


 Figure 1
Figure 1
















« Prev Next »
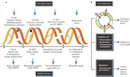
The genome of a cell is continuously damaged, which is inevitable because DNA damage often arises as a result of normal cellular processes. By-products of the cell's own metabolism such as reactive oxygen species can damage DNA bases and cause lesions that can block progression of replication. The result is double-strand breaks (DSBs) in the chromosome. A DSB can also be caused by environmental exposure to irradiation, other chemical agents, or ultraviolet light (UV). The good news is that organisms have evolved checkpoint mechanisms (responses that facilitate repair or damage tolerance by arresting cell cycle progression) that inspect the genome for damage. The cell then goes through a series of repair pathways such as base excision repair (BER), nucleotide excision repair (NER), and double-strand break repair (DSBR) (Hoeijmakers 2001; Aguilera & Gomez-Gonzalez 2008). DSBs are particularly troublesome because they can lead to cell death if not repaired. And, if not repaired correctly, DSBs can cause deletions, translocations, and fusions in the DNA. These consequences are collectively referred to as genomic rearrangements, and they are commonly found in cancerous cells (Figure 1) (Aplan 2006). Trying to understand how these DNA repair pathways work, which proteins are involved, and how the pathways perform are just some of the topics that researchers in genetics, biochemistry, and molecular biology are working on today.
What Are the Factors Involved in Repairing Damaged DNA?
How do we find out which protein works in a particular DNA repair pathway? We know that if a DNA repair gene is mutated, cells become more sensitive to DNA damaging agents. Researchers in the 1960s isolated the first radiation-sensitive mutants in yeast. By the 1970s, researchers had identified a great number of genes that, when mutated, increased the sensitivity of the mutant yeast to radiation.
How did researchers obtain these mutant strains? They randomly mutagenized yeast cells by exposing them to an agent, such as ionizing radiation (IR) or an alkylating agent, that causes mutations in DNA. Those mutant cells were then screened for sensitivity to DNA damaging agents. Many mutant cells were identified, and careful genetic studies allowed scientists to determine the functional relationships of these radiation-sensitive (rad) mutants.
Today, that is how we know which genes code for proteins that work together in a specific DNA repair pathway. The approach used is a typical genetic strategy in which you take something away from the system you are studying (create a mutant strain deficient in one gene) and see how the component behaves and what phenotype you observe. Then you can take out more than one component at a time (you can cross two different mutant strains to obtain a double mutant) and see if the system behaves in the same way or if the phenotype worsens. For example, if the protein responsible for step 1 of a linear pathway is eliminated, the pathway will not be functional and there may be a phenotype you can measure. Eliminating proteins responsible for step 2, then, should not have any additional effect: The pathway is still nonfunctional, and the phenotype observed should be the same in the double mutant as in the single mutant; these two mutants are referred to as being epistatic to each other. If, instead, the second protein you eliminate belongs to another pathway that is involved in a related cellular process, the phenotype in the double mutant is exacerbated because there are two different pathways being inactivated; this situation is referred to as the mutants having a synergistic interaction. The RAD52 epistasis group was identified with respect to radiation sensitivity through this strategy. In fact, the RAD52 epistasis group consists of several proteins that are involved in DSB repair by homologous recombination (HR) (Figure 2) (Friedberg 1988).
The genetic evidence suggested that all these genes coded for proteins with roles in DSB repair, but what exactly is the role of each at the molecular level? The actual function of each protein is then determined using a combination of biochemical, cellular, genetic, and molecular biology approaches. Knowledge of the functions of each of these proteins in the repair process has expanded greatly in recent years. However, the more we know, the more questions arise, and research continues (West 2003; Hoeijmakers 2007; Aguilera & Gomez-Gonzalez 2008; Shrivastav et al. 2008; Huertas 2010).
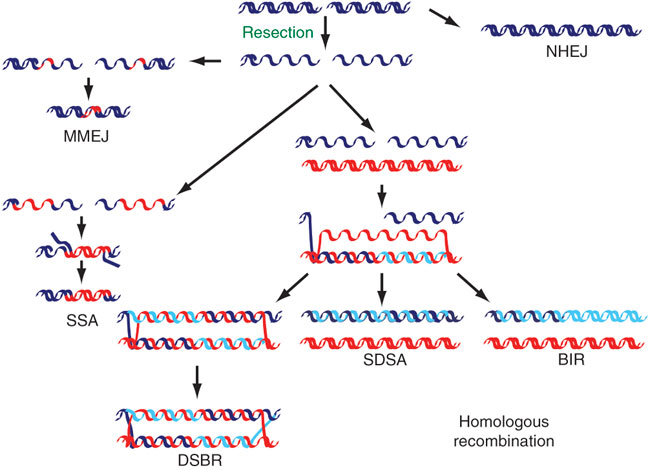
Figure 2: The repair of DNA double-strand breaks (DSBs)
DSBs can be repaired using several different mechanisms. Both ends can be simply rejoined with little or no further processing (nonhomologous end joining, or NHEJ) or can be repaired using homologous sequences (red DNA; homologous recombination) after 5'-3' degradation has occurred (resection). The 3'-OH group exposed after resection can be used to prime DNA synthesis using a homologous region as a template after DNA strand invasion. The newly synthesized DNA (light blue) can then be joined with the 5' end of the resected strand, forming a double Holliday junction (double-strand break repair; DSBR), or can be displaced and reannealed (synthesis-dependent strand annealing; SDSA); or DNA synthesis can continue to the end of the chromosome (break-induced replication; BIR). If two homologous regions flank the DSB, they can anneal after being exposed by DNA resection (single-strand annealing; SSA), which causes the deletion of the intervening region. An additional mechanism that shares components with both SSA and NHEJ and that uses short homology stretches (usually 2-3 bp) flanking the DSB can also be used (microhomology-mediated end joining; MMEJ).
© 2010 Nature Publishing Group Huertas, P. DNA resection in eukaryotes: deciding how to fix the break. Nature Structural & Molecular Biology 17, 11–16 (2010). All rights reserved. 
Why Is It So Important to Understand the Proteins Involved in the DNA Repair Process?
Figuring out how things work at the molecular level is important because those molecular events result in an observable and measurable phenotype in the actual organism. There are several examples of human hereditary diseases that are the consequence of single point mutations in DNA repair genes. When the activity of one protein is altered because of a single amino acid change, that one change can wreck havoc and result in a human hereditary disease such as xeroderma pigmentosum, trichothiodystrophy, or Fanconi anemia (Garfinkel & Bailis 2002; Hoeijmakers 2007; Hashimoto & Egly 2009; Moldovan & D'Andrea 2009). A large number of these diseases are triggered by issues with normal cellular process. It is therefore important to identify the cause, the molecular event within the cell that malfunctioned that resulted in disease development. To understand how these diseases come about, sometimes the same cellular process affected by the mutations can be studied in a eukaryotic cell of a simpler organism, even in a simple unicellular organism like yeast. Basic cellular processes such as DNA repair pathways are highly conserved from lower to higher eukaryotes; thus, basic research in simple model organisms has been instrumental for the advancement of science. Most of what is described here comes from research done in yeast cells, a great model organism that is helping us understand how some human diseases arise at the molecular level (Aguilera & Gomez-Gonzalez 2008; Gitler 2008).
Detecting and Studying DSBs Using Pulsed-Field Gel Electrophoresis
DSBs are rare events during the normal life of a cell. Scientists have successfully used IR, radiomimetic agents (agents that produce effects similar to radiation), and UV to study systems that are involved in DSB repair. Besides these tools, scientists are also trying to perfect an assay to detect DSBs in DNA.
Gel electrophoresis is an essential tool in any molecular biology laboratory used to analyze DNA, but one of its limitations is that it only works well for resolving DNA fragments smaller than 50 Kb (the smallest yeast chromosome is about 200 Kb). In 1983, Schwartz and Cantor (Schwartz et al. 1983; Herschleb et al. 2007) developed a new method, called pulsed-field gel electrophoresis (PFGE), for separating (or resolving) DNA molecules of high molecular weight, such as chromosomes. Using this method, whole yeast chromosomes can be isolated intact and can be resolved due to differences in molecular weight. If you analyze intact yeast chromosomal DNA with PFGE, you can clearly see about twelve to fifteen distinct bands in an agarose gel. You do not see sixteen bands (budding yeast have sixteen chromosomes) because some chromosomes are of similar length and, depending on the gel conditions, do not resolve as separate bands. Why can we "see" these chromosomes without a microscope? The trick is that we typically isolate DNA in large samples, from about 60 million yeast cells. Therefore, we have 60 million copies of each chromosome. The original PFGE system used nonuniform electric fields, which resolved chromosomal bands well but resulted in crooked lanes, making it difficult to compare different samples (Contopoulou et al. 1987). In 1986, Chu invented a new system that could produce uniform electric fields, resulting in gels with samples running in straight lanes. This method is called contour-clamped homogeneous electric fields (CHEF) (Chu et al. 1986). Most examples of PFGE mentioned below are actually use CHEF gels, but most researchers refer to the method as PFGE.
How Do We Detect DNA Breaks with PFGE?
When cells are treated with a DNA damaging agent that causes DSBs, the original chromosomal bands get broken into smaller fragments. This process translates into an increase in intensity of smears between chromosomal bands, a smear toward the bottom of the gel, or both. Because bigger chromosomes are more susceptible to breakage, slowly migrating bands (bigger chromosomes) clearly visible in the control samples are the first ones to disappear or to appear as less intense in the treated samples (Westmoreland et al. 2009). Another approach to introducing DSBs in yeast cells is by using phenolic compounds. Figure 3 shows an example of this type of PFGE data. Compare lanes 1 and 3 at the top of the figure, where the largest chromosomal bands would appear, and note that damaged DNA is visible in one of the phenolic-treated samples. The effect of the phenolic treatment is not as drastic as that observed when treating cells with IR, as mentioned above.
PFGE is not only used to detect DSBs; it can also be used to follow the repair process. To study DSB repair using this system, researchers treat cells with a dose of IR high enough so that the genomic DNA is shattered, leaving no intact chromosomal bands. Cells are then allowed time to recuperate before their DNA is extracted and resolved by PFGE. When cells are allowed time for repair, the original band pattern, instead of the smear associated with DSBs, reappears in PFGE. These astonishing results indicate that DNA that was completely broken down can revert back to the original band pattern in a very accurate way.
Which factors are involved in putting the DNA back together in a precise way? Performing the above experiment with the same technique but with different DNA repair mutant strains allows us to see how the restoration takes place and check its accuracy. Researchers who have performed similar experiments have concluded that HR plays a main role in accomplishing this highly accurate repair (Contopoulou et al. 1987; Moore et al. 2000; Singh & Krishna 2005).
How are these breaks repaired? Can their repair lead to genome rearrangements? We know that X-rays increase mutation rates and also lead to chromosome rearrangements, but we do not known in detail which mechanisms of DNA repair lead to chromosomal rearrangements after this kind of perturbation. Using genetic assays that select for a particular outcome, several research groups have investigated the types of rearrangements induced by DNA damaging agents (Myung & Kolodner 2003; Pannunzio, Manthey et al. 2008). Researchers at the Petes and Resnick lab used PFGE to investigate chromosomal rearrangements induced by IR. First, the researchers irradiated diploid cells arrested in G2 phase, a cell cycle phase that occurs when the cell is naturally creating DSBs for HR, and also when the HR repair mechanism is quite active. The researchers induced approximately 250 DSBs per cell, and when the cells were allowed to recuperate, they observed that yeast were extremely efficient at reconstituting their genome from completely fragmented (Argueso et al. 2008). These results validated other researchers' previous observations (Contopoulou et al. 1987; Moore et al. 2000; Singh & Krishna 2005).
Then the scientists at the Petes and Resnick lab used PFGE to analyze genomic DNA from approximately one hundred different single colonies originated from cells that survived the irradiation. (A single colony arises from one cell going through multiple rounds of mitotic divisions, with each cell in the colony being a clone, an exact copy, of the original cell.) To their surprise, most genomes analyzed showed the presence of at least one new chromosomal band that was not present in the unirradiated control. As it turns out, these results were mainly due to HR events between dispersed repetitive sequences. These findings are exciting because several research groups think that recombination between dispersed repetitive sequences, which are highly abundant in human cells, could be the origin of genomic instability, leading ultimately to cancer (Rothstein et al. 1987; Strout et al. 1998; Aplan 2006; Pannunzio et al. 2008).
Modifying the PFGE Method to Meet Different Research Needs
Another advantage of the PFGE technique is that it can be easily modified in clever ways to address different research questions. The high-molecular-weight DNA isolated from cells is prepared in agarose plugs or inserts and can then be incubated with DNA-modifying enzymes such as restriction endonucleases. The agar matrix in which the DNA is embedded allows most enzymes to diffuse freely (Ma et al. 2008; Westmoreland et al. 2009).
Many protocols are being developed, so with some creativity and luck, it is possible to design a variation of the technique to suit a particular experiment. For example, one problem is that scientists want to detect DNA breaks with more sensitivity than that observed in the PFGE example (Figure 3A). One way to achieve this goal is to incubate isolated chromosomal DNA inserts from treated and untreated cells with the TUNEL (terminal deoxynucleotidyl transferase dUTP nick end labeling) enzyme and fluorescently labeled deoxynucleotides (dNTPs). This enzyme is a template-independent polymerase that can add nucleotides to free 3' DNA ends (with free 3'OH groups). If there are clean DNA breaks, these damaged chromosomal ends will be labeled with fluorescent nucleotides. The treated samples are resolved by PFGE, and the gel is scanned to detect fluorescence. The fluorescent smear observed in the treated samples results from specific labeling of DNA breaks with fluorescence. One advantage of this method is that there is no background due to intact chromosomal DNA (Figure 3B; compare lanes 3 and 4 with control lane 1). Once the PFGE is scanned for fluorescence, we can view all the DNA in the gel using ethidium bromide solution (another fluorescent DNA intercalator) (Figure 3B, lanes 5-8). With this technique, the difference between the treated and untreated samples becomes more evident (compare Figure 3A with Figure 3B).
There are still many discrepancies in the field of DSB repair regarding which factors are involved in the processing of DNA ends. The types of breaks being generated to study DSB repair differ depending on the agent used. IR or reactive oxygen species induce what is referred to as DSBs with "ragged" ends, which means that the ends need to be trimmed down before they can be repaired by a polymerase or be ligated.
Another limitation of inducing DSBs with a radiomimetic drug, IR, or other chemical agent is that the damage occurs randomly. If we want to study the detailed events taking place at each break, we need a different experimental system. For example, in yeast, an inducible endonuclease system, was developed. In this system, a single DSB can be induced synchronously in a large number of cells, allowing the mechanism of DSB repair to be studied in more detail at the molecular level.
Summary
The use of systems such as PFGE of whole yeast chromosomes has allowed scientists who study DNA repair to understand the genetic requirements for the precise repair of DSBs. In addition, PFGE is a great technique for observing DSB formation, processing, and repair. Despite some answers about mechanism, a slew of unanswered questions remains. For instance, how does the degradation of DNA ends take place? Does the extent of degradation have an effect on the repair pathway chosen? How is the degradation of DNA ends regulated? Is it coordinated with the cell cycle? These questions and others continue to drive the curiosity and interest of researchers.
References and Recommended Reading
Aguilera, A. & Gomez-Gonzalez, B. Genome instability: A mechanistic view of its causes and consequences. Nature Reviews Genetics 9, 204–217 (2008).
Aplan, P. D. Causes of oncogenic chromosomal translocation. Trends in Genetics 22, 46–55 (2006).
Argueso, J. L. et al. Double-strand breaks associated with repetitive DNA can reshape the genome. Proceedings of the Naitonal Academy of Sciences 105, 11845–11850 (2008).
Chu, G. et al. Separation of large DNA molecules by contour-clamped homogeneous electric fields. Science 234, 1582–1585 (1986).
Contopoulou, C. R. et al. Analysis of DNA double strand breakage and repair using orthogonal field alternation gel electrophoresis. Yeast 3, 71–76 (1987).
Friedberg, E. C. Deoxyribonucleic acid repair in the yeast Saccharomyces cerevisiae. Microbiology Reviews 52, 70–102 (1988).
Garfinkel, D. J. & Bailis. A. M. Nucleotide excision repair, genome stability, and human disease: New Insight from model systems. Journal of Biomedicine and Biotechnology 2, 55–60 (2002).
Gitler, A. D. Beer and bread to brains and beyond: Can yeast cells teach us about neurodegenerative disease? Neurosignals 16, 52–62 (2008).
Hashimoto, S. & Egly, J. M. Trichothiodystrophy view from the molecular basis of DNA repair/transcription factor TFIIH. Human Molecular Genetics 18, R224–R230 (2009).
Herschleb, J. et al. Pulsed-field gel electrophoresis. Nature Protocols 2, 677–684 (2007).
Hoeijmakers, J. H. Genome maintenance mechanisms for preventing cancer. Nature 411, 366–374 (2001).
Hoeijmakers, J. H. Genome maintenance mechanisms are critical for preventing cancer as well as other aging-associated diseases. Mechanisms of Ageing and Development 128, 460–462 (2007).
Huertas, P. DNA resection in eukaryotes: Deciding how to fix the break. Nature Structural and Molecular Biology 17, 11–6 (2010).
Ma, W. et al. Apn1 and Apn2 endonucleases prevent accumulation of repair-associated DNA breaks in budding yeast as revealed by direct chromosomal analysis. Nucleic Acids Reseach 36, 1836–1846 (2008).
Moldovan, G. L. & D'Andrea, A. D. How the fanconi anemia pathway guards the genome. Annual Review of Genetics 43, 223–249 (2009).
Moore, C. W. et al. DNA damage-inducible and RAD52-independent repair of DNA double-strand breaks in Saccharomyces cerevisiae. Genetics 154, 1085–1099 (2000).
Myung, K. & Kolodner, R. D. Induction of genome instability by DNA damage in Saccharomyces cerevisiae. DNA Repair (Amsterdam) 2, 243–258 (2003).
Pannunzio, N. R. et al. RAD59 is required for efficient repair of simultaneous double-strand breaks resulting in translocations in Saccharomyces cerevisiae. DNA Repair (Amsterdam) 7, 788–800 (2008).
Rothstein, R. et al. Concerted deletions and inversions are caused by mitotic recombination between delta sequences in Saccharomyces cerevisiae. Molecular and Cellular Biology 7, 1198–1207 (1987).
Schwartz, D. C. et al. New techniques for purifying large DNAs and studying their properties and packaging. Cold Spring Harbor Symposia on Quantative Biology 47, Part 1, 189–195 (1983).
Shrivastav, M. et al. Regulation of DNA double-strand break repair pathway choice. Cell Research 18, 134–147 (2008).
Singh, R. K. & Krishna, M. DNA strand breaks signal the induction of DNA double-strand break repair in Saccharomyces cerevisiae. Radiation Research 164, 781–790 (2005).
Strout, M. P. et al. The partial tandem duplication of ALL1 (MLL) is consistently generated by Alu-mediated homologous recombination in acute myeloid leukemia. Proceedings of the National Academy of Sciences 95, 2390–2395 (1998).
West, S. C. Molecular views of recombination proteins and their control. Nature Reviews Molecular Cell Biology 4, 435–445 (2003).
Westmoreland, J. et al. RAD50 is required for efficient initiation of resection and recombinational repair at random, gamma-induced double-strand break ends. PLoS Genetics 5, e1000656 (2009).










