


 Figure 2: The energetic funnel
Figure 2: The energetic funnel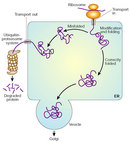
















« Prev Next »
Current advances in medicine and technology are making our lives longer. Sadly, as our life expectancy increases, the chances of getting a degenerative disease like Alzheimer's, Parkinson's, or diabetes also increases. Why is this? As incredible as it might sound, these diseases are caused not by bacteria or viruses but rather by something conceptually quite simple: incorrect protein folding. Introductory biology courses teach us that proteins are essential for the organism because they participate in virtually every process within the cell. Therefore, if their function is impaired, the consequences can be devastating. As we age, mutations and thermodynamics (as well as some external factors) conspire against us, resulting in the misfolding of proteins. How does this happen? What are the genetic and molecular causes for incorrect folding of proteins, and what is their relationship to aging?
Protein Function and Three-Dimensional Structure
Our modern understanding of how proteins function comes from almost 200 years of biochemical studies. Biochemistry is the science that studies the chemical processes in living organisms. Using different experimental models, biochemists demonstrated that most of the cell's chemical reactions and structural components are mediated or supplied by proteins. These experiments revealed that proteins are crucial for proper cell function. Actually the word "protein" comes from the Greek proteios, which means "first" or "foremost," reflecting the importance of these
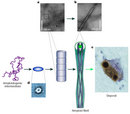
Figure 1: Proteins are long polymers made of amino acids.
(A) Part of the amino acid sequence of a spider silk protein. (B) The three-dimensional configuration of the same protein.
© 2010 Nature Publishing Group Askarieh, G. et al. Self-assembly of spider silk proteins is controlled by a pH-sensitive relay. Nature 465, 236–238 (2010). All rights reserved. 
How are proteins made in the cell? The answer to this question took decades of study and the birth of a new scientific discipline: molecular biology. Many experiments had shown that DNA is the vehicle of genetic information, and that DNA contains the information to make proteins. While discovering that DNA is itself a long polymer made out of four different types of small molecules called nucleotides, scientists realized that genetic information is transferred from a language system of four letters (nucleotides) in DNA to a language system of twenty (amino acids) in proteins.
The Energetic Funnel
The structure of a gene is one-dimensional. This means that a linear sequence of nucleotides codes for a specific linear sequence of amino acids linked to each other in a head-to-tail (amino-carboxyl) manner. The process of converting the information contained in the nucleotides to amino acids using the genetic code is called translation. Conceptually, translation "expands" the concentrated single dimension of the genetic code into a fully realized three-dimensional protein structure. From this point of view, DNA and the genome are very similar to a highly compressed digital file, such as an MP3, in which a lot of information is packed very efficiently. How is this possible? The Nobel laureate Christian B. Anfinsen postulated an answer. He proposed that all the information needed for a protein to fold into its three-dimensional conformation is contained in the amino acid sequence.
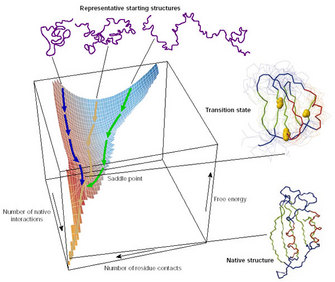
© 2000 Elsevier Adapted from Dinner, A. R., et al. Understanding protein folding via free energy surfaces from theory and experiment. Trends in Biochemical Sciences 25, 331–339 (2000). All rights reserved.
Amino acids have different side chains (R groups), which give them different properties. Some of these side chains are big, some are small, some are hydrophilic (interact with water), and some are hydrophobic (tend not to interact with water molecules); some are positively charged, and some are negatively charged. In a properly folded protein, hydrophobic amino acid residues are together, shielding each other from water molecules; hydrophilic residues are exposed on the surface of the protein, interacting with the water of the cytoplasm; and big amino acids make nooks and crannies for small ones. This kind of tight folding and packing minimizes the overall free energy of the protein.
An average protein has about 300 amino acid residues. If we consider that there are twenty different amino acids, the combinatorial number of protein sequences that can be made is astronomically high; by the most conservative calculation, the human body synthesizes at least 30,000 different kinds of proteins. Furthermore, the number of possible minimal-energy configurations of a single protein sequence is also unimaginably enormous, and usually only a few may have normal activity. Surprisingly, newly synthesized proteins usually fold correctly in the appropriate minimal-energy configuration, and thus they are able to do their job correctly.
As Anfinsen demonstrated, the information needed for proteins to fold in their correct minimal-energy configuration is coded in the physicochemical properties of their amino acid sequence. Usually a protein is capable of finding its functional or native state just by itself, in a matter of microseconds. The concept of how proteins explore the enormous structural conformational space is known as Levinthal's paradox. In 1968, Levinthal proposed that a protein folds rapidly because its constituent amino acids interact locally, thus limiting the conformational space that the protein has to explore and forcing the protein to follow a funnel-like energy landscape that allows it to fold into the most stable configuration possible (Figure 2; Levinthal 1968).
Most proteins follow the correct funnel, but some of them have bifurcating pathways that can make them fold in very different but energetically minimal structures, and only one of these is the native conformation (Dill & Chan 1997). In these cases, something must come to their aid, helping them find the correct native form. Amazingly the rescuer is nothing less than a protein itself.
Chaperones
Proteins that have a particularly complicated or unstable conformation sometimes have difficulty achieving their native state. In these cases other, specialized proteins called molecular chaperones help them find their native functional conformation. Molecular chaperones were first mentioned in 1978 by Ron Laskey, who found that nucleoplasmin (a protein found in the nucleus of the cell) is able to bind to histones. Histones are nuclear proteins whose major function is to interact with DNA to form structures known as nucleosomes (Laskey et al. 1978). Laskey observed that nucleoplasmin acted like a chaperone, accompanying and supervising the activity of the histones and preventing inappropriate interactions. Later, John Ellis extended the term chaperone to describe proteins that help other proteins fold or assemble into protein complexes (Ellis 1987). Interestingly, the existence of chaperones implies that some proteins have inherently unstable conformations that can "flip" from a functional minimal-energy state to a state that is nonfunctional or even toxic. Why is the final conformation of a protein so important? The three-dimensional structure of a protein is what allows it to do its work, to connect with reactive sites on other proteins and molecules within the cell. In other words, the multidimensional structure determines the function, and this concept is one of the most fundamental in biology.
Stable and Unstable Proteins
When native folded proteins are synthesized in a healthy cell, usually everything is right and well. However, our genome also codes for proteins that, as mentioned before, are inherently unstable because they have the property of folding in alternative minimal-energy states. Only very few of these alternative structures are functional and useful to the cell; the overwhelming majority are useless or even toxic. The functional or native conformation of non-membrane-bound proteins is typically water soluble. Chaperones will help unstable proteins fold correctly, although some proteins misfold anyway. Misfolded proteins (also called toxic conformations) are typically insoluble, and they tend to form long linear or fibrillar aggregates known as amyloid deposits. But how can a protein change so radically by folding differently, if the sequence of amino acids is the same? The answer is in the way the amino acids interact.
Protein Conformation and the Concept of Misfolding
For many proteins, the most prominent structural motif of the functional protein in its native conformation is known as the alpha helix, a right-handed spiral coil (Pauling et al. 1951). When a protein becomes toxic, an extensive conformational change occurs and it acquires a motif known as the beta sheet. Note that the beta sheet conformation also exists in many functional native proteins, such as the immunoglobulins, but the transition from alpha helix to beta sheet is characteristic of amyloid deposits. The abnormal conformational transition from alpha helix to beta sheet exposes hydrophobic amino acid residues and promotes protein aggregation.
As discussed already, misfolded proteins result when a protein follows the wrong folding pathway or energy-minimizing funnel, and misfolding can happen spontaneously. Most of the time, only the native conformation is produced in the cell. But as millions and millions of copies of each protein are made during our lifetimes, sometimes a random event occurs and one of these molecules follows the wrong path, changing into a toxic configuration. This kind of conformational change is most likely to occur in proteins that have repetitive amino acid motifs, such as polyglutamine; such is the case in Huntington's disease.Remarkably, the toxic configuration is often able to interact with other native copies of the same protein and catalyze their transition into the toxic state. Because of this ability, they are known as infective conformations. The newly made toxic proteins repeat the cycle in a self-sustaining loop, amplifying the toxicity and thus leading to a catastrophic effect that eventually kills the cell or impairs its function. A prime example of proteins that catalyze their own conformational change into the toxic form is the prion proteins, discussed below.
Under normal circumstances, the cell has mechanisms to prevent proteins from folding incorrectly, as well as to get rid of misfolded proteins. Proteins that have problems achieving their native configuration are helped by chaperones to fold properly, using energy from ATP. Chaperones can avoid the conformational change to beta sheet structure and the aggregation of these altered proteins; thus they seem fundamental to the prevention of protein misfolding. Despite chaperone actions, some proteins still misfold, but there is a remedy: The misfolded proteins can be detected by quality-control mechanisms in the cell that tags them to be sent to the cytoplasm, where they will be degraded (Figure 3).
Infectious Proteins
The concept of an infectious protein, or prion, was proposed in the 1960s to explain scrapie infection. Researchers found that the infectious agent that transmits scrapie is resistant to ultraviolet radiation (which typically destroys nucleic acids), and they proposed that this agent was actually protein based (Alper et al. 1967; Griffith 1967). The idea that proteins could be infectious by themselves was highly controversial because it appeared to challenge the central dogma of molecular biology. Eventually Stanley B. Prusiner and his team purified the prion protein responsible for scrapie, and they were able to show that proteins can indeed be infectious (Prusiner 1982). For this work, Prusiner was awarded the Nobel Prize in Physiology or Medicine in 1997. Prions are also responsible for transmissible spongiform encephalopathies, or TSEs, that include infectious diseases such as scrapie in sheep; bovine spongiform encephalopathy (mad cow disease), whose infective form can cause Creutzfeldt-Jakob disease in humans; and kuru, the only epidemic human prion disease known.
In the late 1950s, before the idea of prions was even proposed, an epidemic of the neurodegenerative disease called kuru suggested that proteins could be infectious. Kuru was discovered among populations of the Fore tribe of the eastern highland of Papua New Guinea, and the disease was associated with their cannibalistic funeral practices. With experimental testing, researchers showed that kuru could be infective in chimpanzees after intercerebral inoculation with brain suspension from kuru patients (Gajdusek et al. 1967). Years later, after kuru was recognized as a prion disease, the discovery that in some conditions prions can be infectious across species led to the naming of a similar neurodegenerative disease, Creutzfeldt-Jakob disease. This affliction could be caused by the ingestion of beef containing toxic protein particles. The conformational error in the toxic protein can also be caused by a mutation, thus making the disease familial. Prions are not an exclusive phenomenon of mammals; they also occur naturally in unicellular organisms such as yeast, which therefore have become good experimental models for studying these protein conformational changes.
Misfolded Proteins and Neurodegenerative Diseases
Accumulation of misfolded proteins can cause disease, and unfortunately some of these diseases, known as amyloid diseases, are very common. The most prevalent one is Alzheimer's disease, which affects about 10 percent of the adult population over sixty-five years old in North America. Parkinson's disease and Huntington's disease have similar amyloid origins. These diseases can be sporadic (occurring without any family history) or familial (inherited). Regardless of the type, the risk of getting any of these diseases increases dramatically with age. The mechanistic explanation for this correlation is that as we age (or as a result of mutations), the delicate balance of the synthesis, folding, and degradation of proteins is perturbed, resulting in the production and accumulation of misfolded proteins that form aggregates (Figure 4; Finkel 2005).
Among the environmental factors known to increase the risk of suffering degenerative diseases is exposure to substances that affect the mitochondria, increasing the amount of oxidative damage to proteins. However, it is clear that no single environmental factor determines the onset of these disorders. In addition, there are genetic factors. For example, in the simplest forms of familial Parkinson's disease, mutations are associated with dominant forms of the disease. This means that an individual with a single copy of a defective gene will develop the disease, yet two copies of the defective gene are required for recessive forms of the disease to develop. In the case of Alzheimer's disease, and for other less common neurodegenerative diseases, the genetics can be even more complicated, since different mutations of the same gene and combinations of these mutations may differently affect disease risk (Dobson 2002, 2003; Chiti & Dobson 2006).
Misfolding in Nonneurological Diseases
Protein aggregation diseases are not exclusive to the central nervous system; they can also appear in peripheral tissues. In general, the genes and protein products involved in these kinds of diseases are called amyloidogenic. Such diseases include type 2 diabetes, inherited cataracts, some forms of atherosclerosis, hemodialysis-related disorders, and short-chain amyloidosis, among many others. All these diseases have in common the expression of a protein outside its normal context, leading to an irreversible change into a sticky conformation rich in beta sheets that make the protein molecules interact with each other.
The general pattern that emerges in all these diseases is an abnormal tendency of proteins to aggregate as a result of misfolding. The aggregation can be caused by chance; by protein hyperphosphorylation (a condition where multiple phosphate groups are added to the protein), by prion self-catalytic conformational conversion, or by mutations that make the protein unstable. Aggregation can also be caused by an unregulated or pathological increase in the intracellular concentration of some of these proteins. Such imbalances in protein concentration can be a consequence of mutations such as duplications of the amyloidogenic gene or changes in the protein's amino acid sequence. Imbalances can also be caused by deficiencies in the proteasome, the cellular machinery involved in the degradation of aging proteins. Inhibition of autophagy (a process by which cells engulf themselves) also promotes amyloid aggregation. In addition, some evidence suggests that the severity of these diseases correlates with an increase in oxidative stress, mitochondrial dysfunction, alteration of cytoplasmic membrane permeability, and abnormal calcium concentration (Table 1; Lin & Beal 2006).
| Table 1 | ||
| Disease | Genetic causes | Function |
| Alzheimer's disease | APP | Gives rise to Aβ, the primary component of senile plaques |
| Parkinson's disease | PS1 and PS2 | A component of γ-secretase, which cleaves APP to yield Aβ |
| Parkinson's disease | α-Synuclein | The primary component of Lewy bodies |
| Parkinson's disease | Parkin | A ubiquitin E3 ligase |
| Parkinson's disease | DJ-1 | Protects the cell against oxidant-induced cell death |
| Parkinson's disease | PINK1 | A kinase localized to mitochondria. Function unknown. Seems to protect against cell death |
| Parkinson's disease | LRRK2 | A kinase. Function unknown |
| Parkinson's disease | HTRA2 | A serine protease in the mitochondrial intermembrane space. Degrades denatured proteins within mitochondria. Degrades inhibitor of apoptosis proteins and promotes apoptosis if released into the cytosol |
| Amyotrophic lateral sclerosis | SOD1 | Converts superoxide to hydrogen peroxide. Disease-causing mutations seem to confer a toxic gain of function |
| Huntington's disease | Huntingtin | Function unknown. Disease-associated mutations produce expanded polyglutamine repeats |
| Lin, M. T. & Beal, M. F. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 443, 787–795 (2006) doi:10.1038/nature05292. | ||
Looking Forward
At the moment there is no treatment for any of the known amyloid diseases. However, there is hope. The increasing knowledge of the causes of amyloid accumulation is beginning to pay off with possible pharmacological treatments. Therapeutic inhibition of precursor protein synthesis is within reach, with the expanding use of RNA interference (RNAi) technologies. Drugs that induce chaperone expression are also being tested, as well as inhibitors that prevent protein hyperphosphorylation. And as the number of known amyloid beta sheet structures grows, scientists have more options to find common structures for the design of specific chemical inhibitors of aggregation. Finally, vaccines against the aggregates are being developed (Chiti & Dobson 2006). Although we are at risk of accumulating misfolded proteins every day we age, and to function properly our cells must continually make proteins, understanding misfolding will ultimately help protect us from serious diseases.
References and Recommended Reading
Alper, T. et al. Does the agent of scrapie replicate without nucleic acid? Nature 214, 764–766 (1967)
Anfinsen, C. B. The formation and stabilization of protein structure. Biochemical Journal 128, 737–749 (1972)
Beadle, G. W. & Tatum, E. L. Genetic control of biochemical reactions in Neurospora. PNAS 27, 499–506 (1941)
Chiti, F. & Dobson, C. M. Protein misfolding, functional amyloid, and human disease. Annual Review of Biochemistry 75, 333–366 (2006)
Dill, K. A. & Chan, H. S. From Levinthal to pathways to funnels. Nature Structural Biology 4, 10–19 (1997)
Dobson, C. M. Protein folding and misfolding. Nature 426, 884–890 (2003) doi:10.1038/nature02261
Dobson, C. M. Protein misfolding diseases: Getting out of shape. Nature 418, 729–730 (2002) doi:10.1038/418729a
Ellis, J. Proteins as molecular chaperones. Nature 328, 378–379 (1987) doi:10.1038/328378a0
Fändrich, M. & Dobson, C. M. The behaviour of polyamino acids reveals an inverse side chain effect in amyloid structure formation The EMBO Journal 21, 5682-5690 (2002).
Finkel, T. Radical medicine: Treating ageing to cure disease. Nature Reviews Molecular Cell Biology 6, 971–976 (2005)
Gajdusek, D. C., Gibbs, C. J., Jr. & Alpers, M. Transmission and passage of experimental "kuru" to chimpanzees. Science 155, 212–214 (1967)
Gamow, G. & Ycas, M. Statistical correlation of protein and ribonucleic acid composition. PNAS 41, 1011–1019 (1955)
Griffith, J. S. Self-replication and scrapie. Nature 215, 1043–1044 (1967)
Kaufman, R. J., et al. The unfolded protein response in nutrient sensing and differation. Nature Reviews Molecular Cell Biology 3, 411-421 (2002)
Laskey, R. A. et al. Nucleosomes are assembled by an acidic protein which binds histones and transfers them to DNA. Nature 275, 416–420 (1978) doi:10.1038/275416a0
Levinthal, C. Are there pathways for protein folding? Journal de Chimie Physique et de Physico-Chimie Biologique 65, 44–45 (1968)
Lin, M. Y. & Beal, M. F. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 443, 787–795 (2006)
Nirenberg, M. W. & Matthaei, H. The dependence of cell-free protein synthesis in E. coli upon RNA prepared from ribosomes. Biochemical and Biophysical Research Communications 4, 404–408 (1961)
Pauling, L., Corey, R. B. & Branson, H. R. The structure of proteins: Two hydrogen-bonded helical configurations of the polypeptide chain. PNAS 37, 205–211 (1951)
Prusiner, S. B. Novel proteinaceous infectious particles cause scrapie. Science 216, 136–144 (1982)
Smith, M. A. et al. Effect of polyadenylic acid chain length on the size distribution of lysine peptides. Acta Biochimica Polonica 13, 361–365 (1966)










