
Abstract
Background:
Recombinant human erythropoietin (EPO) is the leading biotechnology engineered hormone for treatment of anaemia associated with chronic conditions including kidney failure and cancer. The finding of EPO receptors on cancer cells has raised the concern that in addition to its action in erythropoiesis, EPO may promote tumour cell growth. We questioned whether EPO-induced signalling and consequent malignant cell manifestation is mediated by polyADP-ribosylation.
Methods:
Erythropoietin-mediated PARP (polyADP-ribose polymerase-1) activation, gene expression and core histone H4 acetylation were examined in UT7 cells, using western blot analysis, RT–PCR and immunofluorescence. Erythropoietin-driven migration of the human breast epithelial cell line MDA-MB-435 was determined by the scratch assay and in migration chambers.
Results:
We have found that EPO treatment induced PARP activation. Moreover, EPO-driven c-fos and Egr-1 gene expression as well as histone H4 acetylation were mediated via polyADP-ribosylation. Erythropoietin-induced cell migration was blocked by the PARP inhibitor, ABT-888, indicating an essential role for polyADP-ribosylation in this process.
Conclusions:
We have identified a novel pathway by which EPO-induced gene expression and breast cancer cell migration are regulated by polyADP-ribosylation. This study introduces new possibilities regarding EPO treatment for cancer-associated anaemia where combining systemic EPO treatment with targeted administration of PARP inhibitors to the tumour may allow safe treatment with EPO, minimising its possible undesirable proliferative effects on the tumour.
Similar content being viewed by others

Main
Erythropoietin (EPO) is a 34-kDa cytokine that regulates the survival, proliferation and differentiation of erythroid precursors (Wu et al, 1995). Erythropoietin binding to its receptor (EPO-R) initiates signalling by activating Janus kinase (JAK)2, which binds to an EPO-R dimer at the conserved box1 motif (Witthuhn et al, 1993). Janus kinase2 activation leads to phosphorylation of tyrosine residues in EPO-R and activation of signalling proteins that include protein kinase C, the mitogen-activated protein kinase, extracellular signal-regulated kinase (ERK), and the phosphatidylinositol 3-kinase (PI3K) pathway that leads to activation of protein kinase B/Akt (Bergelson et al, 1998; Richmond et al, 2005).
Recombinant human EPO (rHuEPO) is a worldwide leading biotechnology engineered hormone replacement therapy in the clinic. It is endorsed for the treatment of anaemia associated with common chronic diseases, such as kidney failure, chronic inflammation or cancer, and for elevating the red blood cell mass in patients prior to autologous blood transfusions, as well as in cancer patients undergoing chemotherapy (Aapro et al, 2012). However, evidence has accumulated during the past 20 years that expression of the EPO-R is not restricted to the erythropoietic lineage. Hence, it has been detected in non-hematopoietic tissues, including the cardiovascular (Heidenreich et al, 1991; Anagnostou et al, 1994; Beleslin-Cokic et al, 2004), central nervous (Masuda et al, 1993; Morishita et al, 1997) and the immune systems (Lifshitz et al, 2009; Rocchetta et al, 2011). While these findings promise beneficial effects of endogenous EPO and its therapeutic analogues as tissue-protective factors (e.g., in ischaemic and degenerative heart (Wright et al, 2004; Ogino et al, 2010) and brain diseases (Brines and Cerami, 2006)), the detection of functional EPO-Rs on certain tumour cells (Yasuda et al, 2001; Lombardero et al, 2011), raises the concern that EPO treatment may actually promote tumour cell survival and stimulate tumour growth (Lappin et al, 2007; Sytkowski, 2007). A current dilemma thus concerns the validity of rHuEPO treatment for various cancers, ranging from the risk of possible deleterious effects of the hormone on cancer progression (Leyland-Jones et al, 2005; Abhold et al, 2011; Kumar et al, 2011), to the potential benefit of its anti-neoplastic effect in certain types of cancer (Mittelman et al, 2004).
PolyADP-ribose polymerases (PARPs) catalyse a posttranslational modification of proteins by polyADP-ribosylation. Using NAD+ as a substrate, PARPs catalyse the covalent polymerisation of ADP-riboses on the glutamic or aspartic acid residues of target proteins, and generate long linear and branched poly(ADP-ribose) (PAR) chains (Schreiber et al, 2006).
PolyADP-ribose polymerase-1 is composed of three major structural and functional domains: a DNA-binding domain, an automodification domain and a catalytic domain (Kraus, 2008). During apoptosis, cleavage of PARP-1 into fragments of 89 and 24 kDa is mediated by caspases 3 and 7 and has become a hallmark of this type of cell death (Gobeil et al, 2001).
PolyADP-ribose polymerase-1, the most abundant member in the PARP family, and PARP-2, have long been studied as DNA damage responsive enzymes required for the maintenance of genome integrity. They both form heterodimers upon activation in response to DNA single-strand breaks (Schreiber et al, 2006). These PARP proteins share common nuclear substrates and are active players of the single-strand break repair/base excision repair process (Quenet et al, 2009). In addition, accumulating studies over the past decade demonstrated a role for PARP-1 in the regulation of gene expression under basal, signal-activated, and stress-activated conditions (Kraus, 2008). Recent findings in quiescent cells and in cell-free systems disclosed an alternative mechanism, inducing an intense and long-lasting activation of PARP-1 in the absence of DNA damage (Cohen-Armon, 2007; Cohen-Armon et al, 2007). In this process, PARP-1 interaction with phosphorylated ERK2 (pERK2) resulted in PARP-1 activation. Moreover, activated PARP-1 up-regulated the activity of pERK in the nucleus, enhancing the phosphorylation of ERK-targeted transcription factors, core histone acetylation and the expression of ERK target genes (Cohen-Armon, 2007, 2008; Cohen-Armon et al, 2007; Carbone et al, 2008).
Recently, PARP-1 has been considered a potential target for the development of pharmacological strategies to enhance the antitumor efficacy of radio- and chemotherapy. Clinical trials introducing PARP inhibitors for the treatment of tumours bearing mutations in the tumour-suppressor genes BRCA1 and BRCA2 are currently underway (Bryant et al, 2005; Farmer et al, 2005; Grant et al, 2007; Javle and Curtin, 2011; Rios and Puhalla, 2011).
In the present study, we investigated the involvement of polyADP-ribosylation in EPO-induced activation. Our findings indicating that EPO signalling is mediated by polyADP-ribosylation, opens a new avenue for using PARP-1 inhibitors in combination with rHuEPO administration for preventing the undesired effects of EPO in cancer patients.
Materials and methods
Antibodies
Antibodies against phosphorylated forms of STAT5, Akt, ERK1/2 (extracellular-signal-regulated kinase 1/2) and against c-fos were purchased from Cell Signaling Technology (Danvers, MA, USA). The antibodies against phosphorylated EPO-R (pTyr479) and Egr-1 were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). The antibody against ADP-ribose polymers (anti-PAR) was purchased from Alexis Biochemicals (Farmingdale, NY, USA). The antibody against PARP was purchased from Serotec (Kidlington, Oxford, UK). The antibody against acetylated histone H4 was purchased from Upstate, Millipore (Billerica, MA, USA). The anti-actin antibody was purchased from Chemicon, Millipore.
PARP inhibitors
ABT-888 (benzimidazole-4-carboxamide) and PJ-34 (N-(6-oxo-5,6 dihydrophenanthridin-2-yl)-N, N-dimethyl-acetamide) were purchased from Alexis Biochemical.
Cell lines
The UT7 EPO-dependent human leukaemic cell line was cultured in IMDM medium containing 10% fetal calf serum (FCS) and 2 U ml−1 of rHuEPO (Epoetin alfa, Ortho Biotech, Janssen Biotech, Horsham, PA, USA) at 37 °C and 5% CO2. The human breast epithelial cell line, MDA-MB-435 (Cailleau et al, 1978; Chambers, 2009) kindly provided by Professor TJ Lappin (Centre for Cancer Research and Cell Biology, Queen's University Belfast) was cultured in DMEM containing 10% FCS at 37 °C and 5% CO2.
Erythropoietin activation and cell lysis
UT7 cells were starved overnight at 37 °C, by incubation in IMDM medium without supplements. Subsequently, rHuEPO (10 U ml−1) was added to the cells for different time points. Cells (2 × 106) were lysed at 4 °C in lysis buffer (50 mM Tris pH 7.4, 1% Triton X-100, 5 mM EDTA, 150 mM NaCl) containing phosphatase inhibitors (1 mM vanadate, 50 mM sodium fluoride, 10 mM sodium pyrophosphate and 1 mM ZnCl2, final concentrations) and protease inhibitors (Complete Protease Inhibitor Cocktail, Roche Diagnostics, Basel, Switzerland). For nuclear extraction, cells (6 × 106) were lysed at 4 °C in lysis buffer (10 mM Tris–HCl pH 7.5, 10 mM KCl, 0.1 mM EDTA, 1.5 mM MgCl2, 0.2% NP-40, 1 mM DTT) containing phosphatase and protease inhibitors (as above). Lysates were spun at 6000 r.p.m. for 5 min and resuspended for 30 min in 40–100 μl of extraction buffer (10 mM Tris–HCl pH 7.5, 0.55 M NaCl, 0.1 mM EDTA, 1.5 mM MgCl2, 20% glycerol, 1 mM DTT) containing phosphatase and protease inhibitors (as above). Cell lysates were separated on SDS/PAGE gels (7.5% gel) and subjected to western blot analysis (Lifshitz et al, 2009).
Colorimetric MTT assay
UT7 cells (3 × 103 cells per well) were seeded in triplicate into 96-well, flat-bottom culture plates and cultured in the presence of rHuEPO with or without PARP inhibitors at different concentrations, for 24, 48 and 72 h. Cell viability was determined using the colorimetric methylthiazole tetrazolium bromide (MTT) assay (Mosmann, 1983). Briefly, cells were incubated for 4 h at 37 °C with MTT added to a final concentration of 5 μg ml−1. After complete solubilisation of the dye by acid/alcohol (0.04 N HCl in 2-propanol), plates were read at 570 nm in an ELISA reader.
Cell-cycle assay
UT7 cells (2 × 105 cells per sample) were cultured for 72 h in the presence of 2 U ml−1 rHuEPO with or without 20 μ M ABT-888. Subsequently, the cells were collected and washed three times in cold PBS and stained with 50 μg ml−1 propidium iodide, in the presence of 0.1% Triton X-100 and 0.1% citric acid. Samples were then incubated in the dark on ice for 1 h, and analysed on a FACSort flow cytometer (BD Biosciences, San Jose, CA, USA) with WinMDI software (version 2.9, The Scripps Research Institute, La Jolla, CA, USA).
Immunofluorescence assay
UT7 cells (1 × 106 cells per sample) were washed thrice with PBS and fixed with 3% paraformaldehyde diluted in PBS for 30 min. Cells were washed with washing buffer (0.2% saponin, 3% BSA in PBS), and incubated in blocking solution (5% FCS in PBS) for 30 min. Antibody dilutions were performed in the buffer provided by the manufacturer (Biomeda Corp., Cat No. M35), and washed thrice with washing buffer.
Real-time PCR
To quantify c-fos and Egr-1 expression in the UT7 cell line, we used RT–PCR. Total RNA from UT7 cells was isolated using PerfectPure RNA tissue kit (5 prime, Gaithersburg, MD, USA) according to the manufacturer’s protocol. The cDNA was prepared using high capacity cDNA reverse transcription kit, Applied Biosystems (Foster City, CA, USA). RT–PCR was performed using the StepOnePlus Real-time PCR System and the SYBR Green PCR Master Mix, Applied Biosystems. Primer sequences used for amplification were as follows: c-fos upstream primer, 5′-CTG TCA ACG CGC AGG ACT T-3′ downstream primer, 5′-CGG ACT GGT CGA GAT GGC-3′, Egr-1 upstream primer, 5′-GAG CAC CTG ACC GCA GAG TC-3′ downstream primer, 5′-GCG GCC AGT ATA GGT GAT GG-3′, control gene- HPRT upstream primer, 5′-TGA CAC TGG CAA AAC AAT GCA-3′ downstream primer, 5′-GGT CCT TTT CAC CAG CAA GCT-3′.
Scratch assay
MDA-MB-435 cells were plated on a six-well dish in DMEM containing 10% FCS and cultured to confluence (∼100%). The cells were then washed twice in serum-free DMEM. Using a sterile 200 μl pipette tip the cells were scratched horizontally. The cells were then washed and transferred to starvation medium (0.1% FCS in DMEM) for overnight incubation at 37 °C, with or without 10 U ml−1 of rHuEPO, in the presence or absence of 20 μM ABT-888. Pictures of a marked area, for each treatment, were taken at the first day (day 0) and on two following days (‘day 1’ and ‘day 2’) using × 10 magnification (Nikon eclipse Ti-S microscope and the integrated NIS-Elements imaging software, Tokyo, Japan). The width of a scratch was calculated at five different points for each picture taken, using adobe illustrator software. Cell migration was calculated as the per cent of width of the scratch at days 1 and 2, relating to the width of the scratch at day 0.
Migration assay
Cell migration assay was performed using Transwell (8 μ M polycarbonate Med plate, 6.5 mm inserts, Costar). MDA-MB-435 cells were washed thrice with PBS and then plated overnight in serum-free DMEM. In all experiments, 5 × 104 cells were resuspended in serum-free DMEM (100 μl) and incubated with or without rHuEPO, and the PARP-1 inhibitor, ABT-888. Cells were added to the top of each migration chamber and allowed to migrate to the underside of the chamber for 24 h in the presence of 10% FCS in the lower chamber. The upper surfaces of the filters were wiped clean of cells and the filters were then fixed by immersion in 100% methanol and stained with Hemacolor (Merck, Darmstadt, Germany). The migrated cells were captured using bright-field microscopy (Nikon eclipse Ti-S microscope and the integrated NIS-Elements imaging software). Migrated cells from the captured image were counted using ImageJ software (National Institutes of Health (NIH), Bethesda, MD, USA). Cell migration was calculated as percentage of migrating cells, regarding the number of cells in the absence of EPO as 100%.
Results
EPO-induced polyADP-ribosylation of PARP-1
To address whether EPO activates PARP-1, we subjected the EPO-dependent human leukaemic cell line, UT7 to stimulation with rHuEPO (10 U ml−1, 37 °C) for 5, 10 and 30 min as described (Lee et al, 2004; Shi et al, 2010). Nuclear lysates and fixated cells were subjected to western blot analysis (Figure 1A) and immunofluorescence labelling (Figure 1B), using an antibody directed against polyADP-ribosylated (PAR) proteins to detect activation of PARP-1. Western blot analysis demonstrated a peak of elevated polyADP-ribosylated proteins after 5 min of EPO treatment and a decrease after 10 and 30 min of EPO. Immunofluorescence analysis confirmed these findings, as depicted in Figure 1C. Figure 1B presents a representative immunofluorescence staining detected by confocal microscopy. Figure 1C depicts quantification of the results, which were obtained by measuring the fluorescence intensity of each cell for all fields, relative to fluorescence intensity in the absence of EPO. The confocal images show enhanced fluorescence intensity of stained polyADP-ribosylated proteins (PAR, green) in the nuclei after treatment with EPO. PolyADP-ribosylation significantly increased within 10 min and declined after 30 min of EPO treatment (Figure 1B and C). Nuclear size was calculated by measuring the diameter of the nucleus of each cell for all fields, relative to nuclear size in the absence of EPO (Figure 1D). Nuclear size was significantly increased within 10 and 30 min of EPO treatment.

EPO activates PARP-1 in UT7 cells. (A) Western blot analysis of EPO-induced polyADP-ribosylated proteins. UT7 cells (6 × 106) were starved of serum and EPO for 18 h at 37 °C, and subsequently activated with 10 U ml−1 rHuEPO for the indicated times. Nuclear protein extracts were prepared and subjected to western blot analysis with an antibody directed against polyADP-ribosylated proteins (PAR). Total PARP levels were used as a loading control. The molecular mass in kDa is indicated. (B) Immunofluorescence of EPO-induced polyADP-ribosylated proteins. UT7 cells (1 × 106) were starved of serum and EPO for 18 h at 37 °C, and subsequently activated with 10 U ml−1 rHuEPO for the indicated times. PolyADP-ribosylated proteins (green) were immunodetected by confocal imaging. Nuclei were stained by DAPI. The micrograph depicts a representative experiment. Magnification × 63, bar=25 μm. (C) Quantitative analysis of EPO-induced polyADP-ribosylated proteins. Results were quantified by measuring the fluorescence intensity of each cell for all fields, using zeiss, LSM microscope software (Version 3.98, Zeiss, Oberkochen, Germany). The graph depicts fluorescence intensities of the treated cells relative to non-treated cells ±s.e.m., n=3 independent experiments quantifying at least 40 cells in each experiment. Statistical analysis was obtained by a two-tailed Student’s t-test. *P<0.05, EPO treatment vs no EPO treatment. (D) Analysis of nuclear size. Results were obtained by measuring the diameter of each nucleus for all fields. The graph depicts nucleus size in the treated cells relative to the non-treated cells ±s.e.m., n=3 independent experiments quantifying at least 30 cells per treatment. Statistical analysis was obtained by a two-tailed Student’s t-test. *P<0.05, EPO treatment vs no EPO treatment. The colour reproduction of this figure is available at the British Journal of Cancer online.
EPO-induced gene expression is regulated by polyADP-ribosylation
We then addressed the effect of the PARP inhibitor ABT-888 on cell survival and proliferation using the MTT method and cell-cycle analysis. Incubation of the UT7 cells with the inhibitor showed no significant impairment of cell viability after 72 h (Figure 2A). However, at this time period, ABT-888 treatment led to G2/M arrest in the UT7 cells (Figure 2B). These findings are in line with a previous report of G2/M arrest mediated by PARP inhibition in cancer cells (Gaymes et al, 2009). To further confirm the involvement of polyADP-ribosylation in EPO-induced signalling, we activated the UT7 cells with rHuEPO (10 U ml−1), in the presence or absence of ABT-888 (20 μ M) as described (Cohen-Armon et al, 2007; Patel et al, 2011). Erythropoietin-induced signalling was monitored by immunofluorescence staining for acetylated core histone H4 (Figure 3), known to induce gene transcription (Gregory et al, 2001; Kramer et al, 2001; Eberharter and Becker, 2002; Felsenfeld and Groudine, 2003). The upper panel of Figure 3 shows a representative immunofluorescence staining detected by confocal microscopy. The lower panel depicts quantification of the fluorescence intensity of each cell, for all fields, relative to the fluorescence intensity in the absence of EPO. As shown in Figure 3, EPO elevated the acetylation of histone H4. This EPO-mediated effect was not evident in the presence of heat-denatured rHuEPO. Moreover, it was suppressed by the PARP inhibitor ABT-888. ABT-888 alone had no effect on histone H4 acetylation (Figure 3). Similar results were obtained using the PARP-1 inhibitor, PJ-34 (data not shown). Collectively, these data indicate that EPO-induced gene expression is mediated by polyADP-ribosylation.

Viability and cell-cycle analysis of UT7 cells in the presence of ABT-888. (A) UT7 cells were grown in the presence of rHuEPO (2 U ml−1) for 24–72 h. The PARP inhibitor (ABT-888) was added at the indicated concentrations. Viability was measured daily, using the MTT method. Results were calculated relative to O.D. at 570 nm of non-treated cells at 24 h, ±s.d., n=3. There was no difference in cell viability between EPO-treated groups in the presence of ABT-888 to EPO-treated groups in the absence of the inhibitor, as determent by two-tailed Student’s t-test. (B) UT7 cells were grown in the presence of rHuEPO (2 U ml−1) for 72 h with or without 20 μ M ABT-888. Subsequently, cells were stained with PI, and subjected to flow cytometry analysis. Samples were analysed with WinMDI software (version 2.9). The right panel of Figure 2B shows a representative cell-cycle analysis of UT7 cells, in the presence or absence of ABT-888 (20 μ M). The left panel of Figure 2B summarises the cell-cycle profile of three independent experiments ±s.d. Statistical analysis was obtained by a two-tailed Student’s t-test. *P<0.05, EPO treatment in the presence of ABT-888 vs EPO treatment.

PARP inhibitors block EPO-induced acetylation of core histone H4. UT7 cells (1 × 106) were starved of serum and EPO for 18 h at 37 °C, and subsequently activated with 10 U ml−1 rHuEPO for 30 min at 37 °C, in the presence or absence of ABT-888 (20 μ M, 30 min of incubation before activation with EPO). Acetylated core histone H4 (green) was immunodetected by confocal imaging. Nuclei were stained by DAPI. Quantification of the results was obtained by measuring the fluorescence intensity of each cell for all fields, using Zeiss, LSM microscope software (Version 3.98). The micrograph depicts a representative experiment. The graph depicts fluorescence intensities of treated cells relative to non-treated cells, ±s.e.m., n=3 independent experiments quantifying at least 40 cells in each experiment. Statistical analysis was obtained by a two-tailed Student’s t-test. *P<0.05, EPO treatment vs no EPO group. **P<0.05, EPO in the presence of ABT-888 vs EPO. Magnification × 63, bar=50 μm. The colour reproduction of this figure is available at the British Journal of Cancer online.
C-fos and Egr-1 (early growth response-1) are early-response genes reported to be induced by EPO. Their expression is associated with EPO-induced proliferation and differentiation (Janknecht et al, 1993; Westphal et al, 2002; Schulze et al, 2008). To further investigate the involvement of polyADP-ribosylation in EPO-induced gene expression, we measured the expression levels of c-fos (Figure 4A) and Egr-1 (Figure 4B) transcripts in EPO-activated UT7 cells in the presence or absence of ABT-888 (20 μ M). C-fos and Egr-1 gene expression was assayed by real-time PCR (RT–PCR). As shown in Figure 4A and B, c-fos and Egr-1 gene expression significantly increased after 15 and 30 min treatment with rHuEPO (10 U ml−1). Notably, ABT-888 suppressed EPO-induced elevation of c-fos and Egr-1 mRNA (Figure 4A and B), whereas transcript levels of the housekeeping gene, HPRT, were not affected. Of note, both c-fos and Egr-1 transcript levels were not completely reduced by ABT-888. This could result from polyADP-ribosylation independent signal transduction pathways involved in their induction by EPO. Western blot analysis confirmed EPO-mediated induction of c-fos and Egr-1 protein levels in UT7 cells, which were also suppressed by ABT-888 (Figure 4C and D). Similar results were obtained using a different PARP inhibitor, PJ-34 (data not shown). Taken together, these findings indicate that polyADP-ribosylation mediates EPO-induced expression of c-fos and Egr-1 in UT7 cells.

ABT-888 inhibits EPO-induced c-fos and Egr-1 expression in UT7 cells. Quantification of c-fos (A) and Egr-1 (B) expression using RT–PCR. Levels of c-fos (A) and Egr-1 (B) transcripts were measured 10, 15 and 30 min after treatment with rHuEPO (10 U ml−1) by RT–PCR, relative to the expression levels of the reference gene, HPRT. Before treatment with EPO, cells were incubated with 20 μ M ABT-888 for 30 min. Data from three independent experiments were averaged and shown as normalised gene expression ±s.e.m. Statistical analysis was obtained by a two-tailed Student’s t-test. *P<0.05, EPO treatment vs no EPO treatment. **P<0.05, EPO treatment in the presence of ABT-888 vs EPO treatment. (C) Western blot analysis of c-fos levels. Before treatment with EPO, cells were incubated with 20 μ M ABT-888 for 30 min. Nuclear protein extracts were prepared 60 min after treatment with rHuEPO (10 U ml−1, 37 °C) and subjected to western blot analysis using anti-c-fos antibody. PMA, the PKC activator (100 nM, 90 min), was used as a positive control for c-fos activation. Histone levels were used as a loading control. (D) Western blot analysis of Egr-1 levels. Cells were incubated with 20 μ M ABT-888 for 30 min prior to addition of rHuEPO (10 U ml−1, 37 °C). Nuclear protein extracts were prepared 60 and 90 min after treatment with rHuEPO and subjected to western blot analysis using anti Egr-1 antibody. Histone levels were used as a loading control.
While EPO-induced polyADP-ribosylation peaked at 10 min (Figure 1), c-fos and Egr-1 mRNA expression was still increasing at 30 min, and the levels of these corresponding proteins continued to increase at 90 min of EPO stimulation (Figure 4). A reasonable interpretation of these data is that EPO-driven polyADP-ribosylation triggers gene transcription which is then followed by protein expression.
Inhibition of PARP-1 does not affect signalling upstream to PARP-1
To verify that the PARP inhibitor does not affect EPO-induced signalling upstream of PARP-1, we examined EPO driven phosphorylation of the EPO-R and phosphorylation of ERK and STAT5 in EPO-activated UT7 cells (Figure 5). After activation with rHuEPO (10 U ml−1), in the presence or absence of ABT-888 (20 μ M), cells were lysed and proteins in the lysates were analysed by SDS–PAGE. Phosphorylated EPO-R, ERK1/2 and STAT5 were detected on western blots by immunolabelling. Results indicated that the PARP inhibitor ABT-888 did not interfere with the phosphorylation of EPO-R, ERK or STAT5, which are all activated by EPO, upstream to PARP. Similar results were obtained by using the PARP-1 inhibitor, PJ-34 (data not shown).

ABT-888 does not affect EPO-driven signalling (pEPO-R, pSTAT5 and pERK) upstream to PARP-1. UT7 cells were incubated with medium with or without ABT-888, 20 μ M for 30 min, prior to addition of rHuEPO (10 U ml−1, 37 °C for the indicated times). Cells were lysed and subjected to western blot analysis using antibodies against pEPO-R, pSTAT5 and pERK. Antibodies against actin, total STAT5 and total ERK were used as a loading control.
EPO promotes MDA-MB-435 breast cancer cell migration via polyADP-ribosylation-dependent signal transduction pathway
EPO-induced migration of cancer cells has recently been reported (Hamadmad and Hohl, 2008; Fu et al, 2009). To determine whether polyADP-ribosylation affects EPO-induced cell migration, we treated MDA-MB-435 breast cancer cells with the PARP inhibitor (Figure 6).

ABT-888 inhibits EPO-induced cell migration. (A) Cell migration analysis–‘scratch assay’. MDA-MB-435 cells were grown to 100% confluence in a six-well plate and a scratch was created using a sterile tip. Cells were washed and subsequently cultured in medium containing 0.1% FCS with or without EPO in the presence or absence of ABT-888. The scratched area was marked and photographed on day 0 and on 2 consecutive days. (Left panel) A representative display of migrated MDA-MB-435 cells after 24 h. Magnification × 10, bar=50 μm. (Right panel) Quantification of % of wound repair obtained from six measurements of every treatment from three independent experiments ±s.d. Statistical analysis was obtained by a two-tailed Student’s t-test, *P<0.05, EPO treatment vs no EPO group. **P<0.05, EPO in the presence of ABT-888 vs EPO alone. (B) Cell migration analysis–migration chambers. MDA-MB-435 cells were incubated in FCS-free medium over night. Cells were then placed at the top chamber and incubated with or without rHuEPO (10 U ml−1), and ABT-888 (20 μ M), and were allowed to migrate for 24 h. Migrated cells were captured and counted; the percentage of migration was normalised to the percentage of migrating cells without EPO treatment calculated as 100%. (Left panel) A representative display of migrated MDA-MB-435 cells after 24 h. Magnification × 10, bar=50 μm. (Right panel) Quantification of % of migrated cells. Statistical analysis was obtained by a two-tailed Student’s t-test.*P<0.05, EPO treatment vs no EPO treatment group. **P<0.05, EPO in the presence of ABT-888 vs EPO alone. Data are obtained from three independent experiments ±s.d. (C) ABT-888 blocks EPO-induced acetylation of core histone H4. MDA-MB-435 cells were activated with 10 U ml−1 rHuEPO for 30 min at 37 °C, in the presence or absence of ABT-888 (20 μ M, 30 min of incubation before activation with EPO). Histone deacetylase inhibitor (sodium butyrate, 5 mM, 30 min of incubation before activation with EPO) was added to the cells. Acetylated core histone H4 (green) was immunodetected by confocal imaging. Nuclei were stained by DAPI. Quantification of the results was obtained by measuring the fluorescence intensity of each cell for all fields, using Zeiss microscope software. The micrograph depicts a representative experiment. The graph depicts fluorescence intensities of treated cells relative to non-treated cells, ±s.e.m., n=3 independent experiments quantifying at least 40 cells in each experiment. Statistical analysis was obtained by a two-tailed Student’s t-test. *P<0.05, EPO treatment vs no EPO treatment group. **P<0.05, EPO treatment in the presence of ABT-888 vs EPO alone. Magnification × 63, bar=50 μm. The colour reproduction of this figure is available at the British Journal of Cancer online.
In these experiments, cell cultures were ‘scratch-wounded’ with a pipette tip, and stimulated with rHuEPO (10 U ml−1) in the absence or presence of ABT-888 (20 μ M) for 2 days. To enable migration in the absence of proliferation, cells were cultured in DMEM containing 0.1% FCS. The results of these experiments indicated that EPO-driven cell migration was blocked by ABT-888, suggesting an essential role for polyADP-ribosylation in EPO-driven migration of the MDA-MB-435 cells (Figure 6A), and of MDA-MD-231 (data not shown). It should be noted that EPO-induced cell migration in the presence of lower ABT-888 concentrations (10 μ M), was also inhibited, albeit to a lower extent (data not shown), suggesting that this is a dose-dependent effect. Erythropoietin-driven cell migration was also measured using multiwell inserts (Figure 6B). Cells were washed thrice with PBS and plated in FCS-free DMEM overnight before EPO treatment. Then, cells were resuspended in FCS-free DMEM and placed in the upper migration chamber (see Materials and Methods). The cultures were incubated in the absence or presence of rHuEPO (10 U ml−1) with or without ABT-888 (20 μ M) for 24 h. The left panel of Figure 6B shows a representative display of migrated cells after 24 h of treatment using the migration chamber assay. The right panel of Figure 6B depicts quantification of cell migration relative to cell migration in the absence of EPO. In these experiments too, EPO-driven cell migration was blocked in the presence of ABT-888 (Figure 6B).
Acetylation of core histone H4 was detected in MDA-MB-435 cells after activation with rHuEPO (10 U ml−1); this acetylation significantly declined in the presence of ABT-888 (Figure 6C). The left panel of Figure 6C shows a representative immunofluorescence staining detected by confocal microscopy. The right panel depicts quantification of the fluorescence intensity of each cell, for all fields, relative to the fluorescence intensity in the absence of EPO. As shown in Figure 6, EPO treatment of the MDA-MB-435 cells resulted in elevated histone H4 acetylation, similarly to the results observed for the UT7 cells (Figure 3). Moreover, this EPO-driven histone acetylation was suppressed by ABT-888.
Discussion
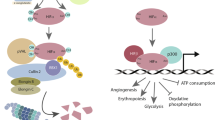
The present study was driven by the controversy regarding the benefits of treatment of cancer patients with erythropoiesis stimulating reagents, for example, multiple myeloma patients (Mittelman et al, 2004), vs the possible hazards of such treatment in patients with certain solid tumours, for example, head and neck and melanoma (Abhold et al, 2011; Kumar et al, 2011), respectively. Our results show for the first time that application of EPO caused activation of PARP proteins including PARP-1, and that polyADP-ribosylation subsequently mediated EPO-induced histone H4 acetylation and expression of c-fos and Egr-1 that mediate EPO-induced proliferation (Erickson-Miller et al, 2000). In addition, EPO-mediated cell migration of the breast cancer cell line MDA-MB-435 was blocked by the PARP inhibitor, ABT-888, suggesting a possible role for polyADP-ribosylation in EPO-induced migration of these cells (Figure 6). On the basis of our results, we propose a novel mechanism for EPO-induced effects, which are mediated by PARP-1 activation (Figure 7).

A scheme depicting EPO-induced PARP-1 activation.
It has been reported that PARP-1 mediates histone acetylation and expression of c-fos in cerebral neurons treated with nerve growth factors (Cohen-Armon et al, 2007). This mechanism involved DNA-independent PARP-1 activation, apparently induced by interaction with phosphorylated ERK2. PolyADP-ribose polymerase-1 activation up-regulated ERK-activity, inducing Elk1 phosphorylation and thereby core histone H4 acetylation due to histone acetyltransferase (HAT) activity (Cohen-Armon et al, 2007). It was also shown that decondensation of the chromatin is a consequence of polyADP-ribosylation of linker histone H1, a prominent PARP-1 substrate (Kraus and Lis, 2003) and core histones (Messner and Hottiger, 2011). Our finding that EPO stimulation was associated with nuclei enlargement (Figure 1D) is thus in line with nuclei enlargement associated with enhancement of gene transcription (Sato et al, 1994) and with EPO-mediated induction of gene transcription (Lombardero et al, 2011). Notably, while EPO-mediated polyADP-ribosylation was reduced after 30 min, nuclei were still enlarged at this time period, pointing to the different time course of polyADP-ribosylation and chromatin decondensation (Figure 1).
C-fos and Egr-1 are transcription factors activated by several growth factors (Berthoud et al, 2001; Cheng et al, 2001; Weisstein et al, 2001; Kang et al, 2007; Tai et al, 2010), including EPO (Erickson-Miller et al, 2000; Mengozzi et al, 2012). Here, we demonstrate for the first time that the EPO-induced expression of these two immediate early genes (Erickson-Miller et al, 2000), is mediated by polyADP-ribosylation in UT7 cells (Figure 4). This is in line with the previous report in neuronal cells demonstrating the essential role of DNA-independent PARP-1 activation in c-fos expression (Cohen-Armon, 2007).
The involvement of PARP-1 in cell migration was demonstrated in numerous experimental setups. For example, PARP activation was found necessary for VEGF-induced angiogenesis (Rajesh et al, 2006) and migration of HUVEC cells (Rajesh et al, 2006; Pyriochou et al, 2008), as deduced from the inhibitory action of the PARP inhibitors on these processes. Similarly, it was shown that inhibition of polyADP-ribosylation abrogates the ability of endothelial cells to migrate in response to VEGF or PlGF (Tentori et al, 2007) and suppresses cell growth and migration in osteosarcoma cells (Zheng et al, 2011). The current study shows for the first time that polyADP-ribosylation is also crucial for EPO-induced breast cancer cell migration. Taken together, our data support the premise that polyADP-ribosylation probably via PARP-1 (which is the most abundant PARP; Schreiber et al, 2006) activation is a pivotal step in EPO-R signalling pathways (Figure 7).
Although PARP inhibitors, which are currently under clinical investigation, inhibit both PARP-1 and PARP-2 (Underhill et al, 2010), our results are causally ascribed to PARP-1, due to its physiological role in regulation of transcription (Kraus, 2008). This result is in accordance with previous findings, indicating that PARP-1 is activated by pERK2 in the absence of DNA damage (Cohen-Armon et al, 2007). According to this scenario, PARP-1 activated and auto-polyADP ribosylated by a direct interaction with pERK2 enhances pERK2-catalysed phosphorylation of the transcription factor Elk1, promoting the HAT activity of CBP/p300 and the expression of Elk1-target genes such as c-fos (Cohen-Armon, 2007; Cohen-Armon et al, 2007). Accumulating data indicate the essential role of ERK phosphorylation in EPO-induced tumour cell proliferation (Bose and Udupa, 2008; Peres et al, 2011), and migration (Lester et al, 2005; Hamadmad and Hohl, 2008; Shi et al, 2010). Our findings are thus in line with EPO-induced polyADP-ribosylation via ERK phosphorylation activating PARP-1. This signal transduction mechanism could be essential for EPO-induced cell migration, strongly implying that EPO-mediated polyADP-ribosylation is conferred by ERK2. Although PARP-1 was classically associated with DNA damage (Bouchard et al, 2003), our suggested model depicts EPO-mediated activation of PARP-1 in the absence of DNA damage (Figure 7).
Notably, it was recently shown that PARP inhibitors induced tumour cell death by down-regulation of AKT phosphorylation (Wang et al, 2011). Moreover, EPO-mediated cell invasion and adhesion in breast cancer cells was shown to be conferred via AKT (Shi et al, 2010), yet, the question of whether PARP activation can also be mediated via JAK/STAT through an AKT pathway is not yet clear. Hence, our data cannot exclude EPO-mediated activation of AKT, which could lead to activation of PARP-1 and its downstream effects. The current study thus provides a working platform to further investigate the mechanism by which PARP-1 is involved in EPO-induced signalling, gene transcription and cell migration.
The pivotal role of ERK activation in cancer progression was also demonstrated in experimental in vivo models (Watanabe et al, 2010; Kandala et al, 2011; Kim et al, 2012) and in clinical studies (Wang et al, 2007; Kohno et al, 2011). Nevertheless, the introduction of MEK inhibitors (Wang et al, 2007) does not seem to provide the desired anti-tumour outcome (Rinehart et al, 2004; Kohno et al, 2011; Poulikakos and Solit, 2011) and causes deleterious side effects. Although in clinical trials PARP inhibitors are being administrated intravenously or orally (Underhill et al, 2010), several studies in mice have shown the feasibility of in vivo local administration of PARP inhibitors (Conrad et al, 2006; Inbar-Rozensal et al, 2009). Our current study may thus set the scene for targeted administration of PARP inhibitors to counteract possible deleterious EPO-associated in vivo effects on the tumour cells (Leyland-Jones et al, 2005; Sytkowski, 2007; Wright et al, 2007; Szenajch et al, 2010). The current lack of EPO-specific antagonists renders inhibition of selected EPO-mediated downstream targets a plausible approach.
Conclusions
The present study identifies PARP-1 as a novel EPO target. Accumulating data regarding the introduction of PARP inhibitors for the treatment of ovarian (Banerjee and Kaye, 2011; Sessa, 2011), prostate (Antonarakis and Carducci, 2010; Sebastian de Bono et al, 2011) and breast cancer (Glendenning and Tutt, 2011; Irshad et al, 2011) coupled to our current data may enable targeted introduction of PARP inhibitors along with the administration of EPO, in those cases where EPO treatment for the anaemia may enhance cancer progression.
Change history
04 October 2012
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
Aapro M, Cornes P, Sun D, Abraham I (2012) Comparative cost efficiency across the European G5 countries of originators and a biosimilar erythropoiesis-stimulating agent to manage chemotherapy-induced anemia in patients with cancer. Ther Adv Med Oncol 4 (3): 95–105
Abhold E, Rahimy E, Wang-Rodriguez J, Blair KJ, Yu MA, Brumund KT, Weisman RA, Ongkeko WM (2011) Recombinant human erythropoietin promotes the acquisition of a malignant phenotype in head and neck squamous cell carcinoma cell lines in vitro. BMC Res Notes 4: 553
Anagnostou A, Liu Z, Steiner M, Chin K, Lee ES, Kessimian N, Noguchi CT (1994) Erythropoietin receptor mRNA expression in human endothelial cells. Proc Natl Acad Sci USA 91: 3974–3978
Antonarakis ES, Carducci MA (2010) Future directions in castrate-resistant prostate cancer therapy. Clin Genitourin Cancer 8: 37–46
Banerjee S, Kaye S (2011) The role of targeted therapy in ovarian cancer. Eur J Cancer 47 (Suppl 3): S116–S130
Beleslin-Cokic BB, Cokic VP, Yu X, Weksler BB, Schechter AN, Noguchi CT (2004) Erythropoietin and hypoxia stimulate erythropoietin receptor and nitric oxide production by endothelial cells. Blood 104: 2073–2080
Bergelson S, Klingmuller U, Socolovsky M, Hsiao JG, Lodish HF (1998) Tyrosine residues within the intracellular domain of the erythropoietin receptor mediate activation of AP-1 transcription factors. J Biol Chem 273: 2396–2401
Berthoud HR, Patterson LM, Zheng H (2001) Vagal-enteric interface: vagal activation-induced expression of c-Fos and p-CREB in neurons of the upper gastrointestinal tract and pancreas. Anat Rec 262: 29–40
Bose C, Udupa KB (2008) Erythropoietin enhancement of rat pancreatic tumor cell proliferation requires the activation of ERK and JNK signals. Am J Physiol Cell Physiol 295: C394–C405
Bouchard VJ, Rouleau M, Poirier GG (2003) PARP-1, a determinant of cell survival in response to DNA damage. Exp Hematol 31: 446–454
Brines M, Cerami A (2006) Discovering erythropoietin's extra-hematopoietic functions: biology and clinical promise. Kidney Int 70: 246–250
Bryant HE, Schultz N, Thomas HD, Parker KM, Flower D, Lopez E, Kyle S, Meuth M, Curtin NJ, Helleday T (2005) Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 434: 913–917
Cailleau R, Olive M, Cruciger QV (1978) Long-term human breast carcinoma cell lines of metastatic origin: preliminary characterization. In Vitro 14: 911–915
Carbone M, Rossi MN, Cavaldesi M, Notari A, Amati P, Maione R (2008) Poly(ADP-ribosyl)ation is implicated in the G0-G1 transition of resting cells. Oncogene 27: 6083–6092
Chambers AF (2009) MDA-MB-435 and M14 cell lines: identical but not M14 melanoma? Cancer Res 69: 5292–5293
Cheng RX, Feng DY, Zheng H, Tan Y (2001) [Effect of activation of p-MAPK on activating c-fos and c-jun proteins in breast cancer]. Hunan Yi Ke Da Xue Xue Bao 26: 10–12
Cohen-Armon M (2007) PARP-1 activation in the ERK signaling pathway. Trends Pharmacol Sci 28: 556–560
Cohen-Armon M (2008) PARP-1 activation mediates the expression of immediate early genes implicated in long-term memory formation. FEBS J 275 (Suppl 1): 92
Cohen-Armon M, Visochek L, Rozensal D, Kalal A, Geistrikh I, Klein R, Bendetz-Nezer S, Yao Z, Seger R (2007) DNA-independent PARP-1 activation by phosphorylated ERK2 increases Elk1 activity: a link to histone acetylation. Mol Cell 25: 297–308
Conrad MF, Albadawi H, Stone DH, Crawford RS, Entabi F, Watkins MT (2006) Local administration of the Poly ADP-Ribose Polymerase (PARP) inhibitor, PJ34 during hindlimb ischemia modulates skeletal muscle reperfusion injury. J Surg Res 135: 233–237
Eberharter A, Becker PB (2002) Histone acetylation: a switch between repressive and permissive chromatin. Second in review series on chromatin dynamics. EMBO Rep 3: 224–229
Erickson-Miller CL, Pelus LM, Lord KA (2000) Signaling induced by erythropoietin and stem cell factor in UT-7/Epo cells: transient versus sustained proliferation. Stem Cells 18: 366–373
Farmer H, McCabe N, Lord CJ, Tutt AN, Johnson DA, Richardson TB, Santarosa M, Dillon KJ, Hickson I, Knights C, Martin NM, Jackson SP, Smith GC, Ashworth A (2005) Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 434: 917–921
Felsenfeld G, Groudine M (2003) Controlling the double helix. Nature 421: 448–453
Fu P, Jiang X, Arcasoy MO (2009) Constitutively active erythropoietin receptor expression in breast cancer cells promotes cellular proliferation and migration through a MAP-kinase dependent pathway. Biochem Biophys Res Commun 379: 696–701
Gaymes TJ, Shall S, MacPherson LJ, Twine NA, Lea NC, Farzaneh F, Mufti GJ (2009) Inhibitors of poly ADP-ribose polymerase (PARP) induce apoptosis of myeloid leukemic cells: potential for therapy of myeloid leukemia and myelodysplastic syndromes. Haematologica 94: 638–646
Glendenning J, Tutt A (2011) PARP inhibitors--current status and the walk towards early breast cancer. Breast 20 (Suppl 3): S12–S19
Gobeil S, Boucher CC, Nadeau D, Poirier GG (2001) Characterization of the necrotic cleavage of poly(ADP-ribose) polymerase (PARP-1): implication of lysosomal proteases. Cell Death Differ 8: 588–594
Grant SG, Das R, Cerceo CM, Rubinstein WS, Latimer JJ (2007) Elevated levels of somatic mutation in a manifesting BRCA1 mutation carrier. Pathol Oncol Res 13: 276–283
Gregory PD, Wagner K, Horz W (2001) Histone acetylation and chromatin remodeling. Exp Cell Res 265: 195–202
Hamadmad SN, Hohl RJ (2008) Erythropoietin stimulates cancer cell migration and activates RhoA protein through a mitogen-activated protein kinase/extracellular signal-regulated kinase-dependent mechanism. J Pharmacol Exp Ther 324: 1227–1233
Heidenreich S, Rahn KH, Zidek W (1991) Direct vasopressor effect of recombinant human erythropoietin on renal resistance vessels. Kidney Int 39: 259–265
Inbar-Rozensal D, Castiel A, Visochek L, Castel D, Dantzer F, Izraeli S, Cohen-Armon M (2009) A selective eradication of human nonhereditary breast cancer cells by phenanthridine-derived polyADP-ribose polymerase inhibitors. Breast Cancer Res 11: R78
Irshad S, Ashworth A, Tutt A (2011) Therapeutic potential of PARP inhibitors for metastatic breast cancer. Expert Rev Anticancer Ther 11: 1243–1251
Janknecht R, Ernst WH, Pingoud V, Nordheim A (1993) Activation of ternary complex factor Elk-1 by MAP kinases. EMBO J 12: 5097–5104
Javle M, Curtin NJ (2011) The potential for poly (ADP-ribose) polymerase inhibitors in cancer therapy. Ther Adv Med Oncol 3: 257–267
Kandala PK, Wright SE, Srivastava SK (2011) Blocking EGFR activation suppresses ovarian tumor growth in vitro and in vivo. J Pharmacol Exp Ther 341 (1): 24–32
Kang JH, Kim MJ, Jang HI, Koh KH, Yum KS, Rhie DJ, Yoon SH, Hahn SJ, Kim MS, Jo YH (2007) Proximal cyclic AMP response element is essential for exendin-4 induction of rat EGR-1 gene. Am J Physiol Endocrinol Metab 292: E215–E222
Kim HS, Kim MJ, Kim EJ, Yang Y, Lee MS, Lim JS (2012) Berberine-induced AMPK activation inhibits the metastatic potential of melanoma cells via reduction of ERK activity and COX-2 protein expression. Biochem Pharmacol 83: 385–394
Kohno M, Tanimura S, Ozaki K (2011) Targeting the extracellular signal-regulated kinase pathway in cancer therapy. Biol Pharm Bull 34: 1781–1784
Kramer OH, Gottlicher M, Heinzel T (2001) Histone deacetylase as a therapeutic target. Trends Endocrinol Metab 12: 294–300
Kraus WL (2008) Transcriptional control by PARP-1: chromatin modulation, enhancer-binding, coregulation, and insulation. Curr Opin Cell Biol 20: 294–302
Kraus WL, Lis JT (2003) PARP goes transcription. Cell 113: 677–683
Kumar SM, Zhang G, Bastian BC, Arcasoy MO, Karande P, Pushparajan A, Acs G, Xu X (2011) Erythropoietin receptor contributes to melanoma cell survival in vivo. Oncogene 31 (13): 1649–1660
Lappin TR, Maxwell AP, Johnston PG (2007) Warning flags for erythropoiesis-stimulating agents and cancer-associated anemia. Oncologist 12: 362–365
Lee SM, Nguyen TH, Park MH, Kim KS, Cho KJ, Moon DC, Kim HY, Yoon DY, Hong JT (2004) EPO receptor-mediated ERK kinase and NF-kappaB activation in erythropoietin-promoted differentiation of astrocytes. Biochem Biophys Res Commun 320: 1087–1095
Lester RD, Jo M, Campana WM, Gonias SL (2005) Erythropoietin promotes MCF-7 breast cancer cell migration by an ERK/mitogen-activated protein kinase-dependent pathway and is primarily responsible for the increase in migration observed in hypoxia. J Biol Chem 280: 39273–39277
Leyland-Jones B, Semiglazov V, Pawlicki M, Pienkowski T, Tjulandin S, Manikhas G, Makhson A, Roth A, Dodwell D, Baselga J, Biakhov M, Valuckas K, Voznyi E, Liu X, Vercammen E (2005) Maintaining normal hemoglobin levels with epoetin alfa in mainly nonanemic patients with metastatic breast cancer receiving first-line chemotherapy: a survival study. J Clin Oncol 23: 5960–5972
Lifshitz L, Prutchi-Sagiv S, Avneon M, Gassmann M, Mittelman M, Neumann D (2009) Non-erythroid activities of erythropoietin: functional effects on murine dendritic cells. Mol Immunol 46: 713–721
Lombardero M, Kovacs K, Scheithauer BW (2011) Erythropoietin: a hormone with multiple functions. Pathobiology 78: 41–53
Masuda S, Nagao M, Takahata K, Konishi Y, Gallyas F, Tabira T, Sasaki R (1993) Functional erythropoietin receptor of the cells with neural characteristics. Comparison with receptor properties of erythroid cells. J Biol Chem 268: 11208–11216
Mengozzi M, Cervellini I, Villa P, Erbayraktar Z, Gokmen N, Yilmaz O, Erbayraktar S, Manohasandra M, Van Hummelen P, Vandenabeele P, Chernajovsky Y, Annenkov A, Ghezzi P (2012) Erythropoietin-induced changes in brain gene expression reveal induction of synaptic plasticity genes in experimental stroke. Proc Natl Acad Sci USA 109: 9617–9622
Messner S, Hottiger MO (2011) Histone ADP-ribosylation in DNA repair, replication and transcription. Trends Cell Biol 21 (9): 534–542
Mittelman M, Zeidman A, Kanter P, Katz O, Oster H, Rund D, Neumann D (2004) Erythropoietin has an anti-myeloma effect - a hypothesis based on a clinical observation supported by animal studies. Eur J Haematol 72: 155–165
Morishita E, Masuda S, Nagao M, Yasuda Y, Sasaki R (1997) Erythropoietin receptor is expressed in rat hippocampal and cerebral cortical neurons, and erythropoietin prevents in vitro glutamate-induced neuronal death. Neuroscience 76: 105–116
Mosmann T (1983) Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods 65: 55–63
Ogino A, Takemura G, Kawasaki M, Tsujimoto A, Kanamori H, Li L, Goto K, Maruyama R, Kawamura I, Takeyama T, Kawaguchi T, Watanabe T, Moriguchi Y, Saito H, Fujiwara T, Fujiwara H, Minatoguchi S (2010) Erythropoietin receptor signaling mitigates renal dysfunction-associated heart failure by mechanisms unrelated to relief of anemia. J Am Coll Cardiol 56: 1949–1958
Patel AG, Sarkaria JN, Kaufmann SH (2011) Nonhomologous end joining drives poly(ADP-ribose) polymerase (PARP) inhibitor lethality in homologous recombination-deficient cells. Proc Natl Acad Sci USA 108: 3406–3411
Peres EA, Valable S, Guillamo JS, Marteau L, Bernaudin JF, Roussel S, Lechapt-Zalcman E, Bernaudin M, Petit E (2011) Targeting the erythropoietin receptor on glioma cells reduces tumour growth. Exp Cell Res 317: 2321–2332
Poulikakos PI, Solit DB (2011) Resistance to MEK inhibitors: should we co-target upstream? Sci Signal 4: pe16
Pyriochou A, Olah G, Deitch EA, Szabo C, Papapetropoulos A (2008) Inhibition of angiogenesis by the poly(ADP-ribose) polymerase inhibitor PJ-34. Int J Mol Med 22: 113–118
Quenet D, El Ramy R, Schreiber V, Dantzer F (2009) The role of poly(ADP-ribosyl)ation in epigenetic events. Int J Biochem Cell Biol 41: 60–65
Rajesh M, Mukhopadhyay P, Batkai S, Godlewski G, Hasko G, Liaudet L, Pacher P (2006) Pharmacological inhibition of poly(ADP-ribose) polymerase inhibits angiogenesis. Biochem Biophys Res Commun 350: 352–357
Richmond TD, Chohan M, Barber DL (2005) Turning cells red: signal transduction mediated by erythropoietin. Trends Cell Biol 15: 146–155
Rinehart J, Adjei AA, Lorusso PM, Waterhouse D, Hecht JR, Natale RB, Hamid O, Varterasian M, Asbury P, Kaldjian EP, Gulyas S, Mitchell DY, Herrera R, Sebolt-Leopold JS, Meyer MB (2004) Multicenter phase II study of the oral MEK inhibitor, CI-1040, in patients with advanced non-small-cell lung, breast, colon, and pancreatic cancer. J Clin Oncol 22: 4456–4462
Rios J, Puhalla S (2011) PARP inhibitors in breast cancer: BRCA and beyond. Oncology (Williston Park) 25: 1014–1025
Rocchetta F, Solini S, Mister M, Mele C, Cassis P, Noris M, Remuzzi G, Aiello S (2011) Erythropoietin enhances immunostimulatory properties of immature dendritic cells. Clin Exp Immunol 165: 202–210
Sato S, Burgess SB, McIlwain DL (1994) Transcription and motoneuron size. J Neurochem 63: 1609–1615
Schreiber V, Dantzer F, Ame JC, de Murcia G (2006) Poly(ADP-ribose): novel functions for an old molecule. Nat Rev Mol Cell Biol 7: 517–528
Schulze C, Buchse T, Mikkat S, Bittorf T (2008) Erythropoietin receptor-mediated Egr-1 activation: structural requirements and functional implications. Cell Signal 20: 1848–1854
Sebastian de Bono J, Sandhu S, Attard G (2011) Beyond hormone therapy for prostate cancer with PARP inhibitors. Cancer Cell 19: 573–574
Sessa C (2011) Update on PARP1 inhibitors in ovarian cancer. Ann Oncol 22 (Suppl 8): viii72–viii76
Shi Z, Hodges VM, Dunlop EA, Percy MJ, Maxwell AP, El-Tanani M, Lappin TR (2010) Erythropoietin-induced activation of the JAK2/STAT5, PI3K/Akt, and Ras/ERK pathways promotes malignant cell behavior in a modified breast cancer cell line. Mol Cancer Res 8: 615–626
Sytkowski AJ (2007) Does erythropoietin have a dark side? Epo signaling and cancer cells. Sci STKE 2007: pe38
Szenajch J, Wcislo G, Jeong JY, Szczylik C, Feldman L (2010) The role of erythropoietin and its receptor in growth, survival and therapeutic response of human tumor cells From clinic to bench - a critical review. Biochim Biophys Acta 1806: 82–95
Tai TC, Wong-Faull DC, Claycomb R, Wong DL (2010) Hypoxia and adrenergic function: molecular mechanisms related to Egr-1 and Sp1 activation. Brain Res 1353: 14–27
Tentori L, Lacal PM, Muzi A, Dorio AS, Leonetti C, Scarsella M, Ruffini F, Xu W, Min W, Stoppacciaro A, Colarossi C, Wang ZQ, Zhang J, Graziani G (2007) Poly(ADP-ribose) polymerase (PARP) inhibition or PARP-1 gene deletion reduces angiogenesis. Eur J Cancer 43: 2124–2133
Underhill C, Toulmonde M, Bonnefoi H (2010) A review of PARP inhibitors: from bench to bedside. Ann Oncol 22: 268–279
Wang D, Boerner SA, Winkler JD, LoRusso PM (2007) Clinical experience of MEK inhibitors in cancer therapy. Biochim Biophys Acta 1773: 1248–1255
Wang S, Wang H, Davis BC, Liang J, Cui R, Chen SJ, Xu ZX (2011) PARP1 inhibitors attenuate AKT phosphorylation via the upregulation of PHLPP1. Biochem Biophys Res Commun 412: 379–384
Watanabe K, Tanimura S, Uchiyama A, Sakamoto T, Kawabata T, Ozaki K, Kohno M (2010) Blockade of the extracellular signal-regulated kinase pathway enhances the therapeutic efficacy of microtubule-destabilizing agents in human tumor xenograft models. Clin Cancer Res 16: 1170–1178
Weisstein JS, Majeska RJ, Klein MJ, Einhorn TA (2001) Detection of c-fos expression in benign and malignant musculoskeletal lesions. J Orthop Res 19: 339–345
Westphal G, Niederberger E, Blum C, Wollman Y, Knoch TA, Rebel W, Debus J, Friedrich E (2002) Erythropoietin and G-CSF receptors in human tumor cells: expression and aspects regarding functionality. Tumori 88: 150–159
Witthuhn BA, Quelle FW, Silvennoinen O, Yi T, Tang B, Miura O, Ihle JN (1993) JAK2 associates with the erythropoietin receptor and is tyrosine phosphorylated and activated following stimulation with erythropoietin. Cell 74: 227–236
Wright GL, Hanlon P, Amin K, Steenbergen C, Murphy E, Arcasoy MO (2004) Erythropoietin receptor expression in adult rat cardiomyocytes is associated with an acute cardioprotective effect for recombinant erythropoietin during ischemia-reperfusion injury. FASEB J 18: 1031–1033
Wright JR, Ung YC, Julian JA, Pritchard KI, Whelan TJ, Smith C, Szechtman B, Roa W, Mulroy L, Rudinskas L, Gagnon B, Okawara GS, Levine MN (2007) Randomized, double-blind, placebo-controlled trial of erythropoietin in non-small-cell lung cancer with disease-related anemia. J Clin Oncol 25: 1027–1032
Wu H, Liu X, Jaenisch R, Lodish HF (1995) Generation of committed erythroid BFU-E and CFU-E progenitors does not require erythropoietin or the erythropoietin receptor. Cell 83: 59–67
Yasuda Y, Musha T, Tanaka H, Fujita Y, Fujita H, Utsumi H, Matsuo T, Masuda S, Nagao M, Sasaki R, Nakamura Y (2001) Inhibition of erythropoietin signalling destroys xenografts of ovarian and uterine cancers in nude mice. Br J Cancer 84: 836–843
Zheng YD, Xu XQ, Peng F, Yu JZ, Wu H (2011) The poly(ADP-ribose) polymerase-1 inhibitor 3-aminobenzamide suppresses cell growth and migration, enhancing suppressive effects of cisplatin in osteosarcoma cells. Oncol Rep 25: 1399–1405
Acknowledgements
We gratefully acknowledge Drs Terry Lappin and Mohamed El-Tanani for critically reviewing this manuscript and for their insightful suggestions. This work was supported by the Israel Science Foundation administered by the Israel Academy of Sciences and Humanities (Grant number 244/10 to DN) and by the FP7 European commission grant; 282551 EpoCan to DN. This work was carried out in partial fulfillment of the requirements for a PhD degree for DI from the Sackler Faculty of Medicine, Tel Aviv University, Tel Aviv, Israel.
Author information
Authors and Affiliations
Corresponding author
Additional information
This work is published under the standard license to publish agreement. After 12 months the work will become freely available and the license terms will switch to a Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License.
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Inbar, D., Cohen-Armon, M. & Neumann, D. Erythropoietin-driven signalling and cell migration mediated by polyADP-ribosylation. Br J Cancer 107, 1317–1326 (2012). https://doi.org/10.1038/bjc.2012.395
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/bjc.2012.395
