


 Figure 2: Transformed cells exhibit different growth characteristics than non-transformed cells.
Figure 2: Transformed cells exhibit different growth characteristics than non-transformed cells.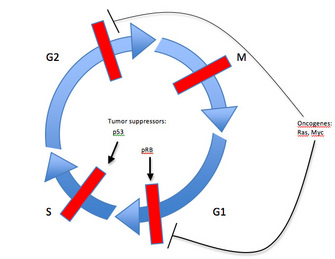

















« Prev Next »
How is a normal cell transformed into a cancerous cell? The proteins involved in regulating cell division events no longer appropriately drive progression from one cell cycle stage to the next. Rather than lacking function, cancer cells reproduce at a rate far beyond the normally tightly regulated boundaries of the cell cycle. Cancer can be distinguished from many other human diseases because its root cause is not a lack of, or reduction in, cell function. For example, individuals with diabetes may lack insulin production or the ability to respond to insulin. With coronary heart disease, poor blood supply to the heart can cause the organ to eventually fail. In the case of acquired immune deficiency syndrome (AIDS), the immune system loses the cells it needs to fend off infection. And with many infectious diseases, foreign microorganisms wreak havoc on the host they have invaded, causing a loss of function within cells, tissues or entire organ systems. Cancers, however, occur due to an alteration of a normal biological process — cell division.
Cells that progress through the cell cycle unchecked may eventually form malignant tumors, where masses of cells grow and divide uncontrollably, then develop the ability to spread and migrate throughout the body. Fortunately, cancer prevention usually occurs through the strict regulation of the cell cycle by groups of proteins that interact with each other in a very specific sequence of events. It is these events that determine whether the cell cycle will go forward or remain stalled between stages.

Figure 1: Cells growing in a tissue culture petri dish, adhered to dish bottom and immersed in liquid medium (pink)
© 2010 Nature Education All rights reserved. 
The answers to these questions serve as the basis for fundamental discoveries made by researchers in tumor cell biology. Abnormal or cancerous cells, grown in vitro have been transformed from their normal phenotype due to genetic changes affecting proteins involved in cell cycle control. Historically, these transformation assays have led to the identification of the genes and proteins important for driving the cell cycle forward. As a class, these genes have been named oncogenes. More recently, creative scientists have used advances in methods of genetic manipulation in combination with the transformation assay to identify the genes and proteins important for restricting cell cycle progression. As a class, these genes are called tumor suppressors.
Here we focus on the transformation assay, which scientists first used to identify oncogenes and tumor suppressors and study their affect on cell cycle progression. Even more important will be understanding the specific sequence of events in which multiple oncogenes and tumor suppressors must act in combination to promote cancerous cell growths (Kinzler 1996; Hahn 2002).
Early Work in Tumor Biology Led to the Discovery of a Standard Technique: The Transformation Assay
As early as 1911, Peyton Rous demonstrated through his studies of tumorous growths in chickens that the potential for tumor generation could be transferred from animal to animal in cell-free extracts. These extracts were eventually shown to contain viruses, whose ability to promote abnormally increased cell division in their hosts served to enhance their own replication. Thus the same processes stimulated by viral replication could lead to tumor production. This field of tumor virology was instrumental in developing the "cellular transformation assay" still used today to assess tumor growth.

© 2009 Nature Publishing Group Hagos, E. G., A. M. Ghaleb, et al. Mouse embryonic fibroblasts null for the Krüppel-like factor 4 gene are genetically unstable. Oncogene 28, 1197-1205 (2009). All rights reserved.
Researchers made use of these results from early virology studies with cellular assays. In these experiments, they infected the cultured cells with various viruses and then looked for "transformations" to occur (Todaro 1966). Since viral genomes are relatively small, researchers could more easily determine the genetic components responsible for transformation. In fact, the first oncogenes they identified were derived from viruses and called viral oncogenes. Remarkably, researchers soon also realized that the source of these viral oncogenes came from cellular counterparts that had been transferred by viruses from one cell to another (Varmus 1983).
The Accelerator Gets Stuck: Activated Oncogenes Drive Uncontrolled Cell Cycling
What, then, are oncogene products? These are the proteins involved in cell cycle regulation that operate by stimulating cellular growth and division (Figure 3). A common analogy equates oncogenes to an automobile's gas pedal stuck in the acceleration mode. Though the driver does not have his foot on the pedal, the car continues to speed up. Likewise, oncogenes code for proteins that function to drive the cell cycle forward, typically causing cells to proceed from one of the G (gap) phases to either chromosome replication (S phase) or chromosome segregation (mitosis). Examples include receptors at the cell surface that bind to growth factors, proteins that interact with DNA to initiate replication, and signaling molecules that link the receptors to the replication initiators through various pathways.
In their normal state, genes that code for the normal proteins controlling these critical processes are called proto-oncogenes. However, once they are altered (see below) to become oncogenes, their abnormal protein products exhibit increased activity that contributes to tumor growth. Therefore, instead of stopping within a G phase as it normally should, a tumor cell continues to progress through subsequent phases of the cell cycle, leading to uncontrolled cell division. In addition, oncogenes can also rescue cells from programmed cell death.
How does a proto-oncogene become converted to an oncogene? (Figure 4) Occasionally, mutations will permanently activate proteins that normally interchange between active or inactive states. For example, Ras proteins function as molecular switches that are turned on and off depending on the form of nucleotide (di-phosphate or tri-phosphate) to which it is bound. In an "on" state, the products of these proto-oncogenes relay proliferation-stimulating signals. Problems arise, however, when mutations convert the proto-oncogene to an oncogene, rendering Ras permanently active regardless of the signals the cell receives.
A second type of genetic alteration that converts a proto-oncogene to an oncogene is a chromosomal translocation. This occurs when the pieces of broken chromosomes reattach haphazardly, leading either to the formation of a fusion protein containing the N-terminus of one protein and the C-terminus of another, or leading to altered regulation of protein expression (Figure 4). One example of an oncogene generated by this type of chromosomal translocation is BCR/ABL, whose protein product consists of the N-terminus of Bcr (breakpoint cluster region) and the C-terminus of Abl, a tyrosine kinase that relays proliferative signals. The fused chromosome is known as the Philadelphia chromosome, and it is widely present in patients with chronic myelogenous leukemia, a blood cell cancer. Formation of the fusion protein renders Abl permanently active, leading to unregulated cell cycling.
Yet another means of generating oncogenes does not change the proto-oncogene directly at all. Instead, the proto-oncogene may exist in multiple copies in the cell, resulting in amplified expression. This is the case for c-MYC, for which 8 to 30 copies are present in each HL60 cell, a promyelocytic leukemia cell line. As myc is a transcription factor, its increased expression will, in turn, lead to the increased expression of its transcriptional targets, many of which function to drive the cell cycle forward. Since all of these genetic alterations result in a gain of function, only one affected chromosome is needed to induce the transformed cell phenotype (Vogelstein 2002).
The Broken Brake: Defective Tumor Suppressors Fail to Restrict Cell Cycling
How do tumor suppressors differ? In contrast to the cellular proliferation-stimulating function of proto-oncogenes and oncogenes that drive the cell cycle forward, tumor suppressor genes code for proteins that normally operate to restrict cellular growth and division or even promote programmed cell death (apoptosis). To continue the automobile analogy, tumor suppressors are like the brakes on a car. Examples include inhibitors of cell cycle progression, factors involved in maintenance of cell cycle checkpoints, and proteins required for apoptosis induction.
One of the best-studied factors of this type is a protein known as retinoblastoma protein (pRb) and its corresponding gene, RB1, the first tumor suppressor gene to be identified. Since pRb activity stops the expression of genes required for progression into S phase of the cell cycle, its inactivation allows for uncontrolled cell division (Figure 3). In fact, this principle applies to all tumor suppressors: genetic alterations in the gene leading to tumorigenesis prevent the regulatory protein from inhibiting cell proliferation. In other words, when the brakes on a car don't work, the car cannot stop.
The types of genetic alterations leading to pRb inactivity most often involve frameshifts or deletions in the RB1 gene causing premature introduction of a stop codon and defective protein expression. In some instances, expression of pRb may be normal, but the pathway in which it functions is defective due to inactivity of other pathway components. By definition, then, these other components would also be considered tumor suppressors (Burkhart, 2008).
Yet another example of a tumor suppressor, and the most commonly mutated gene in human tumors, is the p53 gene (Vogelstein, 2004). As a transcription factor that activates expression of proliferation-inhibiting and apoptosis-promoting proteins in response to DNA damage, p53 plays a critical role in maintaining the G1 to S cell cycle checkpoint (Figure 3). Genetic alterations that inactivate p53 will inhibit the DNA damage response that prevents cell cycle progression. When this occurs, a cell continues to divide even in the presence of DNA damage. Since inactivation of tumor suppressors results in a loss of function, both maternal and paternal copies of a gene coding for a tumor suppressor must usually be altered for tumorigenesis to occur — one good copy of the gene may provide sufficient activity for the cell to maintain proper growth and division.
Modern Strategies for Understanding the Molecular Basis for Cancer
How do scientists currently use the transformation assay to identify oncogenes and tumor suppressors? These days, manipulating gene expression in cultured cells is easily done in the lab. This makes the transformation assay extremely useful for dissecting the genetics responsible for cellular change. Insertion of oncogenes into normal cells in vitro, followed by assessment of the transformation criteria, allows for fairly straightforward identification of the genetic changes leading to tumorigenesis. While researchers use this strategy more commonly for oncogene assessment, they are now also applying it to tumor suppressor identification where shRNA expression knocks down tumor suppressor protein expression and induces a transformed cell phenotype (Bric 2009; Sung 2009). Since determining loss-of-function effects is inherently more difficult than evaluating the acquired characteristics of transformed cells, however, identification of tumor suppressor genes has long been more challenging than identification of oncogenes. Nevertheless, many of the modern strategies described below have been instrumental in making strides towards understanding both types of genetic changes.
In contrast to the first oncogenes that were identified through investigations in tumor virology, the first tumor suppressors (e.g., retinoblastoma gene-RB) were described following analysis of pedigrees of children afflicted with familial retinal tumors. In line with the idea that multiple genetic alterations are often necessary for full-blown tumor progression, these individuals also exhibited defects in other pathways affecting cell growth and division. And although it has since been established that having two defective copies of RB is the most critical feature that initiates tumor development in many different types of cancer, the fact that other genetic events must occur highlights the need for further understanding the tumorigenesis process (Burkhart 2008).
With the fully sequenced human genome and a multitude of strategies for genetic analysis of individuals (e.g., single nucleotide polymorphism analysis, microarray analysis, linkage analysis, sequencing individual genomes), discovery of new genes influencing tumor initiation continues at a rapid pace. Delineating the pathways affected by these genes, and the role they play in controlling the cell cycle, may prove more challenging (Vogelstein 2004).
The cellular transformation assay described above was a start, but scientists must also demonstrate that particular genetic alterations are both necessary and sufficient for the spontaneous development and spread of primary tumors within a human body. Transplantation of transformed cells into "humanized" animal models, such as mice whose hematopoietic systems have been reconstituted with human stem cells, has elucidated genes and pathways involved in the development of white blood cell tumors (leukemias and lymphomas) (Yu 2008). Indeed, a wide variety of mouse models in which inactivating mutations of tumor suppressors leads to spontaneous tumor development have already highlighted pathways important for tumorigenesis (Vogelstein 2004). Just as critical as identifying oncogenes and tumor suppressors, understanding the ways in which they interact to bring about uncontrolled cell cycle progression continues to challenge scientists seeking not only to treat and prevent cancer but also to understand how the cell cycle is fundamentally regulated.
Summary
Two classes of genes, oncogenes and tumor suppressor genes, link cell cycle control to tumor formation and development. Oncogenes in their proto-oncogene state drive the cell cycle forward, allowing cells to proceed from one cell cycle stage to the next. This highly regulated process becomes dysregulated due to activating genetic alterations that lead to cellular transformation. Tumor suppressor genes, on the other hand, restrict cell cycle progression. Their control over cell division is lost with genetic alterations leading to their inactivation. The role that both types of genes play in tumor formation can be experimentally determined using in vitro transformation assays or more complex in vivo animal models. These sorts of experiments will lead to a more thorough understanding of the genetic basis for cancer, more effective therapeutics, and a deeper appreciation of the intricacies of cell cycle regulation.
References and Recommended Reading
Bric, A., Miething, C. et al. Functional identification of tumor-suppressor genes through an in vivo RNA interference screen in a mouse lymphoma model. Cancer Cell 16, 324–335 (2009).
Burkhart, D. L. & Sage, J. Cellular mechanisms of tumour suppression by the retinoblastoma gene. Nature Reviews Cancer 8, 671–682 (2008).
Hahn, W. C. & Weinberg, R. A. Rules for making human tumor cells. New England Journal of Medicine 347, 1593–1603.
Kinzler, K. W. & Vogelstein, B. Lessons from hereditary colorectal cancer. Cell 87, 159–170 (1996).
Kopnin, B. P.Targets of oncogenes and tumor suppressors: key for understanding basic mechanisms of carcinogenesis. Biokhimiya 65, 2–27 (2000).
Kufe, D. W., Pollock, R. E., et al. eds. Holland-Frei Cancer Medicine. Hamilton, Ontario: BC Decker, 2003.
Sung, Y.M., Xu, X., et al. Tumor suppressor function of syk in human MCF10A in vitro and normal mouse mammary epithelium in vivo. PLoS ONE 4,
e7445.
Todaro, G. J. & Green, H. High frequency of SV40 transformation of mouse cell line 3T3. Virology 28, 756–759 (1966).
Varmus, H. & Levine, A. J. eds. Readings in Tumor Virology. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory, 1983.
Vogelstein, B. & Kinzler, K. W. Cancer genes and the pathways they control. Nature Medicine 10, 789–799 (2004).
Vogelstein, B. & Kinzler, K. W. The Genetic Basis of Human Cancer. 2nd ed. New York, NY: McGraw-Hill Medical Publishing Division, 2002.
Yu, C. I., Gallegos, M., et al. Broad influenza-specific CD8+ T-cell responses in humanized mice vaccinated with influenza virus vaccines. Blood 112, 3671–3678 (2008).










