
Abstract
The 2016 WHO classifies IDH-mutant gliomas into oligodendroglioma or diffuse astrocytoma based on co-occurring genetic events. Recent literature addresses the concept of stratifying IDH-mutant gliomas based on prognostically significant molecular events. However, the presence of a second class-defining driver alteration in IDH-mutant gliomas has not been systematically described. We searched the sequencing database at our institutions as well as The Cancer Genome Atlas (TCGA) and cBioPortal for IDH-mutant gliomas with other potentially significant alterations. For each case, we reviewed the clinical information, histology and genetic profile. Of 1702 gliomas tested on our targeted exome sequencing panel, we identified 364 IDH-mutated gliomas, four of which had pathogenic FGFR alterations and one with BRAF V600E mutation. Five additional IDH-mutant gliomas with NTRK fusions were identified through collaboration with an outside institution. Also, a search in the glioma database in cBioPortal (5379 total glioma samples, 1515 cases [28.1%] with IDH1/2 mutation) revealed eight IDH-mutated gliomas with FGFR, NTRK or BRAF pathogenic alterations. All IDH-mutant gliomas with dual mutations identified were hemispheric and had a mean age at diagnosis of 36.2 years (range 16–55 years old). Co-occurring genetic events involved MYCN, RB and PTEN. Notable outcomes included a patient with an IDH1/FGFR1-mutated anaplastic oligodendroglioma who has survived 20 years after diagnosis. We describe a series of 18 IDH-mutant gliomas with co-occurring genetic events that have been described as independent class-defining drivers in other gliomas. While these tumors are rare and the significance of these alterations needs further exploration, alterations in FGFR, NTRK, and BRAF could have potential therapeutic implications and affect clinical trial design and results in IDH-mutant studies. Our data highlights that single gene testing for IDH1 in diffuse gliomas may be insufficient for detection of targets with potential important prognostic and treatment value.
Similar content being viewed by others

Introduction
Isocitrate dehydrogenase (IDH) mutations represent an important, class-defining alteration in diffuse infiltrating gliomas. The 2016 World Health Organization (WHO) Classification of Tumors of The Central Nervous System classifies IDH-mutant gliomas into astrocytoma or oligodendroglioma based on specific co-occurring genetic events, including ATRX and TP53 mutations for diffuse astrocytoma and 1p/19q codeletion for oligodendroglioma [1]. Furthermore, WHO recommends stratification of IDH-mutant gliomas in WHO grades 2–4 for astrocytomas and 2–3 for oligodendrogliomas, based on mitotic count and presence of microvascular proliferation or necrosis. Recent studies challenge the WHO grading of IDH-mutant gliomas and underline the importance of associated genetic alterations. While Yoda and colleagues demonstrated that the number of mitoses is not an accurate measure of outcome in WHO grade 2/3 IDH-mutant gliomas [2], other studies demonstrated that specific genetic alterations surpass histology in predicting outcome. Specifically, the presence of CDKN2A/B homozygous deletion was shown to be associated with shorter survival in IDH-mutant gliomas WHO grade 2 and 3 [3, 4]. RB1 homozygous deletion, PIK3CA pathogenic mutations, PDGFRA amplification, and MYCN amplification have also been associated with shorter overall survival in recent studies [5, 6]. In addition, Touat et al. demonstrated worse survival in a subset of hypermutated, mismatch repair-deficient IDH-mutant gliomas [7].
Neuropathology practice varies based on resources available. Diffuse gliomas are often stratified based on a combination of immunohistochemical stains that predict a given genetic alteration (e.g., IDH1 R132H, p53, and ATRX) and fluorescence in situ hybridization for 1p/19q. Institutions with available next generation targeted exome sequencing panels pursue further testing and the completeness of results is dependent on how comprehensive the panels are. In general, the identification of IDH mutations and the presence or absence of 1p/19q codeletion causes a stop in investigational efforts in clinical practice, with the assumption that the tumor has been sufficiently characterized. This approach fails to identify the IDH-mutant gliomas with prognostically significant associated genetic events. The Consortium to Inform Molecular and Practical Approaches to CNS Tumor Taxonomy, Not Official WHO (cIMPACT-NOW), recently addressed this concept, recognizing the strong correlation of certain genetic alterations, and of homozygous deletion of CDKN2A/B in particular, with shorter survival [8, 9].
While the impact of co-occurring genetic events that are not specific for a particular tumor-type is currently being recognized in predicting the outcome of IDH-mutant gliomas, the presence and significance of other class-defining alterations in IDH-mutant gliomas has not been systematically described. Alterations in BRAF, FGFR, NTRK, ALK, and other genes have been described as oncogenic drivers in a subset of IDH-wild type diffuse gliomas. We encountered in practice the case of a young adult with a diffuse glioma in which we found an IDH1 R132S mutation as well as a BRAF V600E mutation and hypothesized that a subset of IDH-mutant diffuse gliomas in our database have second class-defining genetic alterations. Therefore, we undertook a systematic approach to describe them.
Materials and methods
Cohort
With Institutional Board Review approval, we searched the sequencing database at Dana Farber Cancer Institute (DFCI) and analyzed their associated genetic alterations. For each case at DFCI, we reviewed the clinical information, histology, and genetic profile. The genetic profile for cases from DFCI was established by using targeted next generation sequencing (see below).
Also, we systematically reviewed publicly available data from The Cancer Genome Atlas (TCGA) and cBioPortal (www.cbioportal.org) [10]. The following CNS/brain datasets were specifically searched: Brain Lower Grade Glioma (TCGA, Firehose Legacy), Brain, Lower Grade Glioma (TCGA, PanCancer Atlas), Glioma (MSK, Nature 2019), Glioma (MSKCC, Clin Cancer Res 2019), Low-Grade Gliomas (UCSF, Science 2014), Brain Tumor PDXs (Mayo Clinic, 2019), Glioblastoma (TCGA, Cell 2013), Glioblastoma (TCGA, Nature 2008), Glioblastoma Multiforme (TCGA, Firehose Legacy), Glioblastoma Multiforme (TCGA, PanCancer Atlas), Anaplastic Oligodendroglioma and Anaplastic Oligoastrocytoma (MSKCC, Neuro Oncol 2017).
Samples were filtered to include cases with IDH1 or IDH2 mutation. Samples were then subfiltered to find cases with co-occurring alterations in BRAF, FGFR1/2/3, NTRK1/2/3, H3F3A, HIST1H3B to reveal 56 individual patients. MYB and MYBL1 were also queried but there were no cases with both IDH1/2 mutation and MYB or MYBL1 alteration.
Data for each of the 56 individual cases were manually reviewed within cBioPortal to confirm whether or not the additional alterations were pathologically relevant. Cases with mutations of unclear/unknown significance in gliomas were excluded.
In total, we identified 8 unique cases of IDH-mutated glioma with a second class-defining molecular alteration with this approach (Cases #11, 12, 13, 14, 15, 16, 17, 18). For each case, data was abstracted from cBioPortal to include patient age at diagnosis, patient gender, histologic diagnosis, tumor grade, vital status, overall survival and progression free status. We also tabulated the presence of other co-occurring molecular events, and MGMT methylation status (if available). The methods of molecular testing for the cases in cBioPortal are not uniform, as these were performed at various institutions. Per cBioPortal, the data was generated through whole exome sequencing (Case #11, 15), targeted exome sequencing (MSK-IMPACT assay; Case#12, 13, 14, 16), whole genome sequencing (Case #17, 18) [11,12,13,14].
Cases sequenced at Foundation Medicine and part of a published collaboration on NTRK-fused gliomas [15] with DFCI were included, but a systematic analysis of all Foundation Medicine cases was not possible due to agreements between collaborating institutions. For this reason, the incidences calculated in the manuscript are approximate.
Sequencing
Targeted next generation sequencing (Illumina HiSeq) was performed on DNA isolated from formalin-fixed paraffin-embedded tissue using a next-generation hybrid capture targeted exome sequencing assay (Oncopanel) that interrogates the exons of 447 genes and 191 introns across 60 genes for structural rearrangements [16]. The Foundation Medicine next generation sequencing assay evaluates 324 genes for mutations and copy number alterations, as well as select intronic regions of a subset of genes to detect gene rearrangements. Details about the Foundation Medicine next generation sequencing assay can be found at https://www.foundationmedicine.com/genomic-testing/foundation-one-cdx.
Histology review and immunohistochemical stains
The histology of the cases in the DFCI cohort was reviewed by a neuropathologist (SA) and the histology of the Foundation Medicine cases were also reviewed by a neuropathologist (SR).
Pan-IDH (R132/172) (Sigma-Aldrich, clone MsMab-1) and BRAF V600E (Abcam, VE1 clone) was performed on Case#5 in an attempt to explore if there is co-localization in the same neoplastic cells.
Results
Of 1702 gliomas tested on the DFCI/Brigham and Women’s Hospital targeted exome sequencing panel (OncoPanel), we identified 364 IDH1/2-mutated gliomas (incidence 21.4%), of which 5 (incidence 1.4%) had a second class-defining molecular event (see Table 1, cases #1–5): one each with FGFR1 K656E (31-year-old male with anaplastic oligodendroglioma, WHO grade 3) and BRAF V600E (28-year-old male with glioblastoma, WHO grade 4); and three with FGFR3 focal gain (32 and 42-year-old males and a 54-year-old female, each with glioblastoma, WHO grade 4). Five additional IDH-mutant gliomas (cases #6–10) that also had NTRK rearrangements were identified through a prior approved collaboration with Foundation Medicine [15]. These 5 IDH-mutant cases contained NTRK gene fusions: ARGLU1- NTRK1 (55-year-old male with anaplastic astrocytoma, WHO grade 3); NFASC -NTRK1 (26-year-old make with diffuse astrocytoma, WHO grade 2); ARHGEF2-NTRK1 (31-year-old male with glioblastoma, WHO grade 4); PAIP1-NTRK2 (45-year-old male with glioblastoma, WHO grade 4); and FRY-NTRK3 (38-year-old female with anaplastic astrocytoma, WHO grade 3). Finally, we searched the glioma database in cBioPortal (5379 total glioma samples, 1515 cases with either IDH1 or IDH2 mutation [28.1%]) and found 8 additional IDH-mutated gliomas (0.5%) with a second class-defining molecular event (cases #11–18): two with FGFR1 N546K (31-year-old male oligodendroglioma, WHO grade 2; and 41-year-old female with anaplastic astrocytoma, WHO grade 3); and one each with SPRNP1-FGFR2 fusion (16-year-old girl with diffuse astrocytoma, WHO grade 2), FGFR3 focal gain (34-year-old male with anaplastic astrocytoma, WHO grade 3), BRAF V600E (49-year-old male with diffuse astrocytoma, WHO grade 2), BRAF-UBE2H fusion (21-year-old male with diffuse astrocytoma, WHO grade 2), PEAK1-NTRK3 fusion (41-year-old female with diffuse astrocytoma, WHO grade 2), and AKAP13 -NTRK3 fusion (34-year-old male with anaplastic astrocytoma, WHO grade 3). Allelic frequencies for point mutations, where available, are shown in Supplemental Table 1. Allelic frequency data is not available for fusion events and copy number changes (i.e., FGFR3 gain), as those cannot be inferred from assays utilized in this study. In order to explore if the FGFR3 copy number change is a result of a rearrangement, RNA-based Anchor Plex fusion panel was performed on the three tumors in the DFCI cohort; no rearrangement was found. FGFR3 gain has been proposed as a high-grade glioma oncogenic driver in a poster presented at the 2017 United States and Canadian Academy of Pathology by Bale et al. (poster 255, not published). In an attempt to characterize further these gliomas, we returned to the cohort presented in that poster: all gliomas with FGFR3 gains from the DFCI database occurred in adults, were high-grade, and had coexisting alterations involving CDKN2A (homozygous deletion), TERT and ATRX (mutations). IDH1 R132H was present in 3 of the 14 cases. In the tumors without IDH1 mutation, there were no other established glioma drivers.
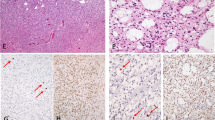
IDH-mutant gliomas with a second class-defining genetic alteration occurred predominantly in adult patients with one 16-year-old pediatric patient (case #13) present in this cohort. The mean age and standard deviation at diagnosis were 36.2 ± 10.5 years (median 34 years) with a range of 16–55 years. Tumors were located in the cerebral hemispheres in all cases in which location data was available (12/18 cases), including seven in the frontal lobe, four in the temporal lobe, and one in the parietal lobe. Twelve cases were diagnosed as high-grade (WHO grade 3 or 4): one anaplastic oligodendroglioma, five anaplastic astrocytoma, and six glioblastoma. Six were diagnosed as low-grade (WHO grade 2): five diffuse astroctyoma and one oligodendroglioma. Histologic features worth noting in the cohort were: in case #5 (BRAF V600E), which had giant cell features; and case #10 (FRY -NTRK3), which had prominent gemistocytic features; cases with FGFR3 gain tended to have an abundant number of giant and bizarre cells. Representative histology is illustrated in Fig. 1. The remainder of the cases displayed classic morphology and diffuse pattern of growth, with no distinguishing morphology.

A Hematoxilin-eosin-stained section of IDH -mutant-oligodendroglioma with FGFR1 K656E mutation showing classic histology. B IDH-mutant glioblastoma with FGFR3 gain showing frequent bizarre and multinucleated giant cells. C IDH-mutant anaplastic astrocytoma with FRY-NTRK3 fusion, showing gemistocytic features. IDH1 R132S-mutant glioblastoma with BRAF V600E showing a high-grade diffusely infiltrative astrocytoma (D), with pan -IDH immunohistochemical stain highlighting neoplastic astrocytes (E) and BRAF V600E expression in occasional tumor cells, particularly in areas that lower grade (F).
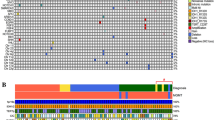
The molecular characteristics of our cohort are summarized in Fig. 2. All IDH mutations involved IDH1, the most common being R132H (15/18 cases). IDH1 R132S mutations were observed in two cases, while IDH1 R132C was seen in one case. Fourteen tumors additionally harbored both TP53 and ATRX mutations, in keeping with astrocytic lineage. In the other four tumors (4 of 18), three had TP53 mutation without ATRX mutations, while one had neither TP53 nor ATRX mutations. 1p/19q codeletion was present in the two oligodendrogliomas, both of which were IDH1 R132H-mutant with either FGFR1 K656E or FGFR1 N546K mutations. CDKN2A/B homozygous deletion was present in seven high-grade astrocytomas. MYCN amplification was observed in four IDH-mutant astrocytomas, three of which had NTRK fusions (two anaplastic astrocytomas and one low-grade diffuse astrocytoma). The fourth glioma with MYCN amplification was an IDH-mutant glioblastoma with BRAF V600E mutation. MGMT methylation status was available for 10/18 cases, seven of which were methylated and three were unmethylated. None of the gliomas in this cohort were hypermutated. Of the cases for which the allelic fraction was available for review (Supplementary Table S1), four had an IDH1 allelic fraction bigger than that of the second genetic event, in keeping with the IDH1 mutation being the primary genetic event. Notably, case# 11 had a FGFR1 N546K allelic fraction bigger than that of the IDH1 R132H (44% vs. 30%). The pan-IDH and BRAF V600E immunostains were performed on Case#5, in an attempt to explore if the IDH1 R132S and BRAF V600E mutations co-localize in the same cells (Fig. 1D–F); the immunostains correlated with the lower allelic fraction of BRAF V600E observed by molecular testing. Also the BRAF V600E immunostain seemed more positive in lower-grade infiltrative areas infiltrating cortex than in high-grade densely cellular areas, while the pan-IDH immunostain was positive in both components. Due to the faint expression of pan-IDH immunostain and to the fact that the this is not a double immunostain, we could not decide with confidence if the mutations co-occur in the same neoplastic cells or in different subclones.

Oncoprint showing the histologic features and genetic events of gliomas in this series.
Survival data was available for 12 of 18 cases, and it is presented here for completeness; however, given the small number of cases, definite conclusions cannot be made. The mean overall survival was 71.9 months (range 17.0–245.1 months) (Fig. 3). Amongst astrocytic tumors, diffuse astrocytoma WHO grade 2 was associated with the best overall survival (mean 81.3 months), followed by anaplastic astrocytoma (51.2 months), and glioblastoma (29.1 months). Case #1, an anaplastic oligodendroglioma, WHO grade 3, with IDH1 R132H and FGFR1 K656E mutations, was associated with the longest overall survival of 245.1 months. Compared to the entire cohort, the IDH-mutant gliomas with FGFR3 gain and NTRK fusion experienced worse overall survival (mean 35.8 months and 54.8 months, respectively). Three of the twelve patients were known to be deceased at the time this manuscript was prepared (cases #12, 14, 18, all anaplastic astrocytomas), while six patients were alive with progressive and/or recurrent disease (cases #1, 2, 3, 13, 15, 17) including: one anaplastic oligodendroglioma; two glioblastomas; and three diffuse astrocytomas, WHO grade 2 (Fig. 3).

Illustration of survival information for the patients included in this study (12 with available outcome data). *indicates a patient that is known to be deceased. ^ indicates a patient with progressive/recurrent disease. DA = diffuse astrocytoma, AA = anaplastic astrocytoma, GBM = glioblastoma, AO = anaplastic oligodendroglioma.
Discussion
IDH mutations are diagnostic hallmarks of infiltrating gliomas, particularly in adults, and are accompanied by TP53 and ATRX mutations in astrocytomas and 1p/19q codeletion and CIC mutations in oligodendroglioma [1]. Current guidelines stress the importance of testing for IDH mutations in adult infiltrating gliomas, which has been aided greatly by the development of a mutation specific antibody against IDH1 R132H. However, other relevant alterations with potential prognostic significance are almost certainly missed due to limited molecular testing.
Other class-defining molecular drivers have been identified in IDH-wild type gliomas, the most common being alterations in BRAF [17, 18], FGFR [19, 20], NTRK [15], and histone H3 [21, 22]. However, the simultaneous presence of second class-defining molecular alterations in IDH-mutated gliomas has not been systematically evaluated, and, given the recent developments in targeted therapy, it may be of interest for some patients. Here, we describe a series of 18 predominantly adult patients with IDH-mutant gliomas with second, co-occurring genetic events that have been described as independent class-defining oncogenic drivers in gliomas.
BRAF, a serine/threonine kinase, is a component of the mitogen-activated protein kinase (MAPK) signaling pathway. Molecular alterations in BRAF are known to drive a variety of cancer types, including gliomas [23, 24]. In particular, KIAA1549-BRAF fusion is a diagnostic feature of pilocytic astrocytomas [25, 26], and BRAF V600E mutations are encountered in many glial and glioneuronal tumors [27]. A small number of studies have examined the co-existence of IDH and BRAF molecular alterations within gliomas. Specifically, two large studies tested a combined 252 glioma samples for KIAA1549-BRAF fusion, BRAF V600E, and IDH1/2 mutations; the conclusion of these studies was that IDH mutations and BRAF alterations were mutually exclusive [28, 29]. Similarly, large-scale next generation sequencing studies in both pediatric and adult gliomas samples demonstrate that IDH and BRAF alterations are mutually exclusive events [11, 30,31,32,33]. In our cohort, we identified three IDH-mutant gliomas with concomitant BRAF alteration out of a total of 1879 IDH-mutant gliomas (0.16%). Only one published article describes the co-existence of IDH mutations with BRAF alterations. In this report, IDH mutation and BRAF-KIAA1549 or BRAF V600E were noted in 17 of 185 (9.2%) adult diffuse gliomas [34]. The high incidence of co-existing IDH and BRAF alterations observed by Badiali et al. is possibly due to a high false positive rate in their molecular methodology.
FGFR alterations have recently become recognized as important molecular drivers in both pediatric and adult low- and high-grade gliomas [31, 35,36,37], but there are no prior reports of IDH-mutated gliomas with co-existing FGFR alteration. We report seven such gliomas with variable histology ranging from WHO grade 2 (diffuse astrocytoma and oligodendroglioma) to WHO grade 4. The most common FGFR alterations observed in glioma samples include FGFR1/2/3 fusions (Particularly with TACC genes), FGFR1 internal tandem duplication, FGFR1 hotspot mutations (N546K and K656E) and isolated FGFR3/FGFR1 gain/amplification. In our series, we observed IDH-mutant gliomas with FGFR1 hotspot mutations, FGFR2 fusion and FGFR3 gain; there were no gliomas with FGFR1/3 fusion in this study.
NTRK gene rearrangements are emerging as important oncogenic drivers in a variety of tumors, including adult and pediatric gliomas [15, 38], both high- and low-grade. However, the presence of co-existent IDH mutation in NTRK-rearranged infiltrating gliomas has only been incidentally described in a handful of cases and without specific discussion as to the relative significance of this finding [39, 40]. We observed seven IDH-mutated tumors with co-occurring NTRK fusion. Five of the seven cases demonstrated high-grade histology (three WHO grade 3 tumors and two WHO grade 4) with two diffuse astrocytomas, WHO grade 2 (case #17: IDH1 R132H with PEAK1-NTRK3 fusion.
In this analysis of IDH-mutated gliomas, we did not detect a single example of a tumor with both IDH mutation and H3 K27M mutation. This is in agreement with other large scale genomic studies demonstrating mutual exclusivity of these two molecular alterations in gliomas[21, 32, 41, 42], including hemispheric H3 G34R/V gliomas [43]. All of the cases described in this manuscript involved IDH1 (the majority IDH1 R132H). We reviewed cases with IDH2 mutations when building the cohort in this study, but none of the IDH2 mutant gliomas had second-class defining alterations.
One limitation of our study is that some of the mutations observed here were not confirmed by secondary means. All cases were screened by immunohistochemistry with an IDH1 R132H mutation specific antibody, which correlated with the presence of IDH1 R132H mutation by sequencing in all cases. In our practice and laboratory, BRAF V600E immunohistochemistry sometimes fails or it is difficult to interpret, which led to the decision to replace it with the results of the next generation sequencing assay or ddPCR. Because the sequencing results of all cases were reviewed by neuropathologists with experience in molecular pathology (DMM, SR) and there was no contamination or other technical issues encountered, we consider it unlikely that the results presented here represent false positives.
While the significance of multiple class-defining molecular alterations within a single tumor remains unclear, the identification of additional molecular driver events has potentially important treatment related implications, as targeted therapies exist for tumors with pathogenic BRAF, FGFR, and NTRK alterations [44,45,46,47]. Although the incidence of multiple class defining molecular alterations is low, infiltrating gliomas are notoriously difficult to treat and the identification of targetable molecular alterations may improve survival in this group of patients. Similarly, almost all clinical trials for infiltrating gliomas utilize molecular data from tumor samples for stratification and analysis purposes, which could have significant impact in tumors with more than one class-defining molecular alteration.
In conclusion, this study highlights that subclonal alterations found clonally in other gliomas can be found in IDH-mutant gliomas, the clinical significance of which is unclear. While this information may be of clinical interest in selected rare cases, wide genetic testing may not always feasible in practice.
Data availability
The data in this manuscript are available from the corresponding author upon reasonable request.
References
Louis DN, Deimling A von, Cavenee WK. Diffuse astrocytic and oligodendroglial tumours. In: Louis DN, Ohgaki H, Wiestler OD, Cavenee WK, editors. WHO classification of tumours of the central nervous system. Revised 4th edition. World Health Organization; 2016. p. 16–77.
Yoda RA, Marxen T, Longo L, Ene C, Wirsching H-G, Keene CD, et al. Mitotic index thresholds do not predict clinical outcome for IDH-mutant astrocytoma. J Neuropathol Exp Neurol. 2019;78:1002–10.
Cimino PJ, Holland EC. Targeted copy number analysis outperforms histologic grading in predicting patient survival for WHO grades II/III IDH-mutant astrocytomas. Neuro Oncol. 2019;21:819–21.
Cimino PJ, Zager M, McFerrin L, Wirsching H-G, Bolouri H, Hentschel B, et al. Multidimensional scaling of diffuse gliomas: application to the 2016 World Health Organization classification system with prognostically relevant molecular subtype discovery. Acta Neuropathol Commun. 2017;5:39.
Aoki K, Nakamura H, Suzuki H, Matsuo K, Kataoka K, Shimamura T, et al. Prognostic relevance of genetic alterations in diffuse lower-grade gliomas. Neuro Oncol. 2017;20:66–77.
Shirahata M, Ono T, Stichel D, Schrimpf D, Reuss DE, Sahm F, et al. Novel, improved grading system(s) for IDH-mutant astrocytic gliomas. Acta Neuropathol. 2018;136:153–66.
Touat M, Li YY, Boynton AN, Spurr LF, Iorgulescu JB, Bohrson CL, et al. Mechanisms and therapeutic implications of hypermutation in gliomas. Nature. 2020;580:517–23.
Brat DJ, Aldape K, Colman H, Figrarella-Branger D, Fuller GN, Giannini C, et al. cIMPACT-NOW update 5: recommended grading criteria and terminologies for IDH-mutant astrocytomas. Acta Neuropathol. 2020;139:603–8.
Louis DN, Wesseling P, Aldape K, Brat DJ, Capper D, Cree IA, et al. cIMPACT‐NOW update 6: new entity and diagnostic principle recommendations of the cIMPACT‐Utrecht meeting on future CNS tumor classification and grading. Brain Pathol. 2020;30:844–56.
Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Disco. 2012;2:401–4.
Ceccarelli M, Barthel FP, Malta TM, Sabedot TS, Salama SR, Murray BA, et al. Molecular profiling reveals biologically discrete subsets and pathways of progression in diffuse glioma. Cell. 2016;164:550–63.
Zehir A, Benayed R, Shah RH, Syed A, Middha S, Kim HR, et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat Med. 2017;23:703–13.
Jonsson P, Lin AL, Young RJ, DiStefano NM, Hyman DM, Li BT, et al. Genomic correlates of disease progression and treatment response in prospectively characterized gliomas. Clin Cancer Res. 2019;25:5537–47.
Johnson BE, Mazor T, Hong C, Barnes M, Aihara K, McLean CY, et al. Mutational analysis reveals the origin and therapy-driven evolution of recurrent glioma. Science. 2014;343:189–93.
Torre M, Vasudevaraja V, Serrano J, DeLorenzo M, Malinowski S, Blandin A-F, et al. Molecular and clinicopathologic features of gliomas harboring NTRK fusions. Acta Neuropathol Commun. 2020;8:107.
Garcia EP, Minkovsky A, Jia Y, Ducar MD, Shivdasani P, Gong X, et al. Validation of OncoPanel: a targeted next-generation sequencing assay for the detection of somatic variants in cancer. Arch Pathol Lab Med. 2017;141:751–8.
Mistry M, Zhukova N, Merico D, Rakopoulos P, Krishnatry R, Shago M, et al. BRAF mutation and CDKN2A deletion define a clinically distinct subgroup of childhood secondary high-grade glioma. J Clin Oncol. 2015;33:1015–22.
Trisolini E, Wardighi DE, Giry M, Bernardi P, Boldorini RL, Mokhtari K, et al. Actionable FGFR1 and BRAF mutations in adult circumscribed gliomas. J Neurooncol. 2019;145:241–5.
Bale TA. FGFR- gene family alterations in low-grade neuroepithelial tumors. Acta Neuropathol Commun. 2020;8:21.
Lasorella A, Sanson M, Iavarone A. FGFR-TACC gene fusions in human glioma. Neuro Oncol. 2017;19:475–83.
Sturm D, Witt H, Hovestadt V, Khuong-Quang D-A, Jones DTW, Konermann C, et al. Hotspot mutations in H3F3A and IDH1 define distinct epigenetic and biological subgroups of glioblastoma. Cancer Cell. 2012;22:425–37.
Meyronet D, Esteban-Mader M, Bonnet C, Joly M-O, Uro-Coste E, Amiel-Benouaich A, et al. Characteristics of H3 K27M-mutant gliomas in adults. Neuro Oncol. 2017;19:1127–34.
Chi AS, Batchelor TT, Yang D, Dias-Santagata D, Borger DR, Ellisen LW, et al. BRAF V600E mutation identifies a subset of low-grade diffusely infiltrating gliomas in adults. J Clin Oncol. 2013;31:e233–236.
Dahiya S, Emnett RJ, Haydon DH, Leonard JR, Phillips JJ, Perry A, et al. BRAF-V600E mutation in pediatric and adult glioblastoma. Neuro Oncol. 2014;16:318–9.
Bar EE, Lin A, Tihan T, Burger PC, Eberhart CG. Frequent gains at chromosome 7q34 Involving BRAF in pilocytic astrocytoma. J Neuropathol Exp Neurol. 2008;67:878–87.
Jacob K, Albrecht S, Sollier C, Faury D, Sader E, Montpetit A, et al. Duplication of 7q34 is specific to juvenile pilocytic astrocytomas and a hallmark of cerebellar and optic pathway tumours. Br J Cancer. 2009;101:722–33.
Schindler G, Capper D, Meyer J, Janzarik W, Omran H, Herold-Mende C, et al. Analysis of BRAF V600E mutation in 1,320 nervous system tumors reveals high mutation frequencies in pleomorphic xanthoastrocytoma, ganglioglioma and extra-cerebellar pilocytic astrocytoma. Acta Neuropathol. 2011;121:397–405.
Cruz GR, Oliveira ID, Moraes L, Paniago MDG, Alves MT, de S, et al. Analysis of KIAA1549–BRAF fusion gene expression and IDH1/IDH2 mutations in low grade pediatric astrocytomas. J Neurooncol. 2014;117:235–42.
Gierke M, Sperveslage J, Schwab D, Beschorner R, Ebinger M, Schuhmann MU, et al. Analysis of IDH1-R132 mutation, BRAF V600 mutation and KIAA1549–BRAF fusion transcript status in central nervous system tumors supports pediatric tumor classification. J Cancer Res Clin Oncol. 2016;142:89–100.
Brennan CW, Verhaak RGW, McKenna A, Campos B, Noushmehr H, Salama SR, et al. The somatic genomic landscape of glioblastoma. Cell. 2013;155:462–77.
Zhang J, Wu G, Miller CP, Tatevossian RG, Dalton JD, Tang B, et al. Whole-genome sequencing identifies genetic alterations in pediatric low-grade gliomas. Nat Genet. 2013;45:602–12.
Frattini V, Trifonov V, Chan JM, Castano A, Lia M, Abate F, et al. The integrated landscape of driver genomic alterations in glioblastoma. Nat Genet. 2013;45:1141–9.
Network CGAR, Brat DJ, Verhaak RGW, Aldape KD, Yung WKA, Salama SR, et al. Comprehensive, integrative genomic analysis of diffuse lower-grade gliomas. N Engl J Med. 2015;372:2481–98.
Badiali M, Gleize V, Paris S, Moi L, Elhouadani S, Arcella A, et al. KIAA1549‐BRAF fusions and IDH mutations can coexist in diffuse gliomas of adults. Brain Pathol. 2012;22:841–7.
Singh D, Chan JM, Zoppoli P, Niola F, Sullivan R, Castano A, et al. Transforming fusions of FGFR and TACC genes in human glioblastoma. Science. 2012;337:1231–5.
Qaddoumi I, Orisme W, Wen J, Santiago T, Gupta K, Dalton JD, et al. Genetic alterations in uncommon low-grade neuroepithelial tumors: BRAF, FGFR1, and MYB mutations occur at high frequency and align with morphology. Acta Neuropathol. 2016;131:833–45.
Jones DTW, Hutter B, Jäger N, Korshunov A, Kool M, Warnatz H-J, et al. Recurrent somatic alterations of FGFR1 and NTRK2 in pilocytic astrocytoma. Nat Genet. 2013;45:927–32.
Gambella A, Senetta R, Collemi G, Vallero SG, Monticelli M, Cofano F, et al. NTRK fusions in central nervous system tumors: a rare, but worthy target. Int J Mol Sci. 2020;21:753.
Zheng Z, Liebers M, Zhelyazkova B, Cao Y, Panditi D, Lynch KD, et al. Anchored multiplex PCR for targeted next-generation sequencing. Nat Med. 2014;20:1479–84.
Ferguson SD, Zhou S, Huse JT, Groot JF, de, Xiu J, Subramaniam DS, et al. Targetable gene fusions associate with the IDH wild-type astrocytic lineage in adult gliomas. J Neuropathol Exp Neurol. 2018;77:437–42.
Schwartzentruber J, Korshunov A, Liu X-Y, Jones DTW, Pfaff E, Jacob K, et al. Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature. 2012;482:226–31.
Korshunov A, Ryzhova M, Hovestadt V, Bender S, Sturm D, Capper D, et al. Integrated analysis of pediatric glioblastoma reveals a subset of biologically favorable tumors with associated molecular prognostic markers. Acta Neuropathol. 2015;129:669–78.
Fontebasso AM, Papillon-Cavanagh S, Schwartzentruber J, Nikbakht H, Gerges N, Fiset P-O, et al. Recurrent somatic mutations in ACVR1 in pediatric midline high-grade astrocytoma. Nat Genet. 2014;46:462–6.
Lang S-S. The role of BRAF-targeted therapy in astrocytomas: a review. Neurosurgery. 2013;60:110–2.
Bautista F, Paci A, Minard‐Colin V, Dufour C, Grill J, Lacroix L, et al. Vemurafenib in pediatric patients with BRAFV600E mutated high‐grade gliomas. Pediatr Blood Cancer. 2014;61:1101–3.
Ardini E, Menichincheri M, Banfi P, Bosotti R, Ponti CD, Pulci R, et al. Entrectinib, a Pan–TRK, ROS1, and ALK inhibitor with activity in multiple molecularly defined cancer indications. Mol Cancer Ther. 2016;15:628–39.
Drilon A, Siena S, Ou S-HI, Patel M, Ahn MJ, Lee J, et al. Safety and antitumor activity of the multitargeted Pan-TRK, ROS1, and ALK inhibitor entrectinib: combined results from two phase I trials (ALKA-372-001 and STARTRK-1). Cancer Discov. 2017;7:400–9.
Acknowledgements
This work could not have been possible without the help of the highly specialized technicians in the Center for Advanced Molecular Diagnostics at The Brigham and Women’s Hospital.
Funding
The manuscript is the result of an IRB-approved search of existing databases. No funding was necessary for this study.
Author information
Authors and Affiliations
Contributions
JTA organized the data, illustrated the results and wrote the manuscript. MT provided data and reviewed molecular results. DMM contributed material and reviewed molecular results. DAR provided clinical information for some of the patients in the manuscript. JLH performed the pan-IDH antibody in his laboratory, provided the methods for it, and provided edits on the manuscript. PYW provided clinical information for some of the patients in the manuscript. RJ helped with the organization of results. SM provided part of the illustrations. SR provided molecular data for a subset of patients. KLL reviewed results and supported the illustrations. SA designed the project and oversaw all aspects of it.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Ethics approval/consent to participate
The study was carried with Dana Farber Cancer Institute Institutional Review Board approval (protocol 10-417). The study was performed in accordance with the Declaration of Helsinki.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Ahrendsen, J.T., Torre, M., Meredith, D.M. et al. IDH-mutant gliomas with additional class-defining molecular events. Mod Pathol 34, 1236–1244 (2021). https://doi.org/10.1038/s41379-021-00795-w
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41379-021-00795-w
This article is cited by
-
Frequent FGFR1 hotspot alterations in driver-unknown low-grade glioma and mixed neuronal-glial tumors
Journal of Cancer Research and Clinical Oncology (2022)
