
Abstract
The grading of gliomas based on histological features has been a subject of debate for several decades. A consensus has not yet been reached because of technical limitations and inter-observer variations. While the traditional grading system has failed to stratify the risk of IDH-mutant astrocytoma, canonical histological and proliferative markers may be applicable to the risk stratification of IDH-wild-type astrocytoma. Numerous studies have examined molecular markers in order to obtain more clinically relevant information that will improve the risk stratification of gliomas. The CDKN2A/B homozygous deletion for IDH-mutant astrocytoma and the following three criteria for IDH-wild-type astrocytoma: the concurrent gain of whole chromosome 7 and loss of whole chromosome 10, TERT promoter mutations, and EGFR amplification, were identified as independent molecular markers of the worst clinical outcomes. Therefore, the 2021 World Health Organization (WHO) Classification of Tumors of the Central Nervous System adopted these molecular markers into the revised grading criteria of IDH-mutant and -wild-type astrocytoma, respectively, as a grading system within tumor types. Of note, several recent studies have shown that some low-grade IDH-wild-type astrocytoma lacking both the molecular glioblastoma signature and genetic alterations typical of pediatric-type gliomas may demonstrate a relatively indolent clinical course, suggesting the existence of lower-grade adult IDH-wild-type astrocytoma. In terms of oligodendroglioma, IDH-mutant, and 1p/19q codeleted, consistent makers that predict poor outcomes have not yet been identified, and, thus, the current criteria have remained unchanged. Molecular testing to fulfill the revised WHO criteria is, however, not always available worldwide, and in that case, an integrated diagnosis combining all available complementary information is highly recommended. This review discusses controversial issues surrounding legacy grading systems and newly identified potential genetic markers of adult diffuse gliomas and provides perspectives on future grading systems.
Similar content being viewed by others

Introduction
The World Health Organization (WHO) adopted a molecular classification for adult diffuse gliomas in the updated 4th edition in 2016 for the first time1,2,3,4. However, the histological grading scale remained unchanged because of the lack of sufficient molecular evidence to revise the traditional histological scale, which was used for many years5. The purpose of tumor grading is to provide clinicians with information to predict outcomes, develop a treatment plan, and engage in clinical studies to establish more appropriate treatment regimens. The grading of diffuse gliomas in children and adults has been a subject of discussion for several decades6. However, a consensus has yet to be reached because grading has been based on the histological appearance of given tumor samples, which do not always reflect the biological behavior of each tumor, and histological assessments are subjective with inter- and intra-observer variabilities. This short review discusses controversial issues surrounding the grading of adult diffuse gliomas according to the 2021 WHO Classification of Tumors of the Central Nervous System (CNS)7 and provides perspectives on future grading systems.
The WHO grading system
Historical aspects
One of the pioneers of the modern grading systems of cancers was Albert Broders at the Mayo Clinic, who for the first time coined a numerical grading system that divided tumors into four histological grades of malignancy, which were independent of any clinical history and based on dissimilarities in the given tumors from the normal tissue from which they originated8. James Watson Kernohan, a colleague of Broders at the Mayo Clinic, who is regarded as one of the early pioneers of neuropathology, adopted a four-tier system for astrocytic gliomas9. This histological grading system of gliomas was broadly accepted and used for the next few decades. However, when the WHO started to publish the classification of tumors series in the 1970s10, they adopted biology-oriented grading under the leadership of Klaus J Zülch, who was a neurologist/neuropathologist11. Even after the publication of the first edition of the WHO Classification of CNS tumors, several different diagnostic schemes were still used in parallel. However, the second edition of the WHO Classification12, incorporating the so-called St. Anne-Mayo grading scheme13, became universally accepted as the standard for glioma grading.
General rules in the current WHO grading system
In the WHO grading system, a biology-oriented grade is generally given to each tumor based on the estimated clinical outcome obtained by epidemiological data. In other words, the grade is fixed to the tumor nomenclature, and variations in the histological appearance of each specimen do not affect the assessment of an individual sample. For example, pilocytic astrocytoma is exclusively assigned to WHO grade I regardless of its histological features. Although a small subset of pilocytic astrocytoma exhibits anaplastic histological features and some patients with such tumors may have a significantly worse prognosis than classic pilocytic astrocytoma, the WHO has not designated a formal grade for these tumors. In addition, the WHO adopted the grading “across tumor types” rather than “within a tumor type”. In this type of grading system, tumors of the same grade roughly have the same clinical outcome regardless of the histological tumor type14. However, the “grading across tumor types” became irrational because the biology of tumors, primarily defined by genetic alterations, is so heterogeneous that it is more reasonable to grade each tumor individually. For example, many studies reported that IDH-mutant glioblastoma, WHO grade IV, has a markedly better prognosis than IDH-wild-type glioblastoma, WHO grade IV, even though they share identical histology. Another regulation of the WHO grading system is that grading is selected by the natural history of a neoplasm with surgical resection alone and not by the outcome of treatment. This rule has been well accepted in the neuro-oncology community because it reflects the intrinsic biology of each tumor, even though current grading criteria are created based solely on retrospective studies.
Nevertheless, after discovering molecularly defined entities, this rule faces an unresolvable dilemma, namely, nearly all patients with malignant gliomas, including anaplastic astrocytoma and glioblastoma, receive adjuvant therapy after resection; therefore, it became essentially impossible to obtain information on the natural history of molecularly defined high-grade gliomas. Furthermore, a randomized control study to verify the grading criteria for high-grade gliomas was not feasible. The ethics do not allow the existence of patients with a high-grade glioma who does not receive any adjuvant therapy.
Issues in the histological grading system
Four histological markers employed by the WHO grading system to evaluate malignancy are nuclear atypia (A), mitosis (M), microvascular proliferation (previously termed endothelial proliferation: E), and necrosis (N), which are often referred to as the ‘AMEN’ score15. A significant mitotic count is a requirement for grade III tumors, and microvascular proliferation or necrosis for grade IV tumors, typically diffuse astrocytic tumors. Although the neuropathology community has accepted this system for more than 25 years, inter- and intra-observer variabilities have never been resolved. The system is subjective and has the following technical limitations: the assessment of atypia depends on individual skills and an investigator’s experience. Mitotic counts also rely on the diligence of the examiner. Since gliomas are permeating neoplasms with marked intra-tumoral heterogeneity, mitoses may accumulate focally or be evenly scattered. Therefore, when mitoses are counted in ten consecutive high-power fields (HPF), the mitotic count captured may vary according to the spreading pattern, either evenly or unevenly distributed. This counting method also has a significant pitfall. The area of each HPF depends on the field number (FN) of the ocular lens used. Typical FNs are 20, 22, and 26.5, which correspond to 0.20, 0.24, and 0.34 mm2, respectively, as a single HPF area. Differences in ocular lenses significantly affect the number of mitoses captured within a single HPF. In addition, mitoses are often difficult to distinguish from apoptosis. In some institutions, the anti-phosphohistone H3 (pHH3) antibody has been used to overcome the aforementioned issues (Fig. 1G, I)16,17,18,19. This mitosis-specific antibody recognizes the phosphorylation of serine 10 in histone H3 and does not react with apoptosis16. Therefore, it allows an investigator to quickly identify mitoses, providing increased sensitivity and reducing inter-observer variability. However, one disadvantage of this antibody is non-specific staining, particularly on an automatic immunostainer.

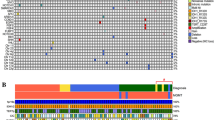
Left panel: Oligodendroglioma, IDH-mutant, and 1p/19q codeleted, WHO grade 2 (A, B, E, G, H, K, L). Right panel: Glioblastoma, IDH-mutant, WHO grade 4 (C, D, F, L, J, M, N). A, B FLAIR images showing an ill-defined, multi-nodular mass bulging from the cortex in the right frontal lobe. Note FLAIR-high nodules within the mass. C, D FLAIR images showing a well-demarcated, heterogenous mass in the right parietal lobe. E A nodule protruding from the cortical surface (H&E stain). F Elongated and oval tumor cells with fine cytoplasmic processes embedded in the microcystic background (H&E stain). G Three mitoses (arrows) positive for the pHH3 antibody in a medium-power field, which does not meet the grade 3 criteria of anaplastic oligodendroglioma. H Tumor cells diffusely positive for the IDH1R132H antibody. I Three mitoses (arrows) positive for the pHH3 antibody in a high-power field, which meets the histological grade 3 criteria of anaplastic astrocytoma. J Tumor cells diffusely positive for the IDH1R132H antibody. K Retained ATRX immunoreactivity. L Loss of immunoreactivity against H3 K27M me3. M Loss of ATRX immunoreactivity in tumor cells and retention in endothelial cells. N Loss of methylthioadenosine phosphorylase (MTAP) immunoreactivity in tumor cells, indicating the CDKN2A homozygous deletion, which corresponds to WHO grade 4 in the WHO 5th classification.
The current grading system was rooted in the 3rd edition of the WHO classification published in 200020. In that edition, the section on diffuse astrocytoma stated “mitotic activity is absent, but a single mitosis does not yet allow the diagnosis of anaplastic astrocytoma.” This notion is based on a retrospective study conducted by the Mayo Clinic showing that the survival of patients with diffuse astrocytoma with a solitary mitosis did not significantly differ from those without mitosis21, which is often referred to as the Modified St. Anne-Mayo scheme. Therefore, a mitotic count greater than 2 in the entire specimen has since been used to designate WHO grade III1.
The use of Ki-67 antibodies to assess the degree of malignancy has been repeatedly proposed;19,22 however, the WHO has never implemented it into the grading system because the immunoreactivity of the Ki-67 antibody is strongly influenced by fixation and the stage duration of formalin-fixed paraffin sections22. Therefore, the Ki-67 index varies with time and location and, thus, is not suitable for assessing biological behaviors across the institution.
The 5th edition of the WHO classification of CNS tumors
cIMPACT-NOW
After the publication of the 2nd edition of the WHO Classification of CNS Tumors, the WHO has revised it every 7 years, which became too long to incorporate the fruit of the latest research into the classification. To update recent and ongoing advances in research on molecular pathology between WHO revisions, cIMPACT-NOW (the Consortium to Inform Molecular and Practical Approaches to CNS Tumor Taxonomy, not officially WHO) was established in 201623. cIMPACT-NOW has since convened its separate working committees to address classification and grading issues24.
WHO grading of IDH-mutant astrocytoma
After the discovery of isocitrate dehydrogenase (IDH) 1 and 2 in diffuse gliomas25, adult diffuse gliomas are now genetically defined by three major genetic events: the IDH mutation, 1p/19q codeletion, and TERT promoter mutations26,27. In astrocytoma, TP53 and ATRX mutations follow IDH mutations. In oligodendroglioma, the 1p/19q codeletion follows IDH mutations26. TERT promoter mutations are involved in glioblastoma and oligodendroglioma27,28. The remaining are glioblastoma and so-called triple-negative gliomas, which lack all three major alterations. IDH mutations occur early in gliomagenesis; mutant IDH genes alter IDH enzymes, causing them to produce 2-hydroxyglutarate. This oncometabolite represses DNA demethylation and leads to genome-wide DNA hypermethylation29.
It subsequently became apparent that survival was longer in patients with IDH-mutant astrocytoma than in those with IDH-wild-type astrocytoma30,31, indicating that an independent grade needs to be given to IDH-mutant astrocytoma and IDH-wild-type even though their histological features are identical. Furthermore, retrospective studies found that the mitotic activity threshold did not reflect progression-free or overall survival in IDH-mutant gliomas, suggesting that the histological grading criteria used in the WHO classification do not sufficiently stratify the risk of IDH-mutant astrocytoma32,33. Nevertheless, these studies demonstrated that WHO grading is still valid for IDH-wild-type astrocytoma32. Similarly, proliferative indices (i.e., Ki-67) did not achieve the sufficient risk stratification of IDH-mutant astrocytoma, but successfully stratified the risk of IDH-wild-type astrocytoma19. Therefore, the WHO adopted a “within a tumor type” grading scheme to resolve the inconsistencies associated with IDH-mutant and -wild-type astrocytoma in the 5th edition in 20217,34. To clarify this revision, the WHO changed all CNS WHO grades to Arabic numerals from Roman numerals in 20217.
In attempts to improve risk stratification, retrospective studies investigated potential molecular markers associated with very poor clinical outcomes that may be incorporated into a more clinically relevant grading scheme. The CDKN2A/B homozygous deletion, amplification of CDK4, RB1 mutations or homozygous deletions, PIK3CA or PIK3R1 mutations, the amplification of PDGFRA, amplification of MYCN, global DNA methylation, genomic instability, and the loss of chromosome 14 were suggested to be strongly associated with a poor prognosis in patients with IDH-mutant astrocytoma (Table 1).
The CDKN2A/B homozygous deletion and CDK4 amplification may both be independent predictors of shorter survival in lower-grade astrocytoma35 and glioblastoma36. They have been associated with a lower level of global DNA methylation37,38,39,40. WHO grade III astrocytoma with the CDKN2A/B homozygous deletion was previously shown to behave almost identically to grade IV astrocytoma; Shirahata and others proposed a novel molecular grading system using the CDKN2A/B homozygous deletion to segregate grade IV lesions from grade II and III lesions regardless of histological findings33,41,42,43,44. Two studies failed to identify the CDKN2A/B homozygous deletion in WHO grade II astrocytoma44,45, while one detected it in 12% of cases35. Previous studies indicated that the CDK4 amplification combined with the loss of chromosome 14 was associated with a poor prognosis41,42, whereas others did not44,45. Similarly, the homozygous deletion of RB1 was strongly associated with inferior overall survival in one study46, but not in other studies44,45. Aoki and others revealed that altered RB pathway genes, including the CDKN2A/B homozygous deletion, amplification of CDK4, or RB1 mutations, were collectively a strong predictor of a poor outcome, but were not when considered independently46. Therefore, further studies are warranted to confirm the significance of the amplification of CDK4 or RB1 mutations. Although the amplification of PDGFRA has repeatedly been associated with a poor prognosis in patients with IDH-mutant astrocytoma, particularly grade II and III tumors35,44,47, one study did not detect this relationship46. The significance of other rare genetic alterations, such as PIK3R or PIK3CA mutations46 and the amplification of MYC44, in IDH-mutant astrocytoma remains unclear and, thus, warrants further study. Based on these findings, the WHO adopted the combination of the CDKN2A/B homozygous deletion and traditional histological criteria (anaplasia, mitoses, microvascular proliferation, and necrosis) into a histomolecular grading system of IDH-mutant astrocytoma (Table 2). In this grading system, when the CDKN2A/B homozygous deletion is present, grade 4 (IV) is automatically given regardless of the histology of IDH-mutant astrocytoma.
WHO grading of IDH-wild-type astrocytoma
In the WHO 2016 classification, diffuse and anaplastic astrocytoma, IDH-wild-type, were listed as a provisional entity1. However, recent studies demonstrated that the vast majority of IDH-wild-type diffuse grade II and anaplastic grade III astrocytoma exhibited an aggressive clinical course equivalent to glioblastoma grade IV48,49,50,51. More precise molecular analyses, including DNA methylation profiling, revealed that the remaining diffuse and anaplastic astrocytoma, IDH-wild-type, which followed an indolent clinical course, were low-grade gliomas, including pilocytic astrocytoma, glioneuronal tumors, and pediatric-type diffuse gliomas52,53. The characteristic molecular features of histologically lower grade, but genetically malignant astrocytoma include the amplification of EGFR, the concurrent gain of whole chromosome 7 and loss of whole chromosome 10, or TERT promoter mutations. TERT promoter mutations are the most prevalent among these three alterations and may overlap with each other48,49,50. Based on these findings, WHO adopted a histomolecular grading scheme for IDH-wild-type astrocytoma that combined traditional histological criteria with three glioblastoma molecular signatures (Table 3)34.
Does adult-type low-grade IDH-wild-type astrocytoma exist?
It currently remains unclear whether true adult-type low-grade IDH-wild-type astrocytoma without pediatric molecular alterations exists. Richardson and others showed that low-grade IDH-wild-type astrocytoma lacking the molecular glioblastoma signature had fewer total copy-number variations (CNV) and less frequent CDKN2A homozygous deletions and PTEN/PIK3CA alterations, but more frequent NF1 alterations, indicating the existence of true low-grade IDH-wild-type astrocytoma54. Berzero et al. examined 517 grade II gliomas with strict radiological and pathological criteria and identified 29 cases of histologically grade II IDH-wild-type diffuse astrocytoma with the molecular features of glioblastoma55. The median overall survival of patients with this group of tumors was 88 months, which was longer than that of glioblastoma. They also demonstrated that grade II IDH-wild-type diffuse astrocytoma is less aggressive than grade III IDH-wild-type diffuse astrocytoma. These findings suggested that the histological grade is still helpful and that strictly defined grade II astrocytoma with TERT promoter mutations alone will not behave as glioblastoma, IDH-wild-type56. Fujimoto et al. also reported that low-grade IDH-wild-type astrocytoma with either TERT promoter mutations or the amplification of PDGFRA mostly clustered with glioblastoma in a DNA methylation analysis. They also found that low-grade IDH-wild-type astrocytoma without the molecular features of glioblastoma were a heterogeneous group of tumors using DNA methylation profiling57.
Molecular prognostic markers in oligodendroglioma, IDH-mutant, and 1p/19q codeleted
The WHO has never officially disclosed the definite threshold between grades II and III for this tumor. However, the criteria of Giannini58,59,60 have frequently been used: increased cellularity, nuclear pleomorphisms, mitotic activity, microvascular proliferation, and necrosis have been associated with a poor prognosis. A mitotic count greater than 6 per 10 HPF or microvascular proliferation showed the strongest correlations with a poor outcome. Previous studies attempted to identify molecular markers to predict the malignant transformation and outcomes of oligodendroglioma. Their findings revealed that NOTCH1 mutations or the NOTCH and PI3K pathways were associated with a poor prognosis in patients with oligodendroglioma39,46,61. Furthermore, TCF12 transcriptional activity was related to a more aggressive tumor type62. However, the CDKN2A homozygous deletion was also linked to shorter survival45. Therefore, further studies are needed to identify molecular markers associated with a poor prognosis in patients with oligodendroglioma. A transcription factor activity signature has also been related to a poor prognosis in patients with molecular oligodendroglioma treated with adjuvant radiotherapy, suggesting that this signature is a predictive biomarker in oligodendrogliomas63.
Grading of pediatric-type diffuse gliomas
Neuroepithelial tumors, particularly low-grade gliomas, are the most common solid tumors in children and adolescents, accounting for ~25% of all tumors64. They are often referred to as pediatric-type gliomas. Although pediatric-type low-grade gliomas share a similar histology with their adult counterparts, they lack IDH mutations and the 1p/19q codeletion and harbor distinctive genetic abnormalities65,66. The histology of pediatric low-grade diffuse gliomas is often non-specific and overlaps with those of other low-grade tumor types, including circumscribed gliomas, preventing confident classification. Nevertheless, the prognosis of pediatric-type diffuse gliomas is generally favorable except for frankly malignant counterparts harboring histone H3 alterations67. Therefore, the WHO grading system of adult gliomas based on histological findings is not applicable to pediatric gliomas. In the WHO 5th edition, pediatric-type gliomas were divided into two categories, low and high grades; however, specific grades have not been given to all entities34.
Epigenetic markers
Glioma epigenetic molecular signatures
Although the epigenetic silencing of O6-methylguanine-DNA methyltransferase (MGMT) by the methylation of its promoter does not correlate with particular subtypes of gliomas, it is a significant biomarker for predicting sensitivity to alkylating agents, such as temozolomide68,69,70. In elderly patients with glioblastoma, the lack of MGMT promoter methylation has been identified as a negative predictor of responses to alkylating agent chemotherapy71,72. Another important epigenetic signature is the glioma cytosine-phosphate-guanine island methylation phenotype (G-CIMP)73. Nearly all IDH-mutant gliomas are positive for G-CIMP, and MGMT promoter methylation is associated with IDH mutations74. More than 75% of G-CIMP-low tumors were found to have alterations in RB pathway genes, including the CDKN2A/B homozygous deletion and amplification of CDK436,39. Patients with G-CIMP-low IDH-mutant astrocytoma had shorter overall survival than those in the G-CIMP-high group. Therefore, the identification of IDH mutations is indispensable in glioma diagnostic practice but is not yet a part of the grading scheme.
DNA methylation profiling
The genome-wide DNA methylation pattern represents both the cell of origin and somatically acquired DNA methylation changes in cancer. These patterns may remain relatively stable during cancer development, and, thus, an analysis of the tumor DNA status represents those of the original cells, thereby allowing for the more precise identification of tumor types than a morphological analysis53,75. An unsupervised learning approach using DNA methylation and NGS data also enables rare tumors without known canonical genetic alterations to be subclassified within IDH-wild-type gliomas46,76. For example, many IDH-wild-type gliomas belong to pediatric-type gliomas have a single driver gene, typically that in the MAP-kinase pathway77. DNA methylation profiling is a reliable and robust approach for the classification of gliomas into molecularly defined subgroups, giving a reasonably accurate estimate of clinical outcomes53.
Grading and CNV
Previous studies reported that IDH-mutant35,36 and -wild-type glioblastomas both have higher total CNV levels and evidence of chromothripsis than their lower-grade counterparts78, suggesting that total CNV levels are a prognostic factor in diffuse astrocytoma and also that mutations in the genes responsible for overall genomic instability may be an underlying mechanism for astrocytoma with a poor clinical outcome. However, this has not yet been verified because the thresholds for high CNV and somatic mutations varied79.
Grading of adult diffuse gliomas in resource-limited settings
In the WHO 5th classification, genetic testing, such as Sanger sequencing and multiplex ligation-dependent probe amplification, is mandatory for reaching an adequate diagnosis that meets the definition of each tumor type in the classification7,34,80,81,82,83. Unfortunately, such genetic testing is not always available, even in developed countries84. Nevertheless, the benefits of scientific advances need to be available to every patient with a brain tumor, and the best possible service in each setting should be given to the patients, even using surrogate diagnostic markers. The WHO updated 4th classification1 and its prior consensus guideline85 have proposed an integrated diagnosis that combines all available complementary information. In the “integrated diagnosis,” diagnosis should be layered to provide a format for displaying multiple types of information85. It helps to visualize the diagnostic process and eventually increases the correctness of diagnoses in resource-limiting settings using surrogate markers83,85,86,87. One type of complementary information is MRI88,89. Diffuse gliomas are morphologically highly heterogeneous, and the pathological findings of a surgically resected specimen may only represent part of the entire tumor. Imaging may depict the whole tumor and compensate for the limitations of a pathological investigation90. Based on an international survey within the International Society of Neuropathology framework, immunohistochemistry (IHC) was available in the majority of countries surveyed, and an IHC surrogate is helpful for reaching the WHO diagnosis84.
Grading of IDH-mutant gliomas (Fig. 1)
When CT and MRI were unavailable, oligodendrogliomas were characterized by mushroom-like, hypertrophic bulging from the cerebral cortex on autopsy (Fig. 1E)11. On MRI, ~90% of genetically defined oligodendroglioma arise from the frontoparietal lobe7,88,91,92. Some studies suggested that oligodendroglioma with 1p/19q codeletion is characterized by an indistinct tumor border and heterogenous signal intensity (Fig. 1A, B)91,92,93,94,95, while IDH-mutant astrocytomas often show a rather discrete border (Fig. 1A, C, D)91 and the T2-FLAIR mismatch sign96,97. The provability of 1p/19q codeleted oligodendroglioma is significantly high when calcification is detected on CT98. Fluorescence in situ hybridization, a microsatellite analysis, or MLAP is often used to detect the 1p/19q codeletion. When these molecular tests are not available, the combination of ATRX and H3K27me3 IHC may provide relevant information equivalent to molecular testing (Fig. 1K, L)99,100,101, although if a codeletion study is not available, “oligodendroglioma, NOS” should be allotted to the diagnosis102. The 1p/19q codeletion is mutually exclusive to the loss of ATRX and is associated with the loss of H3K27me3 immunoreactivity; however, retained or inconclusive H3K27me3 mandates molecular testing. In the WHO 2016 classification, the diagnosis of IDH-mutant astrocytoma requires the presence of IDH-1 or 2 mutations and the absence of the 1p/19q codeletion, whereas the WHO 2021 classification does not require the 1p/19q codeletion once the loss of ATRX and a typical astrocytic histology are confirmed (Fig. 1F, M)103. Although the loss of p16 CDKN2A antibody immunoreactivity does not correlate with the CDKN2A homozygous deletion in malignant mesothelioma, a strong correlation was previously reported between methylthioadenosine phosphorylase (MTAP) IHC and this deletion104. In addition, MTAP IHC facilitated the detection of the CDKN2A homozygous deletion in diffuse astrocytoma (Fig. 1N)105. As mentioned previously, the CDKN2A/B homozygous deletion is often absent in a grade II lesion. Therefore, the WHO grade II histology could be used to exclude the homozygous deletion, particularly in resource-limited settings44,45.
Conclusions
Although the WHO 5th edition of CNS tumor classification has attempted to introduce newly recognized entities, phase out old tumor types, and adjust the taxonomic structure34, a number of grading issues have not yet been resolved. An integrated diagnosis combining histopathological findings with molecular information has provided clearer insights into the classification of brain tumors than a diagnosis by a histopathological assessment alone85. In addition, the unsupervised learning approach using DNA methylation and NGS data, which is free from inter-observer variability, has identified distinctive subgroups with unique molecular characteristics106. However, it has not yet been established whether these subgroups are clinically relevant. Moreover, numerous studies clearly demonstrated that a given tumor often reveals many molecular alterations over space and time, which results in difficulties selecting the most promising treatment regimen for each patient. Therefore, prospective controlled studies are required to clarify this issue.
The current grading scheme based on the expected natural history may not be rational because the clinical course and outcome of each patient may be extensively modified by the tumor location, extent of resection, and type of adjuvant therapy. In that case, grades cannot be assigned unless their designation does not disturb clinical practice. Therefore, in this molecular era of glioma pathology, the grading system needs to evolve to a type of patient-oriented precision medicine, allowing us to not grade a tumor type as a whole, but to provide the most promising guidance to each patient. To achieve this, clinical and histomolecular information needs to be considered and computational pathology assisted by artificial intelligence adopted in order to accurately assess this information107. These advances will ultimately improve the lives of individuals affected by CNS tumors.
References
Louis, D. N., Ohgaki H., Wiestler O. D., Cavenee W. K. WHO Classification of Tumours of the Central Nervous System (International Agency for Research on Cancer, Lyon, 2016).
Komori, T. Updated 2016 WHO classification of tumors of the CNS: turning the corner where molecule meets pathology. Brain Tumor Pathol. 34, 139–140 (2017).
Akagi, Y. et al. Reclassification of 400 consecutive glioma cases based on the revised 2016WHO classification. Brain Tumor Pathol. 35, 81–89 (2018).
Ohba, S., Kuwahara, K., Yamada, S., Abe, M. & Hirose, Y. Correlation between IDH, ATRX, and TERT promoter mutations in glioma. Brain Tumor Pathol. 37, 33–40 (2020).
Komori, T. Updating the grading criteria for adult diffuse gliomas: beyond the WHO2016CNS classification. Brain Tumor Pathol. 37, 1–4 (2020).
Louis, D. N. & von Deimling, A. Grading of diffuse astrocytic gliomas: Broders, Kernohan, Zulch, the WHO… and Shakespeare. Acta Neuropathol. 134, 517–520 (2017).
WHO Classification of Tumours Editorial Board. World Health Organization Classification of Tumours of the Central Nervous System (International Agency for Research on Cancer, Lyon, 2021) in press.
Wright, J. R. & Albert, C. Jr Broders’ paradigm shifts involving the prognostication and definition of cancer. Arch. Pathol. Lab. Med. 136, 1437–1446 (2012).
Etienne, D. et al. James Watson Kernohan (1896-198): frontiers in neuropathology. Clin. Anat. 25, 527–529 (2012).
Scheithauer, B. W. Development of the WHO classification of tumors of the central nervous system: a historical perspective. Brain Pathol. 19, 551–564 (2009).
Zülch, K. J. Histological Typing of Tumours of the Central Nervous System (World Health Organization, Geneva, 1979).
Kleihues, P., Burger, P. C., Scheithauer, B. W. Histological Typing of Tumours of the Central Nevous System (Springer-Verlag, Berlin, 1993).
Daumas-Duport, C., Scheithauer, B., O’Fallon, J. & Kelly, P. Grading of astrocytomas. A simple and reproducible method. Cancer 62, 2152–2165 (1988).
Louis, D. N. et al. The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol. 114, 97–109 (2007).
Perry, A., Brat D. J. Practical Surgical Neuropathology (Elsevier, Philadelphia, 2018).
Takei, H., Bhattacharjee, M. B., Rivera, A., Dancer, Y. & Powell, S. Z. New immunohistochemical markers in the evaluation of central nervous system tumors: a review of 7 selected adult and pediatric brain tumors. Arch. Pathol. Lab. Med. 131, 234–241 (2007).
Habberstad, A. H., Gulati, S. & Torp, S. H. Evaluation of the proliferation markers Ki-67/MIB-1, mitosin, survivin, pHH3, and DNA topoisomerase IIalpha in human anaplastic astrocytomas–an immunohistochemical study. Diagn. Pathol. 6, 43–51 (2011).
Suzuki, A. et al. Modified rapid immunohistochemical staining for intraoperative diagnosis of malignant brain tumors. Brain Tumor Pathol. 34, 141–148 (2017).
Duregon, E. et al. Ki-67 proliferation index but not mitotic thresholds integrates the molecular prognostic stratification of lower grade gliomas. Oncotarget 7, 2190–2198 (2016).
Kleihues, P., Cavenee, W. K. Pathology and Genetics of Tumors of the Nervous System (World Health Organization classification of tumors, IARC Press, Lyon, France, 2000).
Giannini, C. et al. Cellular proliferation in pilocytic and diffuse astrocytomas. J. Neuropathol. Exp. Neurol. 58, 46–53 (1999).
Arima, N. et al. The importance of tissue handling of surgically removed breast cancer for an accurate assessment of the Ki-67 index. J. Clin. Pathol. 69, 255–259 (2016).
Louis, D. N. et al. Announcing cIMPACT-NOW: the consortium to inform molecular and practical approaches to CNS tumor taxonomy. Acta Neuropathol. 133, 1–3 (2017).
Louis, D. N. et al. cIMPACT-NOW update 6: new entity and diagnostic principle recommendations of the cIMPACT-Utrecht meeting on future CNS tumor classification and grading. Brain Pathol. 30, 844–856 (2020).
Parsons, D. W. et al. An integrated genomic analysis of human glioblastoma multiforme. Science 321, 1807–1812 (2008).
Louis, D. N. et al. The 2016 World Health Organization classification of tumors of the central nervous system: a summary. Acta Neuropathol. 131, 803–820 (2016).
Killela, P. J. et al. TERT promoter mutations occur frequently in gliomas and a subset of tumors derived from cells with low rates of self-renewal. Proc. Natl Acad. Sci. USA 110, 6021–6026 (2013).
Arita, H. et al. A combination of TERT promoter mutation and MGMT methylation status predicts clinically relevant subgroups of newly diagnosed glioblastomas. Acta Neuropathol. Commun. 4, 79–93 (2016).
Figueroa, M. E. et al. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell 18, 553–567 (2010).
Eckel-Passow, J. E. et al. Glioma groups based on 1p/19q, IDH, and TERT promoter mutations in tumors. N. Engl. J. Med. 372, 2499–2508 (2015).
Brat, D. J., Verhaak, R. G., Aldape, K. D., Yung, W. K. & Salama, S. R. Comprehensive, integrative genomic analysis of diffuse lower-grade gliomas. N. Engl. J. Med. 372, 2481–2498 (2015).
Olar, A. et al. IDH mutation status and role of WHO grade and mitotic index in overall survival in grade II-III diffuse gliomas. Acta Neuropathol. 129, 585–596 (2015).
Yoda, R. A. et al. Mitotic index thresholds do not predict clinical outcome for IDH-mutant astrocytoma. J. Neuropathol. Exp. Neurol. 78, 1002–1010 (2019).
Louis, D. N. et al. The 2021 WHO classification of tumors of the central nervous system: a summary. Neuro Oncol. 23, 1231–1251 (2021).
Yang, R. R. et al. IDH mutant lower grade (WHO Grades II/III) astrocytomas can be stratified for risk by CDKN2A, CDK4 and PDGFRA copy number alterations. Brain Pathol. 30, 541–553 (2019).
Li, K. K. et al. Identification of subsets of IDH-mutant glioblastomas with distinct epigenetic and copy number alterations and stratified clinical risks. Neurooncol. Adv. 1, 1–11 (2019).
Korshunov, A. et al. Integrated molecular characterization of IDH-mutant glioblastomas. Neuropathol. Appl. Neurobiol. 45, 108–118 (2019).
Binder, H. et al. DNA methylation, transcriptome and genetic copy number signatures of diffuse cerebral WHO grade II/III gliomas resolve cancer heterogeneity and development. Acta Neuropathol. Commun. 7, 59 (2019).
Ceccarelli, M. et al. Molecular profiling reveals biologically discrete subsets and pathways of progression in diffuse glioma. Cell 164, 550–563 (2016).
Ferreyra Vega, S. et al. DNA methylation profiling for molecular classification of adult diffuse lower-grade gliomas. Clin Epigenet. 13, 102–113 (2021).
Cimino, P. J. et al. Multidimensional scaling of diffuse gliomas: application to the 2016 World Health Organization classification system with prognostically relevant molecular subtype discovery. Acta Neuropathol. Commun. 5, 39–53 (2017).
Cimino, P. J. & Holland, E. C. Targeted copy number analysis outperforms histological grading in predicting patient survival for WHO grade II/III IDH-mutant astrocytomas. Neuro Oncol. 21, 819–821 (2019).
Reis, G. F. et al. CDKN2A loss is associated with shortened overall survival in lower-grade (World Health Organization Grades II-III) astrocytomas. J. Neuropathol. Exp. Neurol. 74, 442–452 (2015).
Shirahata, M. et al. Novel, improved grading system(s) for IDH-mutant astrocytic gliomas. Acta Neuropathol. 136, 153–166 (2018).
Appay, R. et al. CDKN2A homozygous deletion is a strong adverse prognosis factor in diffuse malignant IDH-mutant gliomas. Neuro Oncol. 21, 1519–1528 (2019).
Aoki, K. et al. Prognostic relevance of genetic alterations in diffuse lower-grade gliomas. Neuro Oncol. 20, 66–77 (2018).
Phillips, J. J. et al. PDGFRA amplification is common in pediatric and adult high-grade astrocytomas and identifies a poor prognostic group in IDH1 mutant glioblastoma. Brain Pathol. 23, 565–573 (2013).
Weller, M., et al. Molecular classification of diffuse cerebral WHO grade II/III gliomas using genome- and transcriptome-wide profiling improves stratification of prognostically distinct patient groups. Acta Neuropathol. 129, 679–693 (2015)
Stichel, D. et al. Distribution of EGFR amplification, combined chromosome 7 gain and chromosome 10 loss, and TERT promoter mutation in brain tumors and their potential for the reclassification of IDHwt astrocytoma to glioblastoma. Acta Neuropathol. 136, 793–803 (2018).
Tesileanu, C. M. S. et al. Survival of diffuse astrocytic glioma, IDH1/2 wildtype, with molecular features of glioblastoma, WHO grade IV: a confirmation of the cIMPACT-NOW criteria. Neuro Oncol. 22, 515–523 (2020).
Kuwahara, K. et al. Clinical, histopathological, and molecular analyses of IDH-wild-type WHO grade II–III gliomas to establish genetic predictors of poor prognosis. Brain Tumor Pathol. 36, 135–143 (2019).
Reuss, D. E. et al. Adult IDH wild type astrocytomas biologically and clinically resolve into other tumor entities. Acta Neuropathol. 130, 407–417 (2015).
Capper, D. et al. Practical implementation of DNA methylation and copy-number-based CNS tumor diagnostics: the Heidelberg experience. Acta Neuropathol. 136, 181–210 (2018).
Richardson, T. E., Hatanpaa, K. J. & Walker, J. M. Molecular characterization of “True” low-grade IDH-wild-type astrocytomas. J. Neuropathol. Exp. Neurol. 80, 431–435 (2021).
Berzero, G. et al. IDH-wild-type lower-grade diffuse gliomas: the importance of histological grade and molecular assessment for prognostic stratification. Neuro Oncol. 23, 955–966 (2021).
Giannini, C. & Giangaspero, F. TERT promoter mutation: is it enough to call a WHO grade II astrocytoma IDH wild-type glioblastoma? Neuro Oncol. 23, 865–866 (2021).
Fujimoto, K. et al. TERT promoter mutation status is necessary and sufficient to diagnose IDH-wild-type diffuse astrocytic glioma with molecular features of glioblastoma. Acta Neuropathol. 142, 323–338 (2021).
Giannini, C. et al. Anaplastic oligodendroglial tumors: refining the correlation among histopathology, 1p 19q deletion and clinical outcome in Intergroup Radiation Therapy Oncology Group Trial 9402. Brain Pathol. 18, 360–369 (2008).
Giannini, C. et al. Oligodendrogliomas: reproducibility and prognostic value of histologic diagnosis and grading. J. Neuropathol. Exp. Neurol. 60, 248–262 (2001).
Komori, T. et al. Controversies over the diagnosis of oligodendroglioma: a report from the satellite workshop at the 4th international symposium of brain tumor pathology, Nagoya Congress Center, May 23, 2012. Brain Tumor Pathol. 30, 253–261 (2013).
Halani, S. H. et al. Multi-faceted computational assessment of risk and progression in oligodendroglioma implicates NOTCH and PI3K pathways. NPJ Precis. Oncol. 2, 24–33 (2018).
Labreche, K. et al. TCF12 is mutated in anaplastic oligodendroglioma. Nat. Commun. 6, 7207–7216 (2015).
So, J., Mamatjan, Y., Zadeh, G., Aldape, K. & Moraes, F. Y. Transcription factor networks of oligodendrogliomas treated with adjuvant radiotherapy or observation inform prognosis. Neuro Oncol. 23, 795–802 (2021).
Ostrom, Q. T. et al. CBTRUS statistical report: primary brain and other central nervous system tumors diagnosed in the United States in 2013-2017. Neuro Oncol. 22, iv1–iv96 (2020).
Tateishi, K., Nakamura, T. & Yamamoto, T. Molecular genetics and therapeutic targets of pediatric low-grade gliomas. Brain Tumor Pathol. 36, 74–83 (2019).
Komori, T. The molecular framework of pediatric-type diffuse gliomas: shifting toward the revision of the WHO classification of tumors of the central nervous system. Brain Tumor Pathol. 38, 1–3 (2021).
Ellison, D. W. et al. cIMPACT-NOW update 7: advancing the molecular classification of ependymal tumors. Brain Pathol. 30, 863–866 (2020).
Hegi, M. E. et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N. Engl. J. Med. 352, 997–1003 (2005).
Wick, W. et al. MGMT testing–the challenges for biomarker-based glioma treatment. Nat. Rev. Neurol. 10, 372–385 (2014).
Friedman, H. S. et al. DNA mismatch repair and O6-alkylguanine-DNA alkyltransferase analysis and response to Temodal in newly diagnosed malignant glioma. J. Clin. Oncol. 16, 3851–3857 (1998).
Wick, W. et al. Temozolomide chemotherapy alone versus radiotherapy alone for malignant astrocytoma in the elderly: the NOA-08 randomised, phase 3 trial. Lancet Oncol. 13, 707–715 (2012).
Fukai, J. et al. Molecular characteristics and clinical outcomes of elderly patients with IDH-wild-type glioblastomas: comparative study of older and younger cases in Kansai Network cohort. Brain Tumor Pathol. 37, 50–59 (2020).
Noushmehr, H. et al. Identification of a CpG island methylator phenotype that defines a distinct subgroup of glioma. Cancer Cell 17, 510–522 (2010).
Malta, T. M. et al. Glioma CpG island methylator phenotype (G-CIMP): biological and clinical implications. Neuro Oncol. 20, 608–620 (2018).
Aoki, K. & Natsume, A. Overview of DNA methylation in adult diffuse gliomas. Brain Tumor Pathol. 36, 84–91 (2019).
Woo, H. Y. et al. Glioblastomas harboring gene fusions detected by next-generation sequencing. Brain Tumor Pathol. 37, 136–144 (2020).
Ellison, D. W. et al. cIMPACT-NOW update 4: diffuse gliomas characterized by MYB, MYBL1, or FGFR1 alterations or BRAF(V600E) mutation. Acta Neuropathol. 137, 683–687 (2019).
Cohen, A. et al. DNA copy number analysis of grade II–III and grade IV gliomas reveals differences in molecular ontogeny including chromothripsis associated with IDH mutation status. Acta Neuropathol. Commun. 3, 34–46 (2015).
Mirchia, K. et al. Establishing a prognostic threshold for total copy number variation within adult IDH-mutant grade II/III astrocytomas. Acta Neuropathol. Commun. 7, 121–124 (2019).
Adachi, J.-i et al. Droplet digital PCR assay for detecting TERT promoter mutations in patients with glioma. Brain Tumor Pathol. 38, 201–209 (2021).
Miki, S. et al. Highly sensitive detection of TERT promoter mutations in recurrent glioblastomas using digital PCR. Brain Tumor Pathol. 37, 154–158 (2020).
Yokogami, K. et al. Impact of PCR-based molecular analysis in daily diagnosis for the patient with gliomas. Brain Tumor Pathol. 35, 141–147 (2018).
Louis, D. N. et al. Data sets for the reporting of tumors of the central nervous system: recommendations from the International Collaboration on Cancer Reporting. Arch. Pathol. Lab. Med. 144, 196–206 (2020).
Andreiuolo, F., Mazeraud, A., Chretien, F. & Pietsch, T. A global view on the availability of methods and information in the neuropathological diagnostics of CNS tumors: results of an international survey among neuropathological units. Brain Pathol. 26, 551–554 (2016).
Louis, D. N. et al. International Society Of Neuropathology-Haarlem consensus guidelines for nervous system tumor classification and grading. Brain Pathol. 24, 429–435 (2014).
Sonoda, Y. et al. Practical procedures for the integrated diagnosis of astrocytic and oligodendroglial tumors. Brain Tumor Pathol. 36, 56–62 (2019).
Rajeswarie, R. T., Rao, S., Nandeesh, B. N., Yasha, T. C. & Santosh, V. A simple algorithmic approach using histology and immunohistochemistry for the current classification of adult diffuse glioma in a resource-limited set-up. J. Clin. Pathol. 71, 323–329 (2018).
Yamauchi, T. et al. Radiological characteristics based on isocitrate dehydrogenase mutations and 1p/19q codeletion in grade II and III gliomas. Brain Tumor Pathol. 35, 148–158 (2018).
Natsumeda, M., et al. Predicting BRAF V600E mutation in glioblastoma: utility of radiographic features. Brain Tumor Pathol. (2021)
Roux, A. et al. Prognostic relevance of adding MRI data to WHO 2016 and cIMPACT-NOW updates for diffuse astrocytic tumors in adults. Working toward the extended use of MRI data in integrated glioma diagnosis. Brain Pathol. 31, e12929 (2021).
Johnson, D. R. et al. Genetically defined oligodendroglioma is characterized by indistinct tumor borders at MRI. Am. J. Neuroradiol. 38, 678–684 (2017).
Smits, M. Imaging of oligodendroglioma. Br. J. Radiol. 89, 20150857 (2016).
Latysheva, A. et al. Dynamic susceptibility contrast and diffusion MR imaging identify oligodendroglioma as defined by the 2016 WHO classification for brain tumors: histogram analysis approach. Neuroradiology 61, 545–555 (2019).
Fellah, S. et al. Multimodal MR imaging (diffusion, perfusion, and spectroscopy): is it possible to distinguish oligodendroglial tumor grade and 1p/19q codeletion in the pretherapeutic diagnosis? Am. J. Neuroradiol. 34, 1326–1333 (2013).
Chawla, S. et al. Differentiation between oligodendroglioma genotypes using dynamic susceptibility contrast perfusion-weighted imaging and proton MR spectroscopy. Am. J. Neuroradiol. 34, 1542–1549 (2013).
Broen, M. P. G. et al. The T2-FLAIR mismatch sign as an imaging marker for non-enhancing IDH-mutant, 1p/19q-intact lower-grade glioma: a validation study. Neuro Oncol. 20, 1393–1399 (2018).
Jain, R. et al. “Real world” use of a highly reliable imaging sign: “T2-FLAIR mismatch” for identification of IDH mutant astrocytomas. Neuro Oncol. 22, 936–943 (2020).
Saito, T. et al. Calcification on CT is a simple and valuable preoperative indicator of 1p/19q loss of heterozygosity in supratentorial brain tumors that are suspected grade II and III gliomas. Brain Tumor Pathol. 33, 175–182 (2016).
Ammendola, S., et al. H3K27me3 immunostaining is diagnostic and prognostic in diffuse gliomas with oligodendroglial or mixed oligoastrocytic morphology. Virchows Arch. https://doi.org/10.1007/s00428-021-03134-1 (2021).
Filipski, K. et al. Lack of H3K27 trimethylation is associated with 1p/19q codeletion in diffuse gliomas. Acta Neuropathol. 138, 331–334 (2019).
Yamamichi, A. et al. Immunohistochemical ATRX expression is not a surrogate for 1p19q codeletion. Brain Tumor Pathol. 35, 106–113 (2018).
Louis, D. N. et al. cIMPACT-NOW update 1: Not Otherwise Specified (NOS) and Not Elsewhere Classified (NEC). Acta Neuropathol. 135, 481–484 (2018).
Louis, D. N. et al. cIMPACT-NOW update 2: diagnostic clarifications for diffuse midline glioma, H3 K27M-mutant and diffuse astrocytoma/anaplastic astrocytoma, IDH-mutant. Acta Neuropathol. 135, 639–642 (2018).
Chapel, D. B. et al. MTAP immunohistochemistry is an accurate and reproducible surrogate for CDKN2A fluorescence in situ hybridization in diagnosis of malignant pleural mesothelioma. Mod. Pathol. 33, 245–254 (2020).
Satomi, K. et al. Utility of methylthioadenosine phosphorylase immunohistochemical deficiency as a surrogate for CDKN2A homozygous deletion in the assessment of adult-type infiltrating astrocytoma. Mod. Pathol. 34, 688–700 (2021).
Capper, D. et al. DNA methylation-based classification of central nervous system tumours. Nature 555, 469–474 (2018).
Cui, M. & Zhang, D. Y. Artificial intelligence and computational pathology. Lab Investig. 101, 412–422 (2021).
Acknowledgements
This manuscript is dedicated to my late mentor, Dr. Bernd W. Scheithauer from the Mayo Clinic.
Author information
Authors and Affiliations
Contributions
This manuscript is the sole product of the author.
Corresponding author
Ethics declarations
Competing interests
The author declares no competing interests.
Ethical approval
This review did not have any ethical issue.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Komori, T. Grading of adult diffuse gliomas according to the 2021 WHO Classification of Tumors of the Central Nervous System. Lab Invest 102, 126–133 (2022). https://doi.org/10.1038/s41374-021-00667-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41374-021-00667-6
This article is cited by
-
Development of preoperative and postoperative models to predict recurrence in postoperative glioma patients: a longitudinal cohort study
BMC Cancer (2024)
-
Molecular mechanisms of tumour development in glioblastoma: an emerging role for the circadian clock
npj Precision Oncology (2024)
-
Beyond the WHO 2021 classification of the tumors of the central nervous system: transitioning from the 5th edition to the next
Brain Tumor Pathology (2024)
-
SEOM-GEINO clinical guidelines for grade 2 gliomas (2023)
Clinical and Translational Oncology (2024)
-
Quantifying H&E staining results, grading and predicting IDH mutation status of gliomas using hybrid multi-dimensional MRI
Magnetic Resonance Materials in Physics, Biology and Medicine (2024)
