
Abstract
The purpose of this study is to manually and semi-automatically curate a database and develop an R package that will act as a comprehensive resource to understand how biological processes are dysregulated due to interactions with environmental factors. The initial database search run on the Gene Expression Omnibus and the Molecular Signature Database retrieved a total of 90,018 articles. After title and abstract screening against pre-set criteria, a total of 237 datasets were selected and 522 gene modules were manually annotated. We then curated a database containing four environmental factors, cigarette smoking, diet, infections and toxic chemicals, along with a total of 25,789 genes that had an association with one or more of gene modules. The database and statistical analysis package was then tested with the differentially expressed genes obtained from the published literature related to type 1 diabetes, rheumatoid arthritis, small cell lung cancer, COVID-19, cobalt exposure and smoking. On testing, we uncovered statistically enriched biological processes, which revealed pathways associated with environmental factors and the genes. The curated database and enrichment tool are available as R packages at https://github.com/AhmedMehdiLab/E.PATH and https://github.com/AhmedMehdiLab/E.PAGE respectively.
Similar content being viewed by others

Introduction
Organisms are constantly being exposed to a wide range of environmental triggers that influence gene expression, resulting in several diseases. Environmental factors, such as drugs, toxic chemicals, smoke, temperature, dietary components and infections are considered modifiable causes of disease through their effects on biological processes, and in response, the expression of many genes is altered1. It is estimated that environmental factors account for approximately 70% percent of all autoimmune diseases and 80% of all chronic diseases2. These large proportions indicate that environmental exposures are an important contributor to disease, and there is ample evidence to support complex interrelationships between various environmental and genomic factors for disease causation3. Manipulation of environmental triggers and the host immune system during the clinical and preclinical stages of a disease will offer significant insight and guide early intervention for many disorders4.
In the era of Big Data technologies, several genomic databases exist to explore differential expression of genes under various clinical conditions5,6. However, to our knowledge there is currently no computational tool that can use information from existing large-scale databases to predict gene–environment relations. Therefore, in this study we formulated an integrated and comprehensive database that will provide insights of how environmental factors are associated to gene expression and disease, and leading to the identification of potential therapeutic strategies for the prevention and control of diseases attributable to both environmental and genetic factors.
Methods
We followed a two-step approach to conduct this study. First, we conducted a systematic review using a standard approach to identify all studies that used integrated datasets containing comprehensive information about environmental and genetic risk factors for various diseases. Second, we curated a database and developed a statistical analysis package to enable the user to understand the relationships between differentially expressed genes and select environmental factors.
Step 1: Systematic review
The aim of this step was to identify the relevant published literature from where we could obtain existing data pertinent to gene expression changes in response to an environmental factor. In detail the systematic review was conducted as follows:
Search strategy
We undertook a comprehensive literature and database search using PubMed, Gene expression omnibus (GEO), and Gene set enrichment analysis (GSEA) databases7. All databases were searched from their inception until 16th October 2020. The reference lists of all the retrieved studies were examined to identify additional studies.
The search terms and their synonyms related to environmental factors and gene expression. The keywords used included medical subject headings (MeSH) terms, e.g., ("Diet"[MeSH Terms] OR diet [All Fields]) AND ("gene expression"[MeSH Terms] OR gene expression [All Fields]). Table 1 details the search strategy and date of searches for various databases.
Inclusion/exclusion criteria
Pre-set inclusion criterion for studies to be considered eligible were:
-
Only articles written in English
-
Participants of any age group and both genders.
-
Since most of the experimental trials involving environmental factors were carried out in humans or mice, we included hits for Homo sapiens and Mus musculus.
-
Four specific environmental factors were chosen, based on the previous published evidence for major contribution as an environmental factor affecting gene expression8. Specifically,
-
o
Cigarette smoking—Includes data related to the practice of tobacco smoking and inhalation of tobacco smoke.
-
p
Diet—Includes data on the various types and quantities of food consumed by a person.
-
q
Infections—Includes data on infections caused by pathogenic organisms such as viruses, bacteria, fungi, protozoa and parasites.
-
r
Toxic chemicals—Includes data on substances such as metals or other chemical agents that are hazardous to human health if inhaled, ingested or absorbed.
-
o
-
We included published data from datasets, series and platforms. Samples were excluded if they consisted of unpublished data. We did not limit the search specific for any disease.
We did not include any dataset relating to mRNA, protein, CDS or small non-coding RNAs like miRNA or siRNA.
Literature review method
Two reviewers SM and SA screened the abstracts and citations independently at the same date and time and using the same search parameters. We identified articles that met the inclusion criteria. After title and abstract screening, studies were selected for full-text review. After the full length article review, those studies that met the inclusion criteria were selected for data extraction7.
Harmonization step
Names of Differentially expressed genes were extracted from GEO and MSigDB C7 databases. Pre-set inclusion criteria were used to select studies to be included in the database. Overlapping studies from the two databases were considered and coded as one study into the spreadsheet. We have further illustrated the harmonization steps in Supplementary SP4 and Figure S1.
Data extraction
Two reviewers SM and SA independently extracted data. The specific features extracted from each article were: (1) Differential gene expression data; (2) specific description of the type of data collected; (3) specific keywords related to the differentially expressed genes for each dataset, including disease, sample condition and pathways. These were manually searched in the abstract, demographics and result sections of each publication.
Data coding
Data were extracted and coded in a spreadsheet to collate information from each study. The data were combined and any anomalies between reviewers were resolved by a third reviewer (LY).
Differential gene expression data were obtained from the results section as well as from the supplementary section of the article. The differentially expressed genesets were annotated based on the information provided in the results section of the article on specific biological processes and/or molecular function regulated. The differentially expressed genes were coded into a spreadsheet and each geneset was provided a unique geneset number. In another spreadsheet the same geneset numbers were provided with annotations extracted from the article and a short description was given to describe the geneset module.
To remove potential bias of manual annotations, E.PAGE also provides functionality to annoSP1tate each geneset using GO, KEGG and MeSH annotations9,10,11 and users have an option to use either of methods or all. Further description on manual curation is provided in supplementary .
Quality and data validity assessment
The methodological quality was checked before including the data, using the Q-Genie tool12. We recorded whether the study used a standard microarray procedure and descriptions of the sample data, causes of up- and downregulation of genes and any other specific changes in the gene expression.
Step 2: Software generation
The statistical analysis package E.PAGE (Environmental Pathways Affecting Gene Expression) (https://github.com/AhmedMehdiLab/E.PAGE) was written in R version 4.0.313 and developed using RStudio14. Using publicly available packages tidyverse15 , Seurat16 as dependencies, the package performs enrichment analysis as previously described by Mehdi and colleagues17.
Mathematically, we represent the collection of annotated modules as \({\mathrm{M}=\{\mathrm{m}}_{1},{\mathrm{m}}_{2},\ldots{\mathrm{m}}_{\mathrm{n}}\}\) and the universal set of genes (background) as \(\mathrm{U}=\{{\mathrm{g}}_{1 } ,{\mathrm{g}}_{2 },..{\mathrm{g}}_{\mathrm{w}}\}\mathrm{ with}\, \mathrm{total}\, \mathrm{of}\, \mathrm{w}(\mathrm{U})\) genes. For each query list of genes \(\mathrm{g}\subseteq \mathrm{U containing n}(\mathrm{g})\mathrm{ genes}\,\mathrm{ in}\, \mathrm{query}\, \mathrm{list},\) we perform statistical enrichment of each module \(\mathrm{m}\) where \(\mathrm{m }\in \{{\mathrm{m}}_{1},{\mathrm{m}}_{2},..{\mathrm{m}}_{\mathrm{N}}\}\) with \({\mathrm{N}}_{\mathrm{m}}^{\mathrm{tot}}\) genes associated with \(\mathrm{m}\). We compared the number of genes \({\mathrm{N}}_{\mathrm{m}}^{\mathrm{g}}\) that had a specific annotation for gene module \({\text{m}}\) against those that did not. A hypergeometric distribution was used to determine a probability (p-value) that \({\mathrm{N}}_{\mathrm{m}}^{\mathrm{g}}\) or more belong to the module \({\text{m}}\) can be calculated using fisher exact test18. The p-value was corrected using false discovery rate (FDR) for multiple hypothesis testing using the Benjamini and Hochberg correction method19 to determine the adjusted p-value (\({p}_{adj}\)). The results are filtered based on the \({p}_{adj}\) are displayed to the user. Fold enrichment was calculated by taking the ratio of a set of genes containing a specific gene modules, and the total set of genes was obtained by taking the union of all the collected gene modules17 as follows; \(F.E = \frac{{N_{{gm}} /\user2{n}\left( \user2{g} \right)}}{{\user2{N}_{\user2{m}}^{{\user2{tot}}} /\user2{w}\left( \user2{U} \right)}}\). The adjusted fold enrichment was measured as a ratio of the fold enrichment value to the negative log of \({p}_{adj}\). An odds ratio then was measured to determine the probability of finding the set of enriched genes specific to an gene module20. We determined the percentage of interactions for four environmental variables (\({I}_{\widetilde{m}})\) where \(\widetilde{m}=\{\mathrm{cigarette}\,\mathrm{smoking }, \mathrm{diet}, \mathrm{infections },\mathrm{toxic}\, \mathrm{chemicals}\}\), \(\widetilde{m}\subseteq {\varvec{M}}\), as follows; \({\mathrm{I}}_{\widetilde{\mathrm{m}}}=\frac{{\mathrm{N}}_{\widetilde{\mathrm{m}}}^{\mathrm{g}}}{{\mathrm{N}}_{\widetilde{\mathrm{m}}}^{\mathrm{tot}}}\times 100\). We have provided examples of running E.PAGE in supplementary SP2.
Step 3: Case studies
We used six case studies to test our enrichment tool, these studies were not used in database curation. Case study 1 involves gene expression data in peripheral blood mononuclear cells (PBMC) in children with type 1 diabetes21. Gene expression changes were identified using microarray analysis from 43 patients with new onset T1D compared with 24 healthy controls. The gene expression data set in case study 2 is taken from the GEO database (microarray datasets; GSE12021, GSE55457, GSE55584 and GSE55235) that includes samples from 45 patients with rheumatoid arthritis, compared with 29 healthy control samples22. Case study 3 includes gene expression data from 23 small cell lung cancer samples and 42 healthy lung tissues23. The gene expression data from the case study 4 was taken from cobalt-exposed rat liver derived cells24. The final two case studies used differentially expressed genes extracted from single-cell expression data. Case study 5 was based on single-cell RNAseq data from COVID-19 patients, comparing severe and healthy cases in peripheral immune environments25, while case study 6 was based on a single-cell RNAseq-based atlas of epithelial cell-specific responses to smoking26. For single-cell RNA seq data, E.PAGE used a Seurat object (with clustering performed) as an input and performs differential expression analyses between the clusters to uncover lists of genes to compute related enriched gene modules.
Results
Systematic review and E.PAGE structure
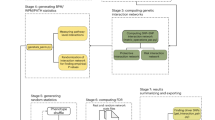
The initial electronic search of GEO and MSigDB database identified a total of 90,018 studies (Fig. 1). Title and abstract screening of retrieved studies resulted in a total of 3547 studies which had potential data related to environmental factors. After full text examination of 3547 studies, 3008 studies were excluded since they did not provide any differential gene expression data associated with any of the four environmental factors. A total of 237 datasets were obtained from 186 studies and the gene expression data were retrieved and collated to form a database. Figure 1 illustrates a flow chart of all the steps taken to obtain the data that satisfy the required parameters. The overall structure of E.PAGE is shown in Fig. 2. After literature screening, a database of 237 datasets was developed by linking each dataset with published lists of differentially expressed genes and the gene modules. Specifically, the text of these 186 publications and associated datasets were manually screened to develop gene modules representing the type of experiment, experimental conditions or disease type, experimental factors, demographics of subjects, and published pathways as previously described by Mehdi and colleagues17. The final database consisting of 237 datasets is obtained through GEO and MSigDB databases and includes 18,015 genes for diet, 13,259 genes for infections, 3841 genes for cigarette smoking and 644 genes for toxic chemicals.

PRISMA flow chart representing the various stages of screening involved in the systematic review process.

Flow chart representing the various parameters and their utilities provided on database query.
Querying E.PAGE
An R package was developed to enable statistical enrichment and gene modules associated with datasets/genes of interest to a user. The package produces various data tables as shown in Fig. 2 and a user can search genes of interest for their statistical enrichment. To test the utility of the statistical analysis package, we performed six case studies as described hereafter.
Case studies 1 and 2: Immune response activation in type-1 diabetes and rheumatoid arthritis
We first tested whether query signatures associated with T1D and RA could recover common pathways associated with these autoimmune disease. We used 291 DE genes uncovered from 43 patients with new-onset T1D as compared to 24 healthy controls8 (Table 2) and 229 DE genes from 45 samples from patients with RA, compared with 29 healthy control samples22 (Table 3). The statistical enrichment using E.PAGE identified that the genes in both datasets are involved in Immune response. Other significant gene modules that were common to both diseases include Interferons, IL-12 and Transcription regulation. These processes are all well known to be involved in RA and T1D27. Insulin resistance and Xenobiotic metabolism, which are both believed to be associated with T1D, were uncovered using E.PAGE and validate the utility of the platform (Table 2). Similarly, for RA, many smoking related gene modules such as Smoking history and Pack years (Smoking Status: Current, Never, Pack-years: (10–20), Pack-years: (20–30; Healthy smoker), (Above 40; Smoker with COPD)), were uncovered indicating an important risk factor for this disease (Table 3). For both T1D and RA, a large number of gene modules related to infections, both viral and bacterial (Lyme disease, Borrelia burgdorferi, HBV Infection, Viral response, Bacterial infection, Zika virus, Influenza A Infection, HIV infection, Echovirus-30, Rhinovirus infection), were significantly associated with disease, indicating that similar responses are occurring in patients suffering from these chronic autoimmune diseases as in responses to infections.
Case study 3: Regulation of the cell-cycle process in small cell lung cancer
We next studied gene modules associated with small cell lung cancer. The query signature containing 71 DE genes was derived from 23 clinical small cell lung cancer samples and 42 healthy control tissues23. We found that several lungs cancer associated gene modules were infections were was the most common environmental factor associated with the DE genes statistically significant (Table 4). The effect of Cigarette smoking (Tumor tissue vs Non tumor tissue in Non-smoker vs Smoker, Cigarette smoking, Smoking Status: Current, Never) was also evident. As expected, Lung tissue gene expression and Adenocarcinoma were amongst the top five gene modules, along with Cytoprotective mechanism, Mitotic spindle formation genes and Cell cycle, which are important pathways dysregulated in cancer (Table 4). Other interesting gene modules that are known to be involved in lung cancer were also identified, including Lung cancer, Cigarette smoking, Airway epithelium and Immune response.
Case study 4: Genotoxicity associated with cobalt exposed gene expression
We next used E.PAGE to understand the gene expression pathways involved in cobalt exposure. We used 27 DE genes uncovered by measuring the effect of cobalt exposure on gene expression in two rat liver derived cell lines using microarray analysis24. Cobalt exposed DE genes were associated with chemical induced gene expression. Other significant gene modules include genotoxicity, carcinogen, non-genotoxic, hepatocarcinogens, and liver-based in vitro models (Table 5).
Case study 5: Single-cell COVID-19 dataset
From a single-cell RNA sequencing dataset25, we first conducted a standard Seurat pipeline to determine the graph based clusters16. We then analysed enrichment of gene modules based on DE genes in Seurat clusters in COVID-19 and healthy cases. As expected, we identified COVID-19, SARS-COV2 modules. Significant enrichment was also observed for the Inflammation, Infection-type: Acute, Immune response, Infection induced gene expression and Cigarette smoking amongst the top modules that were previously shown to be COVID-19-related25,28,29 (Table 6).
Case study 6: Single-cell smoking dataset
As a sixth case study, we attempted to identify enriched gene modules related to smoking using a single cell RNA sequencing dataset which contained data of smokers vs non-smokers26. After processing the data using the Seurat pipeline and analyzing the single-cell expression data, gene set enrichment identified Epithelial gene expression, Cigarette smoking, Airway epithelium, and Chronic obstructive pulmonary disease as the top gene modules with highly significant p-values, confirming that smoking-related pathways were correctly predicted using E.PAGE (Table 7). Furthermore, smoking associated with gene signatures of lung-associated diseases such as Lung cancer, Cystic fibrosis, as well as with Carcinogen and respiratory infections such as Influenza and COVID-19.
User-defined annotations
The E.PAGE do not currently incorporate genetic data. However, to demonstrate its feasibility, we separately used two genetic datasets30,31 associated with Parkinson’s disease (PD) and developed automatic annotations using E.PAGE (Supplementary SP3). An independent transcriptomic dataset associated with PD was queried32. We uncovered annotations such as Genetic Association [Parkinson's Disease, GWAS + eQTL] and cellular response to interferon-gamma.
Discussion
Environmental factors are known to influence the development of disease, with or without combination with genetic factors, however there is currently no curated database and enrichment tool to identify the genes and the corresponding biological processes associated with these environmental conditions. We developed E.PAGE, a database and enrichment tool to understand the gene–environment relationship. Our database was developed based on experimental evidence obtained from the published literature to establish a relationship between environmental factors, differentially expressed genes and specific biological processes associated with the genes.
To set up the database, we used cigarette smoking, infections, toxic chemicals and diet, as they constitute the primary environmental factors influencing disease outcomes4. We made every effort to ensure completeness, accuracy and currency of the database. The current database has 237 datasets which consists of 25,789 genes in total. Traditional methods assume a linear relationship between environment and the genes33. In our study, the annotations such as Cigarette smoking have a direct relationship with environmental variables whereas the Viral response may have direct or indirect relationship with environmental variables depending on each experiment. Thus the annotations included in the study are a combination of linear and non-linear environment variables33. The largest number of datasets relate to diet and infections due to the long research history of these two environmental factors and disease. We manually curated each dataset using specific keywords and a brief description, abstract published with these datasets. We then developed an enrichment tool that uncovers modules associated with genes of interest using the methods we previously published17. In six case studies, we tested E.PAGE with sets of DE genes available from the literature. Specifically, we tested two gene lists associated with autoimmunity—T1D and RA—along with those related to small cell lung cancer, COVID-19 and smoking subjects. To confirm the effect of toxic chemicals on differential gene expression, we also used gene expression data from a study on cobalt exposure.
On testing T1D and RA associated DE genes, we found a large number of gene modules related to immune responses, which supports previous studies on how malfunction in the adaptive immune response results in activation of self-reactive T cells. We also obtained a substantial number of environmental modules associated with viral and bacterial infections, which supports recent findings on how bacterial and viral infections are implicated in immune response signaling in autoimmune disease pathogenesis. The T1D and RA associated DE genes were found to be primarily enriched in infection-associated gene modules and less in gene modules associated with the environmental factors diet, cigarette smoking or toxic chemicals. This information supports the hypotheses that infection-associated immune responses are major contributors to the development of T1D and RA34,35,36. A substantial number of genes involved in the central nervous system were also related to RA, consistent with other evidence37.
When small cell lung cancer genes were tested, we found a large number of environmental modules for DE genes to be related to lung cancer, as expected. We also found an expected link to cell cycle, since cell cycle checkpoints are disrupted leading to tumour development and cancer progression. Genes relating to cytoprotective function, mitotic spindle formation are also generally dysregulated in cancer. Recent studies that show a high incidence of retrovirus in lung small cell cancer suggest a possible direct link between infections and small cell cancer38.
To further assess associations between environmental factors with toxic chemicals, we tested genes differentially expressed due to cobalt exposure against the E.PAGE database. On testing, we found the modules Genotoxicity and Carcinogen to be enriched. We also obtained a substantial number of genes differentially expressed due to toxic chemicals as environmental factors, supporting the validity of the tool to identify potential involvement of toxic chemicals on DE genes involved in critical functions in a relevant datasets.
On testing gene expression data sourced from patients with COVID-19, we found that genes differentially expressed in severe cases were linked to gene modules common between bronchoalveolar and peripheral immune environments25,29. This finding shows how the E.PAGE database can be used to find commonalities between two sets of differentially expressed genes, even if they may not have many genes in common.
On testing the single-cell gene expression data for smoking we found gene modules for Cigarette smoking, Airway epithelium, Epithelial gene expression, and Chronic obstructive pulmonary disease. Additional pathways that are well known to be altered by cigarette smoking were identified. Therefore, E.PAGE was able to find relevant significantly enriched gene modules.
From the above case studies, we found that our database is highly reliable and has the potential to establish a link between environmental factors and important biological processes. In the case studies, we generally obtained a higher number of DE genes related to infection as an environmental factor. Though this link with infection may be valid, there is a possibility of dataset bias due to limited type of input data such as gene list, similarities between infection and tissue damage -associated immune responses. Additionally, our study is limited to four types of environmental variables, therefore to increase usage towards wider community more environmental datasets need to be integrated. Our study is limited to the use of MeSH terms to query GEO database for differential gene expression data. Additional statistical tests such as joint odds ratio and interaction odds ratio could be included to increase the statistical representation of the datasets39. Our study is currently limited to four types of environmental variables, therefore to increase usage in the wider community more environmental datasets will be integrated over time. Further updates will be the addition of other statistical tests to cover genetic data such as Single Nucleotide Polymorphisms, Copy Number Variants and DNA Methylations40,41,42.
A key benefit of this research is to predict gene–environment interactions to identify novel associations between environmental factors and disease, and to inform hypothesis synthesis and target selection. Thereby, it allows scientists and epidemiologists to dissect which genes may be influenced by environmental exposures in different disease conditions. We illustrate this by using examples from type-1 diabetes, rheumatoid arthritis, small cell lung cancer and COVID-19 datasets.
The current study lends itself to future extension to additional environmental variables such as alcohol, physical activities, life-style factors, along with inclusion of other kinds of genetic data which could facilitate the development of disease risk prediction models. Additionally, variable selection methods could be employed to select candidates for gene–environmental variables associated with the disease43.
Data availability
The E.PATH is freely available at https://github.com/AhmedMehdiLab/E.PATH.
Code availability
The R package (E.PAGE) to process E.PATH is available as an R package is openly available at https://github.com/AhmedMehdiLab/E.PAGE.
References
Skinner, M. K. Environmental epigenomics and disease susceptibility. EMBO Rep. 12(7), 620–622 (2011).
Vojdani, A., Pollard, K. M. & Campbell, A. W. Environmental triggers and autoimmunity. Autoimmune Dis. 2014, 798029 (2014).
Rappaport, S. M. Discovering environmental causes of disease. J. Epidemiol. Community Health 66(2), 99–102 (2012).
Vojdani, A. A potential link between environmental triggers and autoimmunity. Autoimmune Dis. 2014, 437231 (2014).
He, K. Y., Ge, D. & He, M. M. Big data analytics for genomic medicine. Int. J. Mol. Sci. 18(2), 412 (2017).
Raghupathi, W. & Raghupathi, V. Big data analytics in healthcare: Promise and potential. Health Inf. Sci. Syst. 2, 3 (2014).
Ennie, N. A. R. K. A systematic review of interventions to improve diabetes care in socially. Diabetes Care 29, 1675–1688 (2006).
Alberti, K. G. & Zimmet, P. Z. Definition, diagnosis and classification of diabetes mellitus and its complications. Part 1: Diagnosis and classification of diabetes mellitus provisional report of a WHO consultation. Diabetes Med. 15(7), 539–553 (1998).
Coletti, M. H. & Bleich, H. L. Medical subject headings used to search the biomedical literature. J. Am. Med. Inform. Assoc. 8(4), 317–323 (2001).
Kanehisa, M., Furumichi, M., Tanabe, M., Sato, Y. & Morishima, K. KEGG: new perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 45(D1), D353–D361 (2017).
Yu, G., Wang, L. G., Han, Y. & He, Q. Y. clusterProfiler: An R package for comparing biological themes among gene clusters. OMICS 16(5), 284–287 (2012).
Sohani, Z. N. et al. Assessing the quality of published genetic association studies in meta-analyses: The quality of genetic studies (Q-Genie) tool. BMC Genet. 16, 50 (2015).
Team RC. R: A Language and Environment for Statistical Computing (R Foundation for Statistical Computing, 2020).
Team R. RStudio: Integrated Development Environment for R (RStudio, PBC, 2021).
Wickham, H. et al. Welcome to the {tidyverse}. J. Open Source Softw. 4(43), 1686 (2019).
Butler, A., Hoffman, P., Smibert, P., Papalexi, E. & Satija, R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol. 36(5), 411–420 (2018).
Mehdi, A. M. et al. A peripheral blood transcriptomic signature predicts autoantibody development in infants at risk of type 1 diabetes. JCI Insight 3, 1–13 (2018).
Winters, R., Winters, A. & Amedee, R. G. Statistics: A brief overview. Ochsner J. 10(3), 213–216 (2010).
Benjamini, Y., Drai, D., Elmer, G., Kafkafi, N. & Golani, I. Controlling the false discovery rate in behavior genetics research. Behav. Brain Res. 125(1–2), 279–284 (2001).
Szumilas, M. Explaining odds ratios. J. Can. Acad. Child Adolesc. Psychiatry 19(3), 227–229 (2010).
Kaizer, E. C. et al. Gene expression in peripheral blood mononuclear cells from children with diabetes. J. Clin. Endocrinol. Metab. 92, 3705–3711 (2007).
Lu, W. & Li, G. Identification of key genes and pathways in rheumatoid arthritis gene expression profile by bioinformatics. Acta Reumatol. Port. 43(2), 109–131 (2018).
Sato, T. et al. PRC2 overexpression and PRC2-target gene repression relating to poorer prognosis in small cell lung cancer. Sci. Rep. 3, 1911 (2013).
Permenter, M. G. et al. Exposure to cobalt causes transcriptomic and proteomic changes in two rat liver derived cell lines. PLoS ONE 8, 1–11 (2013).
Wilk, A. J. et al. A single-cell atlas of the peripheral immune response in patients with severe COVID-19. Nat. Med. 26(7), 1070–1076 (2020).
Goldfarbmuren, K. C. et al. Dissecting the cellular specificity of smoking effects and reconstructing lineages in the human airway epithelium. Nat. Commun. 11(1), 2485 (2020).
Ter Horst, R. et al. Host and environmental factors influencing individual human cytokine responses. Cell 167(4), 1111–24.e13 (2016).
Hopkinson, N. S. et al. Current smoking and COVID-19 risk: Results from a population symptom app in over 2.4 million people. Thorax 76, 714–722 (2021).
Liao, M. et al. Single-cell landscape of bronchoalveolar immune cells in patients with COVID-19. Nat. Med. 26(6), 842–844 (2020).
Li, B. et al. Gene4PD: A comprehensive genetic database of Parkinson’s disease. Front. Neurosci. 15, 679568 (2021).
Pierce, S. & Coetzee, G. A. Parkinson’s disease-associated genetic variation is linked to quantitative expression of inflammatory genes. PLoS ONE 12(4), e0175882 (2017).
Kia, D. A. et al. Identification of candidate Parkinson disease genes by integrating genome-wide association study, expression, and epigenetic data sets. JAMA Neurol. 78(4), 464–472 (2021).
Wu, C. & Cui, Y. A novel method for identifying nonlinear gene–environment interactions in case–control association studies. Hum. Genet. 132(12), 1413–1425 (2013).
Bo, M. et al. Role of infections in the pathogenesis of rheumatoid arthritis: Focus on mycobacteria. Microorganisms 8(10), 1459 (2020).
Mouat, I. C., Morse, Z. J., Shanina, I., Brown, K. L. & Horwitz, M. S. Latent gammaherpesvirus exacerbates arthritis through modification of age-associated B cells. Elife 10, e67024 (2021).
Pino, S. C., Kruger, A. J. & Bortell, R. The role of innate immune pathways in type 1 diabetes pathogenesis. Curr. Opin. Endocrinol. Diabetes Obes 17(2), 126–130 (2010).
Sağ, S. et al. Central nervous system involvement in rheumatoid arthritis: Possible role of chronic inflammation and tnf blocker therapy. Acta Neurol. Belg. 120, 25–31 (2017).
Robinson, L. A. et al. Molecular evidence of viral DNA in non-small cell lung cancer and non-neoplastic lung. Br. J. Cancer 115(4), 497–504 (2016).
Simonds, N. I. et al. Review of the gene–environment interaction literature in cancer: What do we know?. Genet. Epidemiol. 40(5), 356–365 (2016).
Cornelis, M. C. et al. Gene–environment interactions in genome-wide association studies: A comparative study of tests applied to empirical studies of type 2 diabetes. Am. J. Epidemiol. 175(3), 191–202 (2012).
Thomas, D. Gene–environment-wide association studies: Emerging approaches. Nat. Rev. Genet. 11(4), 259–272 (2010).
Winham, S. J. & Biernacka, J. M. Gene–environment interactions in genome-wide association studies: Current approaches and new directions. J. Child Psychol. Psychiatry 54(10), 1120–1134 (2013).
Zhou, F., Ren, J., Lu, X., Ma, S. & Wu, C. Gene–environment interaction: A variable selection perspective. In Epistasis Methods in Molecular Biology Vol. 2212 (ed. Wong, K. C.) 191–224 (Springer US, 2021).
Funding
Supported by NHMRC (Grant No. 1071822, 2008287, 1173927), PA Research Foundation and The Merchant Foundation. RT was supported by Arthritis Queensland and a NHMRC Senior Research Fellowship. AMM was supported by a Postdoctoral Fellowship from JDRF (3-PDF-2016-198-A-N).
Author information
Authors and Affiliations
Contributions
Concept and design of the study contributed by S.M., A.M.M., I.F., and R.T. Data collection contributed by S.M., S.A., L.Y., J.B., S.F.Z., R.A. Data analysis, and interpretation contributed by S.M., S.A., L.Y., A.M.M., J.C., I.F., S.F.Z., R.A. and R.T. Manuscript preparation contributed by S.M., A.M.M., I.F. and R.T.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Muralidharan, S., Ali, S., Yang, L. et al. Environmental pathways affecting gene expression (E.PAGE) as an R package to predict gene–environment associations. Sci Rep 12, 18710 (2022). https://doi.org/10.1038/s41598-022-21988-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-022-21988-6
This article is cited by
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
