
Abstract
Thyroid-like follicular renal cell carcinoma is an uncommon kidney tumor with no distinct molecular alteration described to date. This cohort of eight women with mean and median ages of 45 and 46 years, respectively (range 19–65 years), had unencapsulated, well-circumscribed tumors composed of tightly packed anastomosing follicle-like cysts filled with eosinophilic colloid-like material and lined by cuboidal cells with high nuclear to cytoplasmic ratios, oval to elongated nuclei with perpendicular arrangement toward the lumens, and prominent nuclear overlapping. The stroma between these was minimal with the exception of two tumors. Calcifications and necrosis were absent. Immunohistochemically, the tumors were positive for KRT19 (7/7), PAX8 (5/5), cyclin D1 (6/6), KRT7 (5/7), and AMACR (1/5; focal, weak), and were negative for WT1, TTF1 (transcription termination factor-1), and thyroglobulin. In three of three tumors tested molecularly, EWSR1-PATZ1 fusion was identified by RNA sequencing and confirmed by RT-PCR and Sanger sequencing. Over a follow-up period of 1–7 years, no evidence of recurrence or metastasis has been detected. The EWSR1-PATZ1 fusion has been recognized as a recurrent alteration in a subset of round to spindle cell sarcomas with EWSR1–non-ETS fusions (EWSR1-PATZ1 sarcoma) and in several central nervous system tumors. The finding of an EWSR1-PATZ1 fusion in all three of the thyroid-like follicular renal cell carcinomas for which sufficient tissue was available for genomic profiling provides the first distinct molecular abnormality in thyroid-like follicular renal cell carcinomas, supporting its designation as a distinct diagnostic entity.
Similar content being viewed by others

Introduction
Thyroid-like follicular renal cell carcinoma is rare. It was first described by Amin et al. in 2004 [1] at the United States and Canadian Academy of Pathology annual meeting and the full manuscript of the same study was published in 2009 [2, 3]. To date, fewer than 40 cases have been reported. Due to the small number of reported cases and limited data regarding its pathogenesis, it was considered a provisional entity in the 2016 WHO classification of kidney tumors [4].
Microscopically, these tumors are composed of tightly packed cysts, resembling thyroid follicles; however, they stain negatively for transcription termination factor-1 (TTF1) and thyroglobulin. These tumors appear to occur more frequently in women and seldom progress [5, 6]. Herein, a series of eight tumors are reported with clinicopathologic and immunohistochemical features, of which three of three subjected to molecular testing demonstrated EWSR1-PATZ1 fusion by next-generation sequencing (NGS) and/or reverse-transcription PCR (RT-PCR) analysis. EWSR1-PATZ1 fusion has not been previously described in thyroid-like follicular renal cell carcinoma. We propose that this gene fusion event may be of pathogenic importance in thyroid-like follicular renal cell carcinomas.
Materials and methods
Study population
A search was performed of the institutional databases at the participating institutions following institutional review board approval. For inclusion in the study group, we required the presence of tightly packed, variably sized, anastomosing follicle-like cysts filled with eosinophilic colloid-like material and lined by cuboidal cells with high nuclear to cytoplasmic ratios, oval to elongated nuclei with perpendicular arrangement toward the lumens, and inconspicuous nucleoli. After application of these criteria, eight tumors were identified and retrieved from the pathology department files and the authors’ consultation archives. Tumor 7 was previously published [7]. A control group composed of two oncocytoma-like tumors (dense cytoplasmic eosinophilia or oncocytic renal neoplasm with dilated tubular structures), which contain dense colloid-like secretions, giving them a thyroid-like appearance, was included.
Histopathologic study
Tissues from all ten tumors were fixed in 10% neutral buffered formalin and embedded in paraffin. Cases 1–8 were stained with antibodies directed against PAX8 (rabbit, polyclonal, Cell Marque, CA), keratin (KRT) 7 (OV-TL 12/30; Dako, Carpinteria, CA), KRT19 (PCK108; Dako), cyclin D1 (EO12; Dako), α-methylacyl-CoA-racemase (AMACR/P504S [13H4; Dako]), Wilms tumor 1 (6F-H2, Dako), TTF1, formerly known as thyroid transcription factor-1 (8G7G3/1; Dako), thyroglobulin (Dako), melan-A (A103; Dako), and TFE3 (MRQ-37, Cell Marque) in a Dako automated instrument. The staining intensity was recorded as negative (0), weak (1+), moderate (2+), or strong (3+). Less than 5% weak staining was considered negative. Positive and negative controls gave appropriate results for each stain.
Fluorescence in situ hybridization analysis
Fluorescence in situ hybridization (FISH) was performed on tumors 1 and 2 with the chromosome enumeration probes (CEPs) for chromosomes 3, 7, 10, 12, and 17 (Abbott, Downers Grove, IL) as previously described [8,9,10]. The probes were diluted with tDenHyb-2 (Insitus, Albuquerque, NM) in a ratio of 1 : 25 and 5 µl was applied to each slide under reduced light conditions. The slides were then covered with a 22 × 22 mm cover slip and sealed with rubber cement. Denaturation was achieved by incubating the slides at 83 °C for 10 min in a humidified box and hybridization at 37 °C overnight. FISH signal analysis was described previously [8,9,10].
NGS and data analysis
Formalin-fixed, paraffin-embedded (FFPE) blocks of two thyroid-like follicular renal cell carcinomas (cases 1 and 2) containing more than 60% tumor were submitted for targeted NGS using the University of Chicago Medicine OncoPlus (UCM-OncoPlus) panel, a hybrid-capture panel targeting 1213 cancer-associated genes. Nucleic acid extraction/quantification, library preparation, and sequencing were performed as previously described [11]. Data analysis was performed on a Health Insurance Portability and Accountability Act-compliant high-performance computing system (Center for Research Informatics, University of Chicago) using a University of Chicago-developed bioinformatics pipeline. First, using BWA MEM v0.7.12 [12], the data were aligned to the hg19 reference human genome followed by de novo local assembly and realignment using ABRA [13]. For variant calling, variants were filtered based on depth (>50× base calls with Phred quality score > 30), minor allele frequency (>10%), and location in clinical exonic territory for review. Variant calls were annotated and converted to Human Genome Variation Society nomenclature using Alamut Batch version 1.4.0 software. Further manual variant curation was followed in accordance with the Association for Molecular Pathology recommendations [14].
Genome-wide copy number alterations were calculated using a combination of CNVKit v0.8.2 software [15] and intra-run normalization by comparison with clinical controls, to eliminate run-specific artifacts. Focal gene-level changes were called using the UCM-OncoPlus clinical interpretation criteria [11], whereas the chromosome-level changes were called using a combination of UCM thresholds (1.5-fold change) and manual interpretation [16].
Fusion detection was performed using SvABA v0.2.1 software. De-duplicated ABRA-realigned BAM files were provided as input BAM to the software. The unfiltered vcf file output by SvABA is further filtered based on information provided by the software, such as quality scores, assembly and discordant support, MAPQ, and number of supporting reads (FILTER = PASS, QUAL > 0, and AD > 5 or FILTER = LOWMAPQ, QUAL > 20, and AD > 20), using an in-house script. False-positive calls and results from noisy region of the panel were also removed. Rearrangements from the final filtered vcf file are then manually reviewed [13, 17].
A representative FFPE block from tumor 3 was submitted to Memorial Sloan Kettering Cancer Center (MSKCC) for NGS using the MSK-Fusion assay, a targeted RNA sequencing (RNAseq) panel approved by New York State Department of Health as a clinical assay. MSKCC was blinded to the results identified in cases 1 and 2. Following RNA extraction and quantification using the standard RNeasy FFPE Kit and protocol (Qiagen, Catalog #73504) and Qubit Broad Range RNA Assay Kit (Life Tech., Catalog #Q10211), respectively, the quality of the RNA sample was assessed using the Archer® PreSeq™ RNA QC Assay prior to library preparation and sequencing [18].
For RNAseq, cDNA libraries were prepared using the Archer™ FusionPlex™ standard protocol and supplied reagents including Archer® Universal RNA Reagent Kit for Illumina® (Catalog #AK-0040–8), Archer MBC adapters (Catalog #SA0040–45), and custom-designed Gene Specific Primer Pool kit as previously described. High yield of RNAseq for targetable kinase fusions in lung adenocarcinomas with no mitogenic driver alteration detected by DNA sequencing and low tumor mutation burden was assessed [18]. Data were analyzed using the Archer analysis software (V6.0).
RT-PCR and Sanger sequencing
Total RNA extraction was performed from four unstained FFPE tissue slides of study tumors 1, 2, and 4, and control cases 9 and 10. The concentration of RNA was assessed with Qubit 2.0 Fluorometer (Agilent Technologies) using an RNA HS Assay Kit. The integrity and the average size of RNA was assessed by endpoint RT-PCR assay using two different primer pairs directed against the HPRT gene [HPRT 518/781 (5′-ACT TTG CTT TCC TTG GTC A-3′/5′-TTT ACT GGC GAT GTC AAT AG-3′ and HPRT679/781 (5′-CTT CAG GGA TTT GAA TCA TG-3′/5′-TTT ACT GGC GAT GTC AAT AG-3′)]. For the validation of NGS data and fusion transcript screening, RT-PCR assay was established. Fifty nanograms of total RNA was retro-transcribed to cDNA using the PrimeScript High Fidelity RT-PCR kit (Takara, Mountain View, CA 94043, USA) according to the supplier’s recommendations. PCR amplification of the fusion point in the presence of PCR primers designed based on the analysis of the NGS data set was performed using PrimeSTAR Max DNA Polymerase (Takara, Mountain View, CA 94043, USA). The PCR products were analyzed on a 2% agarose gel in the presence of ethidium bromide and cloned into pCR-4 TOPO vector (Invitrogen, Carlsbad, CA) following the addition of dA to the 3′-ends of the amplified products by the use of TA-cloning Reagent Set for PrimeSTAR (Catalog #6019, Takara). All clones were confirmed by Sanger sequencing at the ASU Genome Sequencing Core facility (Phoenix, AZ) using Big Dye V3.1 chemistry (Applied Biosystem, Foster City, CA) and processed employing an Applied Biosystems 3730XL Capillary electrophoresis Instrument.
Results
Clinical features
Clinicopathological details of the ten patients (including two cases in the control group) are presented in Table 1. For cases 1–8, the patients’ ages ranged from 19 to 65 years (mean 45 years, median 46 years). All were women. Pertinent clinical histories include the following: patient 1 had a history of hypertension, patient 6 received chemoradiation therapy for B-lymphoblastic leukemia at the age of 2 years and had cirrhosis due to hepatitis C viral infection, and patient 7 with leiomyomatosis had a family history of hereditary leiomyomatosis-associated renal cell cancer and underwent nephrectomy for cystic renal dysplasia. Genetic testing for FH mutation was negative.
All tumors were incidental findings. The initial diagnosis was rendered on nephrectomy specimens for six patients (two partial and four radical nephrectomies). Two patients, cases 6 and 8, were diagnosed on biopsy (the biopsy from tumor 6 was obtained at the time of radiofrequency ablation). Four of the resected tumors were stage category pT1a (cases 1, 4, 5, and 7), one (case 3) was pT1b (tumor size 70 mm), and one (case 2) was pT3a, due to invasion of renal sinus fat. Follow-up data were available for five patients. All patients, but one (case 6) were alive, with no evidence of recurrence or metastasis at a median follow-up period of 16 months (mean 37 months, range 10–84 months). Patient 6 died due to complications of cirrhosis at 24 months following renal biopsy. Surveillance radiologic studies showed no evidence of recurrence of metastasis during follow-up.
Pathological and immunohistochemical features
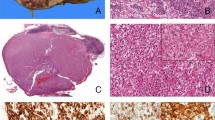
Grossly, the tumors ranged from 8 to 70 mm in diameter (mean 34 mm; median 36 mm). Cases 1 and 3–5 were well-circumscribed and were tan-yellow on cut surface. Tumor 2 was mostly well-circumscribed, yellow-tan with foci of hemorrhage and cystic degeneration (Fig. 1A). Tumor 7 was incidentally found on histologic examination of a nephrectomy specimen resected due to congenital cystic renal dysplasia.

Representative gross image of EWSR1-PATZ1 renal cell carcinoma (case 2) showing a well-circumscribed, tan-yellow cut surface with foci of central fibrosis, hemorrhage, and degenerative changes (A). Microscopically, tumors were formed of closely packed, small and large follicular-like cysts (B, C) or predominantly small follicular-like cysts (C, D), filled by eosinophilic “colloid-like” secretions of variable density. Intra- and inter-follicular cholesterol clefts were seen (B, C). The stroma varied from minimal to prominent (B–F). Case 6 (excisional biopsy) with significant thermal artifact, showing similar morphology.
Microscopically (Fig. 1B–F and 2A–F), the tumors were well-circumscribed and unencapsulated. Tumors 1–3, 5, and 7 were composed of variably sized, small and large follicle-like cysts, whereas cases 4 and 8 featured predominantly small follicle-like cysts. Although tumor 6 was diagnosed in a biopsy with significant thermal artifact, it had a similar morphology to the others with small and large follicle-like cysts. The cysts were filled by eosinophilic secretions of variable density in all cases; however, rare areas lacking the characteristic colloid-like secretions within the cysts were seen in cases 1 and 2. In addition, clear spaces separating the lining epithelial cells and luminal secretions in what appears to be a retraction artifact were evident in all cases. The cells lining the large cysts had minimal eosinophilic to amphophilic cytoplasm with high nuclear to cytoplasmic ratios and predominantly oval to elongated overlapping nuclei that were perpendicularly arranged toward luminal spaces in all cases. The quantity and composition of the intervening stroma varied among the cases with some (tumors 1 and 5–7), demonstrating little to no stroma and others exhibiting relatively abundant paucicellular and collagenized (tumor 2) to densely cellular and spindle cell (tumors 3 and 8) stroma. No calcifications, mitotic figures, or necrosis were present.

Representative sections of tumors showing cysts filled by eosinophilic secretions of variable density with areas lacking the characteristic secretion (A) and some showing clear spaces separating the luminal secretions from lining epithelial cells (B). The cells show minimal eosinophilic to amphophilic cytoplasm with oval to rounded (C), focally cleared (D) overlapping nuclei. These are embedded in a variably cellular (inset), abundant stroma (E, F).
In addition, the two tumors selected as a control group (cases 9 and 10) shared the presence of colloid-like secretions; however, these differed by the presence of non-anastomosing dilated tubular structures, which were lined by cells with moderate amounts of eosinophilic cytoplasm and, more importantly, nonoverlapping round “oncocytoma-like” nuclei (Fig. 3).

Representative sections of both were formed of non-anastomosing follicular-like cysts and tubules, with occasional eosinophilic “colloid-like” secretions. These were lined by cells with moderate amount of eosinophilic cytoplasm and, more importantly, nonoverlapping round “oncocytoma-like” nuclei (A–D).
Using immunohistochemical staining (Table 2), all tumors in the study group with available material showed immunoreactivity for KRT19 (n = 7) and PAX8 (n = 5). Cyclin D1 showed at least focal moderate to strong immunoreactivity in all stained tumors as well (n = 6). KRT7 was immunoreactive in five tumors (5/7) and one tumor exhibited patchy and weak immunoreactivity for AMACR (n = 1/5). None of the tumors was immunoreactive for WT1, thyroglobulin, or TTF1. Tumor 6 was stained for TFE3 and melan-A as a screening for post-therapy MITF-family translocation renal cell carcinoma and both were negative. One tumor (case 7) was stained for fumarate hydratase and showed retained cytoplasmic immunoreactivity (Fig. 4).

Tumors were positive for (A) PAX8, (B) CK7, (C) CK19 (inset: variable CK19 staining), and (D) cyclin D1 was variable, respectively.
Molecular analysis
FISH and copy number alteration results
FISH analysis with the CEPs performed on tumors 1 and 2 showed disomy of chromosomes 3, 7, 10, 12, and 17.
NGS and RT-PCR results
Targeted exome-sequencing analysis revealed a fusion between exon 8 of EWSR1 (NM_005243.3) and intra-exonic sequences of exon 1 of PATZ1 (NM_014323.2) for tumors 1 and 2. RT-PCR analysis using designed primers based on the NGS-implied fusion point (Fig. 5A) was performed, confirming the pre-detected genetic abnormality of cases 1 and 2 suggestive of a fusion event (Fig. 5B, C). Sanger sequencing of the RT-PCR product for case 1 confirmed the in-frame fusion of exon 8 of EWSR1 and intra-exonic sequences of PATZ1 exon 1 at the nucleotide position of 1728 nucleotides, 983 nucleotides downstream from the translational start codon (Fig. 5D). Similarly, in case 2, exon 8 of EWSR1 was fused to intra-exonic sequences of PATZ1 (NM_014323) exon 1 at 1743, 998 n distal of the ATG start codon (Fig. 5E). Tumor 3 was analyzed by NGS using the MSK-Fusion panel. An EWSR1-PATZ1 fusion transcript was detected joining exon 8 of the EWSR1 gene (NM_005243.3) and exon 1 of the PATZ1 (NM_014323.2) gene (Fig. 6A, B). The fusion point of case 3 (at nucleotide position 1800, 1055 nucleotides downstream of the translational start codon) and of cases 1 and 2 are illustrated in Fig. 6C. In all three EWSR1-PATZ1 fusions, the whole N-terminal transcriptional activation domain (TAD, QGSY low-complexity region) from EWSR1 is present in the fusion, in frame with the PATZ1 transcription factor C-terminal DNA-binding domain containing seven Zn++ finger domains.

A Primer design based on the EWSR1-PATZ1 fusion point identified by NGS for case 1. B Reverse-transcriptase PCR (RT-PCR) was performed using RNA extracted from FFPE slides of cases of 1 and 2. Then, 2.0 μl of cDNA were subjected to PCR, to assess the quality and integrity of RNA using an HPRT RT-PCR two-step assay. The oligonucleotide combination of HPRT 518/781 (5′-ACT TTG CTT TCC TTG GTC A-3′/5′-TTT ACT GGC GAT GTC AAT AG-3′ and HPRT 679/781 (5′-CTT CAG GGA TTT GAA TCA TG-3′/5′-TTT ACT GGC GAT GTC AAT AG-3′) was used. Lane M: 100 bp ladder; lanes 1 and 2: case 1 with HPRT 679/781 and HPRT 518/781; lane 3 and 4: case 2 with HPRT 679/781 and HPRT 518/781. C EWSR1-PATZ1 RT-PCR using the oligonucleotides EP10F and EP10R presented in Fig. 3A; M: 100 bp; lane 1: case 1; lane 2: case 2. D, E Sanger sequence of the amplified products from cases 1 and 2, respectively. Partial sequence chromatograms for case 1 (D) and case 2 (E) demonstrate that exon 8 of EWSR1 (NM_005243.3) is fused to intra-exonic sequences of PATZ1 (NM_014323.2). The red arrows indicates the fusion points. The translation of the nucleotide sequence shows the presence of an open reading frame.

A Graphic view of EWSR1 (NM_005243.3) exon 8–PATZ1 (NM_014323.2) exon 1 fusion breakpoint detected by the Archer analysis software. The corresponding transcript sequence is shown on top (light blue: EWSR1; dark blue: PATZ1). B Schematic illustration of the EWSR1 (NM_005243.3) transcript illustrating the exon boundaries and the conserved domains. The TAD domain is the transactivation domain containing a consensus sequence of the repeated QGSY peptide. The three RGG domains are rich in arginine and glycine; they play a role in RNA recognition and binding. RMM represents the RNA recognition domain and Zn, the Zn finger domain. PY is a C-terminal nuclear-localization domain. The vertical bars indicate the exon borders. The start and end of the coding sequence is indicated by dotted vertical lines. The red arrow designates the fusion point. C Schematic illustration of the PATZ1 transcript (NM_014323.2). BTB/POZ is a protein/protein interaction motif, AT is a DNA-binding motif, and Zn is a Zn finger domain. The partial nucleotide and amino acid sequence underneath the PATZ1 transcript shows the position of the breakpoints for cases 1, 2, and 3 in PATZ1 exon 1. The fusion removes the BTB/POZ putative transcriptional repressor domain and the AT-hook domain at the N terminus of PATZ1 (NP_055138.2), and converts a transcriptional repressor into a transcriptional activator.
To further validate our findings, we subjected the two tumors in our control series to RT-PCR analysis using the designed primers described. A different group of renal tumors, including eight acquired cystic disease-associated renal cell carcinomas, five renal cell carcinomas with leiomyomatous stroma, and two atrophic-kidney-like tumors were also subjected to the same NGS performed on cases 1 and 2; however, no similar fusion gene events were identified in any tested tumor (whether subjected to RT-PCR or to NGS).
No known pathogenic gene mutations indexed in the COSMIC (catalog of somatic mutations in cancer) and ClinVar databases or copy number alterations were identified in the study group. Indels and nonsense mutations are tabulated in Supplementary Table 1.
Discussion
Renal tumors are heterogeneous with diverse clinical, histological, and molecular characteristics. Thyroid-like follicular renal cell carcinoma is a rare neoplasm of the epithelium of renal tubules. It was included in the 2013 ISUP Vancouver classification of renal tumors and in the 2016 WHO classification of kidney tumors as an emerging renal tumor [3, 4, 6].
Thyroid-like follicular renal cell carcinoma occurs over a wide age range (10–83 years) and is mostly found in women [19]. Most patients appear to be cured by surgical resection, although metastases have been reported in six patients [2, 20,21,22,23,24]. In the current series of eight women, the mean age at presentation was 45 years (range 19–65 years) and no evidence of recurrent or metastatic disease was identified in follow-up ranging from 10 to 84 months, despite the presence of renal sinus fat invasion in one patient.
On gross examination, thyroid-like follicular renal cell carcinomas appear well-circumscribed or encapsulated, have a tan-brown, solid, or microcystic cut surface, and typically show no hemorrhage and necrosis. Histopathologically, the first series described by Amin et al. [2] included three morphologically different groups of tumors, although they all shared the presence of eosinophilic colloid-like secretions including the following: group 1—a tumor (case 3) with similar histologic features to our series; group 2—tumors that were later described as atrophic-kidney-like tumor/lesion [25]; and group 3—tumors with non-anastomosing microcystic architecture with centrally located, round nonoverlapping “oncocytoma-like” nuclei and inconspicuous occasional nucleoli. The tumors in our control group had similar features to those in group 3.
The presence of a pattern resembling thyroid follicles in a neoplasm detected in the kidney is not restricted to thyroid-like follicular renal cell carcinoma. Included in the differential diagnosis is a metastasis from primary thyroid carcinoma (Fig. 7A). Both neoplasms express PAX8; however, metastasis from the thyroid almost invariably expresses TTF1 and often thyroglobulin. These stains are helpful with this differential diagnosis, as by immunohistochemistry thyroid-like follicular renal cell carcinomas do not express TTF1 [26]. In addition, most of the follicular and papillary, follicular variant, and thyroid carcinomas have NRAS and HRAS or BRAF mutations, respectively, contrasting with thyroid-like follicular renal cell carcinomas that lack these mutations [2, 27, 28]. Morphologic overlap also exists with atrophic-kidney-like tumor/lesion [25, 29, 30]. These are composed of variably sized cysts with dense eosinophilic secretions that may resemble thyroid-like follicular renal cell carcinoma; however, the follicles of atrophic-kidney-like tumor/lesion are typically lined by flattened epithelium with occasional hobnail morphology. Microcalcifications are also common in these lesions (Fig. 7B). Immunohistochemically, these tumors/lesions are consistently positive for WT1 in contrast to thyroid-like follicular renal cell carcinoma, which is negative for WT1 [25]. Papillary renal cell carcinoma type 1 may also show foci that resemble thyroid-like follicular renal cell carcinoma when the papillary cores become markedly edematous imparting a thyroid-like follicular pattern; however, areas of ordinary papillary renal cell carcinoma are invariably present in these tumors (Fig. 7C) [5, 31,32,33]. Lastly, urothelial carcinoma of the renal pelvis with prominent cystitis cystica proliferation may show foci that resemble a thyroid-like follicular pattern (Fig. 7D); however, the distinction between these lesions is usually straightforward and GATA3 immunostaining may be used for pointing toward a urothelial origin.

(A) thyroid carcinoma (inset, TTF1 immunohistochemistry), (B) atrophic-kidney-like tumor (inset, WT1 immunohistochemistry), (C) papillary renal cell carcinoma type 1 (inset, AMACR immunohistochemistry), and (D) urothelial carcinoma (inset, GATA3 [clone L50-823; Biocare medical, Pacheco, CA, USA] immunohistochemistry).
The genetic basis of renal cell neoplasia is complex; however, the identification of shared and subtype-specific molecular features has contributed to classification, correlations with clinical course, and prediction of therapeutic response [34]. For example with respect to classification, several kidney tumor subtypes have distinct genetic abnormalities that may serve as useful diagnostic aids such as gene fusions seen in MITF-family translocation renal cell carcinoma or somatic alterations involving VHL or chromosomal arm 3p deletion in clear cell renal cell carcinoma, MET mutation or gains of chromosomes 7 and 17 in papillary renal cell carcinoma, losses of chromosomes Y, 1, 2, 6, 10, 13, 17, and 21 in chromophobe renal cell carcinoma, or KRAS mutations, in papillary renal neoplasm with reverse polarity [3, 35,36,37]. For thyroid-like follicular renal cell carcinoma, molecular alterations have been reported in a few studies with no distinctive abnormality having been identified [2, 20, 38,39,40,41,42].
For example, Amin et al. [2] reported overexpression of cell cycle regulatory genes and mixed lineage leukemia/trithorax homolog in three tumors. The association between thyroid-like follicular renal cell carcinoma and hematopoietic disease is not understood, despite being found in four patients, including one of our patients (case 6) [5, 38, 40].
Using comparative genomic hybridization analysis, gains and losses of several chromosomal regions or chromosomes have been reported by Jung et al. [41]. These included gains of 7q36, 8q24, 12, 16, 17p11-q11, 17q24, 19q, 20q13, 21q22.3, and Xp, and losses of 1p36, 3, and 9q21–33. Additional molecular cytogenetic studies reported in a few other tumors have shown chromosomal copy number imbalances including monosomy 7 and 17, among others [20, 42]. No chromosomal imbalances were identified by FISH in tumors 1 and 2 in the current series.
In the current study, an in-frame fusion of exon 8 of EWSR1 (NM_0005243) and intra-exonic sequences of PATZ1 exon 1 was detected in all the thyroid-like follicular renal cell carcinomas studied with RNAseq and RT-PCR, with subsequent Sanger sequencing. This is the first recurrent aberration for this tumor. The EWSR1-PATZ1 fusion has been reported previously in round to spindle cell sarcomas classified under “round cell sarcomas with EWSR1–non-ETS fusions” in the 2020 WHO 5th Edition Classification of Soft Tissue and Bone Tumors [43]. In addition, EWSR1-PATZ1 fusion has been found in eight diverse primary central nervous system neoplasms [44,45,46,47,48]. The unique gene expression signature of EWSR1-PATZ1 sarcoma has not been compared with the central nervous system tumors [49]. EWSR1 is a multifunctional protein acting in DNA repair, DNA recombination, mRNA splicing and transport, and G-protein signaling [50]. PATZ1 is an essential transcriptional regulator, repressing developmental genes through its N-terminal BTB domain and C-terminal zinc finger motif-containing domain that mediates DNA binding [51]. The EWSR1-PATZ1 fusion removes the putative transcriptional repressor domain BTB at the N terminus of PATZ1, which might alter a transcriptional repressor into an activator. The chimeric EWSR1-PATZ1 protein is expected to maintain DNA-binding function through the C-terminal zinc finger motifs and modifying the transcription control at PATZ1 target sites, which might be the driving force for tumorigenesis.
In a unique tumor reported by Jenkins et al. [23], the thyroid-like follicular renal cell carcinoma was associated with a primitive small round/spindle cell sarcomatoid component and metastatic disease (mesenteric mass and multiple aortocaval lymph nodes). In contrast, both the primary renal tumor and the lesions of other cases of thyroid-like follicular renal cell carcinomas with metastatic disease have lacked a sarcomatoid component and have been composed almost entirely of follicles [10, 12]. FISH studies conducted on the tumor with a sarcomatoid component were negative for MDM2 amplification and SS18 rearrangement. Assessment of the EWSR1 locus revealed a break-apart signal in 23% of 52 tumor cells analyzed with aneuploidy of EWSR1 signals (up to 4 per cell), findings the authors concluded as negative for EWSR1 rearrangement. As EWSR1 and PATZ1 reside in close proximity to each other on chromosome 22 (both genes localized to 22q12.2 at ~2 Mb distance) and are normally transcribed in opposite directions, the EWSR1-PATZ1 fusion event is most likely the result of a submicroscopic or cryptic intrachromosomal paracentric inversion that poses significant interpretive challenges in visualizing the subtle dissociation or breaking apart of signals by FISH [43, 48]. Moreover, as additional studies would be required for identification of the PATZ1 partnership, other technical approaches instead of FISH are preferable for EWSR1-PATZ1 fusion identification. NGS targeting the exonic sequence of 152 genes of the same tumor revealed variants of uncertain significance (VUS) in RAD51C and SMO, in both the thyroid-like follicular carcinoma and sarcoma components at an allele frequency, suggesting that these are germline variants, while three other VUS mutations (AKT3, NOTCH1, and JAK3) were exclusive to the sarcomatoid component, potentially being acquired somatic mutations [23].
In a molecularly profiled series of paired primary and metastatic papillary renal cell carcinomas and rarer kidney cancer subtypes, one tumor of a primary renal tumor (and associated metastasis) classified as a renal fibrosarcoma was shown to have an EWSR1-PATZ1 fusion [52]. Notably, both this patient and the patient with the thyroid-like follicular renal cell carcinoma with sarcomatoid differentiation experienced progression, succumbing to their disease within a few months of diagnosis [23]. Immunohistochemically, however, the renal fibrosarcoma was negative for PAX8, whereas both the follicular and sarcomatoid components of the thyroid-like follicular renal cell carcinoma with sarcomatoid differentiation expressed PAX8. Moreover, the renal fibrosarcoma also harbored a homozygous deletion of CDKN2A, a secondary alteration that might have prognostic significance in EWSR1-PATZ1 sarcoma [30]. The histopathological and immunophenotypic features described for EWSR1-PATZ1 sarcomas are fairly diverse [43]. Tumor cells are small, round, or spindle-shaped and are often accompanied by a fibrous stroma. Coexpression of muscle markers (desmin, myogenin, and MYOD1) and other markers (S100P, SOX10, MITF, and GFAP) is seen at variable levels. The renal fibrosarcoma tumor reportedly exhibited focal S100P positivity but was negative for desmin and MYOD1. Based on the limited data regarding the thyroid-like follicular renal cell carcinoma with primitive round/spindle cell sarcomatoid differentiation (undetermined EWSR1-PATZ1 fusion status) and the renal fibrosarcoma lacking thyroid-like follicular renal cell carcinoma morphology, it is difficult to draw definitive conclusions or address potential overlap between the two disease processes.
In conclusion, we have identified EWSR1-PATZ1 as a recurrent gene fusion event in thyroid-like follicular renal cell carcinomas. Chromosomal translocations or other structural alterations and associated fusion genes are common in hematopoietic and mesenchymal neoplasia, and more recently have been demonstrated in a number of epithelial neoplasms; however, reports in primary kidney tumors are few [35, 53, 54]. Thyroid-like follicular renal cell carcinoma is a rare tumor that has only been provisionally introduced as an entity and its full histopathologic spectrum has yet to be recognized. We propose that this gene fusion is of a pathogenic importance. Correspondingly, the RT-PCR assay developed in this study could serve as a valuable diagnostic adjunct for this rare disease entity.
Data availability
The datasets generated during and/or analyzed during the current study are available from the first, second and corresponding authors on reasonable request.
References
Amin M, Bichal M, Radhakrishman A, Hes O, McKenney JK, Cheville JC. Primary thyroid-like follicular carcinoma of the kidney: a histologically distinctive primary renal epithelial tumor. Mod Pathol. 2004;17:136–7.
Amin MB, Gupta R, Ondrej H, McKenney JK, Michal M, Young AN, et al. Primary thyroid-like follicular carcinoma of the kidney: report of 6 cases of a histologically distinctive adult renal epithelial neoplasm. Am J Surg Pathol. 2009;33:393–400.
Srigley JR, Delahunt B, Eble JN, Egevad L, Epstein JI, Grignon D, et al. The International Society of Urological Pathology (ISUP) Vancouver classification of renal neoplasia. Am J Surg Pathol. 2013;37:1469–89.
Moch H, Cubilla AL, Humphrey PA, Reuter VE, Ulbright TM. The 2016 WHO classification of tumours of the urinary system and male genital organs-Part A: renal, penile, and testicular tumours. Eur Urol. 2016;70:93–105.
Eble JN, Delahunt B. Emerging entities in renal cell neoplasia: thyroid-like follicular renal cell carcinoma and multifocal oncocytoma-like tumours associated with oncocytosis. Pathology. 2018;50:24–36.
Trpkov K, Hes O. New and emerging renal entities: a perspective post-WHO 2016 classification. Histopathology. 2019;74:31–59.
Tretiakova MS, Kehr EL, Gore JL, Tykodi SS. Thyroid-like follicular renal cell carcinoma arising within benign mixed epithelial and stromal tumor. Int J Surg Pathol. 2020;28:80–6.
Brunelli M, Eble JN, Zhang S, Martignoni G, Cheng L. Gains of chromosomes 7, 17, 12, 16, and 20 and loss of Y occur early in the evolution of papillary renal cell neoplasia: a fluorescent in situ hybridization study. Mod Pathol. 2003;16:1053–9.
Cossu-Rocca P, Eble JN, Zhang S, Bonsib SM, Martignoni G, Brunelli M, et al. Interphase cytogenetic analysis with centromeric probes for chromosomes 1, 2, 6, 10, and 17 in 11 tumors from a patient with bilateral renal oncocytosis. Mod Pathol. 2008;21:498–504.
Cheng L, Zhang S, Wang L, MacLennan GT, Davidson DD. Fluorescence in situ hybridization in surgical pathology: principles and applications. J Pathol Clin Res. 2017;3:73–99.
Kadri S, Long BC, Mujacic I, Zhen CJ, Wurst MN, Sharma S, et al. Clinical validation of a next-generation sequencing genomic oncology panel via cross-platform benchmarking against established amplicon sequencing assays. J Mol Diagn. 2017;19:43–56.
Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–60.
Mose LE, Wilkerson MD, Hayes DN, Perou CM, Parker JS. ABRA: improved coding indel detection via assembly-based realignment. Bioinformatics. 2014;30:2813–5.
Li MM, Datto M, Duncavage EJ, Kulkarni S, Lindeman NI, Roy S, et al. Standards and guidelines for the interpretation and reporting of sequence variants in cancer: a joint consensus recommendation of the Association for Molecular Pathology, American Society of Clinical Oncology, and College of American Pathologists. J Mol Diagn. 2017;19:4–23.
Talevich E, Shain AH, Botton T, Bastian BC. CNVkit: genome-wide copy number detection and visualization from targeted DNA sequencing. PLoS Comput Biol. 2016;12:e1004873.
Parilla M, Alikhan M, Al-Kawaaz M, Patil S, Kadri S, Ritterhouse LL, et al. Genetic underpinnings of renal cell carcinoma with leiomyomatous stroma. Am J Surg Pathol. 2019;43:1135–44.
Benayed R, Offin M, Mullaney K, Sukhadia P, Rios K, Desmeules P, et al. SvABA: genome-wide detection of structural variants and indels by local assembly. Genome Res. 2018;28:581–91.
Benayed R, Offin M, Mullaney K, Sukhadia P, Rios K, Desmeules P, et al. High yield of RNA sequencing for targetable kinase fusions in lung adenocarcinomas with no mitogenic driver alteration detected by DNA sequencing and low tumor mutation burden. Clin Cancer Res. 2019;25:4712–22.
de Jesus LE, Fulgencio C, Leve T, Dekermacher S. Thyroid-like follicular carcinoma of the kidney presenting on a 10 year-old prepubertal girl. Int Braz J Urol. 2019;45:834–42.
Sterlacci W, Verdorfer I, Gabriel M, Mikuz G. Thyroid follicular carcinoma-like renal tumor: a case report with morphologic, immunophenotypic, cytogenetic, and scintigraphic studies. Virchows Arch. 2008;452:91–5.
Dhillon J, Tannir NM, Matin SF, Tamboli P, Czerniak BA, Guo CC. Thyroid-like follicular carcinoma of the kidney with metastases to the lungs and retroperitoneal lymph nodes. Hum Pathol. 2011;42:146–50.
Dong L, Huang J, Huang L, Shi O, Liu Q, Chen H, et al. Thyroid-like follicular carcinoma of the kidney in a patient with skull and meningeal metastasis: a unique case report and review of the literature. Medicine (Baltim). 2016;95:e3314.
Jenkins TM, Rosenbaum J, Zhang PJ, Schwartz LE, Nayak A, Cooper K, et al. Thyroid-like follicular carcinoma of the kidney with extensive sarcomatoid differentiation: a case report and review of the literature. Int J Surg Pathol. 2019;27:678–83.
Chen X, Dou FX, Cheng XB, Guo AT, Shi HY. [Clinicopathologic characteristics of thyroid-like follicular carcinoma of the kidney: an analysis of five cases and review of literature]. Zhonghua Bing Li Xue Za Zhi. 2016;45:687–91.
Herlitz L, Hes O, Michal M, Tretiakova M, Reyes-Mugica M, Nguyen JK, et al. “Atrophic kidney”-like lesion: clinicopathologic series of 8 cases supporting a benign entity distinct from thyroid-like follicular carcinoma. Am J Surg Pathol. 2018;42:1585–95.
Mehra R, Smith SC, Divatia M, Amin MB. Emerging entities in renal neoplasia. Surg Pathol Clin. 2015;8:623–56.
Fanelli GN, Fassan M, Dal Moro F, Soligo M, Munari G, Zattoni F, et al. Thyroid-like follicular carcinoma of the kidney: the mutational profiling reveals a BRAF wild type status. Pathol Res Pr. 2019;215:152532.
Nikiforova MN, Lynch RA, Biddinger PW, Alexander EK, Dorn GW 2nd, Tallini G, et al. RAS point mutations and PAX8-PPAR gamma rearrangement in thyroid tumors: evidence for distinct molecular pathways in thyroid follicular carcinoma. J Clin Endocrinol Metab. 2003;88:2318–26.
Alaghehbandan R, Michal M, Kuroda N, Hes O. Thyroid-like follicular carcinoma of the kidney: an emerging renal neoplasm with curiously misplaced histologic features. A case report. Int J Surg Pathol. 2017;25:379–80.
Muscara MJ, Simper NB, Gandia E. Thyroid-like follicular carcinoma of the kidney. Int J Surg Pathol. 2017;25:73–7.
Delahunt B, Eble JN. Papillary renal cell carcinoma: a clinicopathologic and immunohistochemical study of 105 tumors. Mod Pathol. 1997;10:537–44.
Tosi AL, de Biase D, Leonardi E, Eusebi V. Thyroid-like metastases to the scalp from a papillary renal cell carcinoma: a case report. Tumori. 2012;98:79e–81e.
Delahunt B, Eble JN, McCredie MR, Bethwaite PB, Stewart JH, Bilous AM. Morphologic typing of papillary renal cell carcinoma: comparison of growth kinetics and patient survival in 66 cases. Hum Pathol. 2001;32:590–5.
Ricketts CJ, De Cubas AA, Fan H, Smith CC, Lang M, Reznik E, et al. The Cancer Genome Atlas comprehensive molecular characterization of renal cell carcinoma. Cell Rep. 2018;23:3698.
Quddus MB, Pratt N, Nabi G. Chromosomal aberrations in renal cell carcinoma: an overview with implications for clinical practice. Urol Ann. 2019;11:6–14.
Al-Obaidy KI, Eble JN, Nassiri M, Cheng L, Eldomery MK, Williamson SR, et al. Recurrent KRAS mutations in papillary renal neoplasm with reverse polarity. Mod Pathol. 2020;33:1157–64.
Speicher MR, Schoell B, du Manoir S, Schrock E, Ried T, Cremer T, et al. Specific loss of chromosomes 1, 2, 6, 10, 13, 17, and 21 in chromophobe renal cell carcinomas revealed by comparative genomic hybridization. Am J Pathol. 1994;145:356–64.
Alessandrini L, Fassan M, Gardiman MP, Guttilla A, Zattoni F, Galletti TP, et al. Thyroid-like follicular carcinoma of the kidney: report of two cases with detailed immunohistochemical profile and literature review. Virchows Arch. 2012;461:345–50.
Chougule A, Bal A, Das A, Nayak B. Thyroid-like follicular renal cell carcinoma: an emerging morphological variant. Pathology. 2014;46:657–60.
Wu WW, Chu JT, Nael A, Rezk SA, Romansky SG, Shane L. Thyroid-like follicular carcinoma of the kidney in a young patient with history of pediatric acute lymphoblastic leukemia. Case Rep. Pathol. 2014;2014:313974.
Jung SJ, Chung JI, Park SH, Ayala AG, Ro JY. Thyroid follicular carcinoma-like tumor of kidney: a case report with morphologic, immunohistochemical, and genetic analysis. Am J Surg Pathol. 2006;30:411–5.
Dawane R, Grindstaff A, Parwani AV, Brock T, White WM, Nodit L. Thyroid-like follicular carcinoma of the kidney: one case report and review of the literature. Am J Clin Pathol. 2015;144:796–804.
Le Loarer F, Szuhai K, Tirode F. Round cell sarcoma with EWSR1-non ETS fusions. In: Antonescu CR, Bovee J, Bridge JA, Cunha IW, Dei Tos AP, Flanagan A, et al., editors. WHO classification of tumours of soft tissue and bone, 5th edition. Lyon, France: IARC Press; 2020. p. 326–9.
Qaddoumi I, Orisme W, Wen J, Santiago T, Gupta K, Dalton JD, et al. Genetic alterations in uncommon low-grade neuroepithelial tumors: BRAF, FGFR1, and MYB mutations occur at high frequency and align with morphology. Acta Neuropathol. 2016;131:833–45.
Johnson A, Severson E, Gay L, Vergilio JA, Elvin J, Suh J, et al. Comprehensive genomic profiling of 282 pediatric low- and high-grade gliomas reveals genomic drivers, tumor mutational burden, and hypermutation signatures. Oncologist. 2017;22:1478–90.
Alvarez-Breckenridge C, Miller JJ, Nayyar N, Gill CM, Kaneb A, D’Andrea M, et al. Clinical and radiographic response following targeting of BCAN-NTRK1 fusion in glioneuronal tumor. NPJ Precis Oncol. 2017;1:5.
Siegfried A, Rousseau A, Maurage CA, Pericart S, Nicaise Y, Escudie F, et al. EWSR1-PATZ1 gene fusion may define a new glioneuronal tumor entity. Brain Pathol. 2019;29:53–62.
ridge JA, Sumegi J, Druta M, Bui MM, Henderson-Jackson E, Linos K, et al. Clinical, pathological, and genomic features of EWSR1-PATZ1 fusion sarcoma. Mod Pathol. 2019;32:1593–604.
Watson S, Perrin V, Guillemot D, Reynaud S, Coindre JM, Karanian M, et al. Transcriptomic definition of molecular subgroups of small round cell sarcomas. J Pathol. 2018;245:29–40.
Cantile M, Marra L, Franco R, Ascierto P, Liguori G, De Chiara A, et al. Molecular detection and targeting of EWSR1 fusion transcripts in soft tissue tumors. Med Oncol. 2013;30:412.
Fedele M, Crescenzi E, Cerchia L. The POZ/BTB and AT-hook-containing Zinc finger 1 (PATZ1) transcription regulator: physiological functions and disease involvement. Int J Mol Sci. 2017;18:2524.
Zhu B, Poeta ML, Costantini M, Zhang T, Shi J, Sentinelli S, et al. The genomic and epigenomic evolutionary history of papillary renal cell carcinomas. Nat Commun. 2020;11:3096.
Argani P, Lewin JR, Edmonds P, Netto GJ, Prieto-Granada C, Zhang L, et al. Primary renal sclerosing epithelioid fibrosarcoma: report of 2 cases with EWSR1-CREB3L1 gene fusion. Am J Surg Pathol. 2015;39:365–73.
Ertoy Baydar D, Kosemehmetoglu K, Aydin O, Bridge JA, Buyukeren B, Aki FT. Primary sclerosing epithelioid fibrosarcoma of kidney with variant histomorphologic features: report of 2 cases and review of the literature. Diagn Pathol. 2015;10:186.
Author information
Authors and Affiliations
Contributions
KIA and JAB performed study concept and design, and wrote the manuscript draft. KIA, JAB, LC, JS, RB, MH, JPS, PW, and MN provided acquisition, analysis, and interpretation of data. VER, SRW, OH, and DJG provided material support. VER, SRW, OH, FIA, MTI, and JNE participated in the development of methodology and writing, review, and revision of the paper. JNE and DJG provided supervision to overall work. All authors read and approved the final paper, due to DJG’s unexpected demise.
Corresponding author
Ethics declarations
Ethics statement
This study was approved by the Institutional Review Board (IRB) at Indiana University, Indianapolis, USA, under protocol number: 1301010350. Ethical approval for this study was waived.
Funding statement
The authors received no specific funding for this work.
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Al-Obaidy, K.I., Bridge, J.A., Cheng, L. et al. EWSR1-PATZ1 fusion renal cell carcinoma: a recurrent gene fusion characterizing thyroid-like follicular renal cell carcinoma. Mod Pathol 34, 1921–1934 (2021). https://doi.org/10.1038/s41379-021-00833-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41379-021-00833-7
This article is cited by
-
Genomic alterations and diagnosis of renal cancer
Virchows Archiv (2024)
-
Identifying key genes related to the peritubular capillary rarefaction in renal interstitial fibrosis by bioinformatics
Scientific Reports (2023)
-
Small round cell sarcomas
Nature Reviews Disease Primers (2022)
-
Expanding the clinicopathological spectrum of succinate dehydrogenase-deficient renal cell carcinoma with a focus on variant morphologies: a study of 62 new tumors in 59 patients
Modern Pathology (2022)
