltd. Open Access funding enabled and organized by CAUL and its Member Institutions.
Author information
Authors and Affiliations
Contributions
MF and AS contributed to conception/design; MF, LM, JL, SF, BG, MW, TM, SBH, KB, NM, CF, AC, and AS contributed towards the acquisition of data; MF, LM, RG, and JW contributed towards the analysis of data; MF, LM, BG, RG, and JW contributed towards interpretation of data; MF was lead PI. All authors contributed to the development and writing of the manuscript, and approved the final draft.
Corresponding author
Ethics declarations
Competing interests
MF declares honoraria for advisory boards AstraZeneca, GSK, Incyclix, Lilly, MSD, Novartis, Takeda; consultancy for AstraZeneca, Eisai, Novartis; speaker’s fee and travel from AstraZeneca; speaker’s fees from GSK and ACT genomics; institutional research funding from AstraZeneca, BeiGene, Novartis. JL declares honoraria for advisory boards from AstraZeneca and GSK; financial support for conference attendance: AstraZeneca, GSK, and Novartis. SF declares financial/honoraria: honoraria for advisory board from Akesobio and MSD; financial support for conference attendance from Amgen; institutional research funding from Akesobio, Ambrax, Amgen, AstraZeneca, Aulos, BeiGene, Cullinian, Daiichi Sankyo, Edison Oncology, HaiHe Biopharma, MSD, Takeda, Vivace, and WMS. MW declares Institutional research support from Roche. TM declares honoraria for advisory boards from AstraZeneca, BMS, GSK, MSD, Novartis, Takeda. SBH declares honoraria for advisory boards from AstraZeneca, Eisai, GSK, MSD, Pfizer; speaker fees from Lilly; consultancy for Roche; education travel from Amgen, Novartis. KB declares honoraria for advisory boards/conference attendance from BMS, MSD, Novartis, Pierre Fabre; institutional research funding from BioGene, BMS, Eucure, Lilly, Merck, Regeneron. NM declares honoraria for advisory boards from AstraZeneca, Novartis, Pfizer, Gilead, Roche; financial support for conference from Amgen, Novartis. CF declares institutional research funding from MSD, the National Comprehensive Cancer Network (NCCN) Foundation, the NCCN Oncology Research Program, Pfizer, Taiho Oncology. AC declares institutional research funding from AnHeart, Astellas, Bayer, BeiGene, BMS, FibroGen, Genentech, Lilly, Merck Serono, MSD, Novartis, Roche, Servier, Takeda; advisory board or speaker fees from Amgen, Bayer, Merck Serono, Pierre Fabre, Roche, Servier in the last 5 years. RG and JW declare employment by BeiGene, Ltd. All other authors declare no competing interests.
Ethics approval and consent to participate
Patients voluntarily agreed to participate by giving written informed consent. This study was conducted in full conformance with the ICH E6 guideline for Good Clinical Practice and the principles of the Declaration of Helsinki. Ethics Committee: Melbourne Health Human Research Ethics Committee. • NMA Reference Number: HREC/15/MH/334. • eCTN number: CT-2015-CTN-02675-1 v1.
Consent for publication
Not applicable.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Friedlander, M., Mileshkin, L., Lombard, J. et al. Pamiparib in combination with tislelizumab in patients with advanced solid tumours: results from the dose-expansion stage of a multicentre, open-label, phase I trial. Br J Cancer 129, 797–810 (2023). https://doi.org/10.1038/s41416-023-02349-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41416-023-02349-0
