
Abstract
Intracranial solitary fibrous tumors/hemangiopericytomas (SFT/HPCs) are vascular tumors that have a high rate of local recurrence and extracranial metastases. Intradural extramedullary spinal dissemination of intracranial SFT/HPC is extremely rare. There is a paucity of data available to elucidate the molecular mechanisms of intraspinal dissemination of intracranial SFT/HPC. Herein, we presented a case of intracranial SFT/HPC with intraspinal metastasis. The resected tumor specimens were enrolled in a clinical sequencing program, including whole-exome and transcriptome sequencing. By comparing genomic sequencing data of the intracranial tumors with intraspinal metastasis, we established the somatic mutational profiles of these tumors. Clonality analysis revealed a distinct subclonal structure in the intracranial tumor and its intraspinal metastasis, which might reflect the possibility of intratumoral clonal selection and evolution during the process of tumor dissemination. Through bioinformatics analysis and Sanger sequencing validation, a DSTYK mutation (Met296Ile) was identified as a candidate driver of intraspinal metastasis in this SFT/HPC case. Further, an intracranial tumor-derived SFT/HPC cell line, HPC3, was established to explore the mechanisms of the DSTYK mutation in promoting SFT/HPC metastasis. Based on the HPC3 cell model, we found that the DSTYK mutation promoted cell migration and invasion of HPC3 cells via activation of ERK1/2 signaling, which was inhibited by the MEK/ERK inhibitor AZD6244. The DSTYK mutation was also shown to upregulate the expression of two metastasis-related molecules: MMP2 and MMP9 in HPC3 cells; however, this effect was attenuated by AZD6244 treatment. Therefore, the DSTYK mutation may activate ERK1/2/MMP2/9 signaling to promote tumor cell metastasis in SFT/HPC. In conclusion, our study revealed the potential role of DSTYK mutation in the regulation of intraspinal metastasis of SFT/HPC, which might provide new biological insights into this rare disease.
Similar content being viewed by others
Introduction
Solitary fibrous tumors/hemangiopericytomas (SFTs/HPCs) are relatively rare neoplasms, accounting for 0.5–1% of all tumors of the central nervous system (CNS) [1]. Pathologically, SFT/HPCs are essentially fibrosarcomas derived from the pericytes in the walls of dural capillaries [2]. In 2007, the World Health Organization (WHO) designated HPCs as meningeal tumors of mesenchymal origin most frequently classified as grade II with more aggressive variants receiving a grade III classification (anaplastic HPC) [2]. More recently, HPCs have been characterized by the NAB2-STAT6 fusion gene, leading to nuclear STAT6 expression, and they are now considered part of one SFT/HPC tumor entity by the 2016 WHO classification of tumors of the CNS [3].
While SFTs/HPCs present with imaging characteristics similar to meningiomas, they have a significantly different clinical course with 5-year recurrence rates ranging between 20 and 70% [4]. The main treatments for SFTs/HPCs are surgical resection and radiotherapy. Despite the combination of surgery and adjuvant radiotherapy, the control rates have been disappointing. Nevertheless, SFTs/HPCs present a unique challenge for neurosurgical management due to their notorious ability to metastasize to extracranial locations [4, 5]. The bone, liver, lung, and abdominal cavity are the most commonly reported sites of distant metastasis [4, 5]. However, there are rare reports of metastasis to the spine from the intracranial SFTs/HPCs [6].
In this study, we presented a SFT/HPC case with intracranial lesions and intradural extramedullary spinal metastasis, which was managed successfully by serial neurosurgical resection. The resected specimens were enrolled in a clinical sequencing program, which included whole-exome and transcriptome sequencing. We identified a DSTYK (dual serine/threonine and tyrosine protein kinase) gene mutation as a potential driver promoting tumor cell metastasis in this case of SFT/HPC.
Material and methods
Reagents
Primary antibodies were purchased from the following sources: DSTYK, MMP2, MMP9, and β-actin (Abcam, Cambridge, MA, USA); p-ERK1/2 and ERK1/2 (Cell Signaling Technology, Beverly, MA, USA). Lentiviral plasmids expressing green fluorescent protein (GFP) or DSTYK wild-type (WT) or M296I mutant ORFs were constructed by Genecopoeia (Rockville, MD, USA). The lentiviral packaging and transduction procedure were described previously [7]. MEK/ERK inhibitor AZD6244 was provided by Selleckchem (Houston, TX, USA).
Tissue handling
This study was approved by the Research Ethics Committee of Xiangya Hospital Central South University (Rev No. 2017121022) with written informed consent. Tissue samples were fixed in 10% phosphate-buffered formalin and embedded in paraffin blocks for histological examination and immunohistochemistry. Patient blood samples and fresh tumor tissue specimens from the intracranial (IC) and the intraspinal (IS) lesions were used for the extraction of DNA and RNA and for in vitro culturing.
Whole-exome sequencing
Genomic DNA was isolated from tumor specimens using QIAamp DNA Mini Kit (Qiagen, Germany). Libraries were constructed using KAPA Hyper Prep Kits (KAPA Biosystems, Wilmington, MA, USA) and captured using SureSelect Human All Exon V5 (Agilent, Santa Clara, CA, USA). High-throughput sequencing was performed on an Illumina HiSeq 4000 platform. Sequencing reads were mapped to the human reference genome (hg19) using the Burrows–Wheeler Aligner (BWA) [8]. Duplicate removal, local realignment, and base quality recalibration were performed using PICARD (http://broadinstitute.github.io/picard/) and the Genome Analysis Toolkit (GATK) [9]. Somatic single-nucleotide variations (SNVs) were called using Mutect [10], and small indels were called using strelka [11]. In addition to the build-in filters, the following filtering criteria were applied: (i) total read count in tumor DNA ≥ 15; (ii) presence of variant on both strands; (iii) variant allele frequency in tumor DNA ≥ 10%; (iv) variant allele frequency in germline DNA = 0; and (v) removal of variants in positions listed in the dbSNP129 database [12]. Each variant was confirmed using Integrative Genomics Viewer (IGV). We also assessed whether the somatic variant was previously reported in COSMIC v87 (released 13-NOV-18). Gene-level copy ratios were calculated using a CNVKit [13]. Using a CNVKit algorithm, relative copy ratios for each exon were calculated by correcting for imbalanced library size, GC bias, sequence repeats, and target density.
Pathogenicity prediction of the variants
The unique variants enriched in the IS tumor with an allele frequency ≥ 0.2 were subjected to pathogenicity prediction analysis using Functional Analysis through Hidden Markov Models (FATHMM v2.3, http://fathmm.biocompute.org.uk/cancer.html), a high-throughput web server capable of predicting the functional consequences of both coding variants (i.e., non-synonymous single nucleotide variants (nsSNVs) and non-coding variants) [14]. Predictions with scores less than a default threshold of −0.75 indicated that the variants were potentially cancer-promoting mutations.
RNA sequencing (RNA-seq)
RNA-seq transcriptome analysis was conducted to identify gene fusions in the IC tumor. The mRNA libraries were constructed using Next Ultra II Directional RNA Library Prep Kit for Illumina (NEB, Ipswich, MA, USA), according to the manufacturer’s protocol. High-throughput sequencing was performed on an Illumina HiSeq 4000 platform. Sequencing reads were mapped to a human reference genome (GRCh38) using the Hisat2-2.0.5 [15]. Expression levels of each protein-coding gene were calculated using Stringtie-1.3.1c [16]. Gene fusions were identified by the JAFFA program [17]. The NAB2-STAT6 gene fusion in the IC tumor was confirmed using Sanger sequencing, as described previously [18].
Sanger sequencing
To validate variants by Sanger sequencing, genomic regions containing the mutations of interest were amplified by PCR (10 ng of genomic DNA, 1 × Taq PCR Master Mix (Qiagen), and 200 nM of each primer in a final reaction volume of 20 μl). The cycling conditions comprised 2 min at 94 °C followed by 30 cycles of 94 °C for 10 s, 56 °C for 20 s, and 68 °C for 30 s. Primer sequences for DSTYK were as follows: forward 5′ TCAGGAAATCCGAAAGTA 3′ and reverse 5′ CTCAGATAGCCCAGGTCA 3′. PCR products were purified using High Pure PCR Product Purification Kit (Qiagen). Sequencing analyses were carried out on the ABI 3100 Genetic Analyzer (Applied Biosystems, Austin, TX, USA).
Real-time PCR
Total RNA was extracted from the IC and IS tumor specimens using a RNeasy mini kit (Qiagen), according to the manufacturer’s instructions. First-strand cDNA synthesis was performed using 2 μg of total RNA with oligodT primers and Superscript III, followed by RNase H digestion (Invitrogen, Carlsbad, CA, USA), as previously described [19]. Real-time PCR reactions were performed in an Eppendorf Mastercycler (Eppendorf, Germany). Reactions (20 μl) contained 1 μl of template cDNA, SYBR Green qPCR Master Mix (Qiagen), and 200 nM of each primer. The cycling conditions comprised 2 min at 94 °C followed by 30 cycles of 94 °C for 10 s, 56 °C for 20 s, and 68 °C for 30 s. Primer sequences for DSTYK were as follows: forward 5′ TCAGGAAATCCGAAAGTA 3′ and reverse 5′ CTCAGATAGCCCAGGTCA 3′. PCR products were purified using High Pure PCR Product Purification Kit (Qiagen). Sequencing analyses were carried out on the ABI 3100 Genetic Analyzer (Applied Biosystems).
Subclonal structure analysis
Subclonal structure analysis was conducted to dissect the clonality of the IC and IS tumors. Copy number variations were identified using Control-FREEC [20]. We used ABSOLUTE to infer tumor purity and ploidy for each whole-exome sequencing sample by integrating mutational allele frequencies and copy number calls [20]. Pyclone was used to analyze cellular prevalence (CP) of each mutation. CP represents the proportion of tumor cells with the mutation in tumor [21]. All of the mutations of the two samples were grouped into several clusters by CP using mclust package on R.
SFT/HPC cell line culture
The establishment of SFT/HPC cell line, HPC3, was conducted as described previously [22]. Fresh IC tumor tissues were minced and treated with Type IV collagenase (2 mg/ml) in RPMI-1640 medium for 2 h at 37 °C under agitation. The cell suspension was pelleted by centrifugation, washed once with RPMI-1640 culture medium, plated in RPMI-1640 culture medium supplemented with 10% fetal bovine serum and antibiotics (Penicillin and Streptomycin), and cultured at 37 °C in a humidified atmosphere of 5% CO2. Rapid outgrowth of adherent, spindle-shaped cells was seen within one week of primary culturing. The cultures were passed twice weekly after detaching the cells with trypsin/EDTA solution.
Cell proliferation assay
Cell proliferation was determined using a CCK-8 assay (Dojindo Laboratories, Kumamoto, Japan) as described previously [23]. Briefly, HPC3 cells were seeded at a concentration of 1 × 104 cells/200 μl/well into 96-well plates. The cells were allowed to attach overnight. After different time points, CCK-8 solution (10 μl) was added to the culture, and further incubated for 1 h at 37 °C. The optical density (OD) (absorbance) was measured at 450 nm using a Multiskan MK3 microplate reader (Thermo Fisher Scientific, Waltham, MA, USA)
Soft agar assay
Anchorage-independent growth of HPC3 cells was tested as described previously [7]. The 2 ml culture medium with 0.5% agar was first plated into each well of a 6-cm culture dish. After the agar solidified, each well received another 2 ml of 0.35% agar in culture medium containing 1 × 104 cells. After 2 weeks, colonies were fixed and stained with 0.1% crystal violet. The number of colonies was determined microscopically by manually counting from triplicate wells for each cell line.
Wound healing assay
Cell migration was evaluated using a wound healing assay as described previously [24]. For the cell migration assay, HPC3 cells (5 × 105) were grown in six-well plates for 48 h until the cells were fully confluent. Nonadherent cells were then washed away twice with PBS. A sterile 200-μl pipette tip was used to make a uniform scratch in the center of the well. After 0 and 24 h, the distance between the sides of the wound was measured.
Transwell invasion assay
Cell invasion assays were performed using a transwell chamber with 8-μm pores (Corning, Corning, NY, USA), as described previously [25]. Briefly, HPC3 cells were suspended in DMEM medium at 5 × 105 cells/ml. Each transwell of a 24-well plate was pre-coated with 50 μl Matrigel solution. RPMI 1640 medium (600 μl) containing 10% FBS was added to each well (lower compartment), and 0.1 ml (0.5 × 105 cells) of the cell suspension was added onto each transwell insert (upper compartment). The plates were incubated for 36 h at 37 °C. The cells that invaded the bottom surface of the membrane were fixed by dehydrated alcohol and stained by 0.2% crystal violet solution (Sigma-Aldrich, St. Louis, MO, USA). After washing, the cells were photographed with an Olympus BX43 microscope (Olympus, Tokyo, Japan). Then, the stained cells were eluted with 20% glacial acetic acid and measured with a Multiskan MK3 microplate reader (Thermo Scientific, Waltham, MA, USA) at 570 nm.
Western blot
Western blots were conducted as described previously [26]. Briefly, HPC3 cells were lysed and quantified using a Bicinchoninic Acid Protein Assay Kit (Beyotime Biotechnology, Shanghai, China). Protein lysates (15 μg) was separated by 10% SDS-PAGE and transferred to Hybond-P PVDF membranes. The membranes were blocked with 5% non-fat dry milk in Tris-buffered saline buffer for 2 h at room temperature and then incubated with diluted antibodies overnight at 4 °C. The membranes were then incubated with a horseradish peroxidase-conjugated secondary antibody (Abcam, Cambridge, MA, USA) for 1 h at room temperature. The signal was visualized using an enhanced chemiluminescence detection reagent (Abcam, Cambridge, MA, USA). β-actin served as the loading control.
Immunohistochemistry
Immunohistochemistry on tissue sections was performed as described previously [26]. Briefly, the sections were dewaxed in xylene, rehydrated with graded concentrations of alcohol, and subjected to heat-induced epitope retrieval. The sections were then incubated overnight at 4 °C with a primary antibody or isotype IgG control, followed by incubation with a horseradish peroxidase-conjugated goat secondary antibody (DAKO, Denmark) for 30 min at room temperature. Immunochemical staining was visualized through a reaction with 3,3-diaminobenzidine and hydrogen peroxide chromogen substrate (DAKO, Denmark). Slides were counterstained with hematoxylin and mounted with a coverslip.
Enzyme-linked immunosorbent assay (ELISA)
Expression of matrix metalloproteinases (MMPs), specifically MMP2 and MMP9, were analyzed by ELISA. Cell culture supernatant was centrifuged at 12,000×g for 15 min at 4 °C. MMP2 and MMP9 ELISA kits (Sigma-Aldrich) were used to measure the levels of MMP2 and MMP9 protein secretion by HPC3 cells, respectively, according to the manufacturer’s instructions [19].
Statistical analysis
SPSS 16.0 (SPSS Inc, Chicago, IL, USA) was used for statistical analysis. Error bars throughout the figures indicate standard deviation. The Student’s t-test was used to compare means of two groups. One-way ANOVA was used to compare means of three or more groups. P < 0.05 was considered significant in all of the tests.
Results
Case presentation
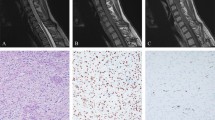
A 53-year-old man suffered from a history of headaches in the right frontotemporal region and gradual progression of back pain. Neurological examination revealed bilateral temporal hemianopia. Brain magnetic resonance imaging (MRI) showed a well-circumscribed, contrast-enhancing lesion in the deep right frontal lobe 3 cm in maximum diameter (Fig. 1a). Coexisting intradural extramedullary spinal metastasis at the sacral S1-2 level was also revealed by MRI (Fig. 1b). Although the intraspinal metastatic tumor was tightly adhered to the cauda equina nerves, total tumor removal was accomplished in February 2017 (Fig. 1b). The IC tumor was also totally excised the same day following spinal surgery (Fig. 1a). Because of the worsening state of mind and body, the patient refused to receive adjuvant radiotherapy. The IC tumor recurred 5 months after the surgery, and the patient deceased 4 months later.

a MRI of the head illustrating the intracranial SFT/HPC case 1-week prior to surgery and 3 days post surgery. The red triangle indicates the intracranial lesions. b MRI of the spine illustrating the intraspinal SFT/HPC 1-week prior to surgery and 3 days post surgery. The red triangle indicates the intraspinal lesions. c Tumor histopathology of the intracranial SFT/HPC and intraspinal metastasis as shown by H&E staining. d Immunohistochemical staining for CD34, STAT6, and Ki-67 in the intracranial tumor and its intraspinal metastasis. Bars: 50 μm
Necrotic and solid areas could be seen in the IC tumor and IS metastasis. Histopathological finding revealed SFT/HPC (WHO grade III), with numerous dilated, staghorn-type vessels with increased cellularity of the tumor cells in both the IC and IS tumors (Fig. 1c). Intertumoral heterogeneity in cellular/nuclei morphology was observed in the IC and IS tumors (Fig. 1c). Mitotic figures were observed in IC and IS tumors (approximately 6–8 mitoses per 10 high-power fields). Immunostaining for CD34 was positive in the IC tumor cells and vascular endothelial cells, but not in the IS tumor cells (Fig. 1d). Strong nuclear STAT6 positivity (specific marker for SFT/HPC) was also revealed in both IC and IS tumor cells (Fig. 1d). The proliferation index, as measured by Ki-67, was similar in the IC and IS tumors (approximately 10–15%, Fig. 1d). The specimens used for genomic analysis had over 70% tumor cell content based on morphologic analysis.
Whole-exome sequencing identified differential somatic mutational profiles in the IC and IS tumors
Genomic DNA was isolated from the IC and IS tumors and was subjected to whole exome sequencing. Whole exome sequencing of the tumors and matched normal blood identified 33 and 28 somatic gene mutations in the IC and IS tumors, respectively (Fig. 2a, Supplementary Table S1). Similar to a previous study [18], no significant germline aberrations or somatic point mutations were identified in hotspot cancer-related genes, such as TP53, KRAS, BRAF, BRCA1/2, or PIK3CA. The EGFR stop-gained mutation (Trp905Ter) was seen in the IC tumor. There were a few regions of substantial copy-number gain or loss (Tables S2). A global landscape of somatic gene mutations was generated from the whole exome sequencing data (Fig. 2b). The IC and IS tumors harbored shared gene mutations, and IC- or IS-specific gene mutations were distributed across the chromosomes (Fig. 2c). Consistent with previous findings, our RNA-Seq analysis also identified the NAB2-STAT6 gene fusion in the IC tumor (Fig. 2d, Table S3), which was further confirmed by Sanger sequencing (Fig. 2e).

a The number and type of somatic mutations identified by whole exome sequencing in the IC and IS tumors. b Circos map showing the distribution of somatic mutations across chromosomes in the IC and IS tumors. c Comparison of somatic mutations identified in the IC and IS tumors. d RNA-Seq identified the NAB2-STAT6 gene fusion in the IC tumor. e Sanger sequencing showing the fusion junction of the NAB2-STAT6 gene
Clonality analysis revealed distinct subclonal structure in the IC tumor and IS metastasis
To find the difference on the clonal structure between the IC and IS tumors, we retrieved all of the mutations in the coding regions and analyzed the subclonal composition for both tumors. A cellular prevalence for each mutation was inferred from the mutational frequency, the copy number at the loci, and the tumor purity, which represented the fraction of tumor cells carrying this mutation. Mutations were clustered into four subclones (Fig. 3a, Table S4). The diversity on the subclonal composition between the IC and IS tumors were found, including one IC-specific subclone (cluster 1) and one IS-specific subclone (cluster 4). In contrast, the CP was relatively stable for those mutations in the shared clusters (cluster 2 and 3, Fig. 3b, c). The high cellular prevalence of IC tumor-specific and metastasis-specific subclones might reflect the possibility of intratumoral clonal selection or evolution during the process of tumor dissemination.

a Subclonal structure of relapsed and metastatic tumors. Mutations were clustered into four groups based on the cellular prevalence of the mutations. The size of the oval represents the average cellular prevalence of the cluster. The numbers of mutations in each cluster are labeled below. b Cellular prevalence distribution of mutations in the relapsed and metastatic tumors. c Dynamics of mutations between relapsed and metastatic tumors. The relationship between mutations in the relapsed and metastatic tumors is indicated by the lines linking them
DSTYK mutation might be the candidate driver of IS metastasis
Searching for the somatic mutations that drive the clonal expansion of metastasis, we identified 11 genes with high allele frequencies ( ≥ 0.2) of functional mutations in the IS tumor (DSTYK, FAM53B, IL12RB1, KLK11, UNC93A, ABCC8, BIRC6, COL6A3, KRT9, PKDREJ, and SPATA31C2). Based on FATHMM analysis, the DSTYK gene mutation (888 G → A; 296Met → Ile, M296I) was predicted to be potentially related to cancer (Fig. 4a). Moreover, DSTYK (M296I) could be a novel mutation, which has not been recorded in the Catalogue of Somatic Mutations in Cancer (COSMIC) database. The DSTYK (M296I) mutation was located in the N-terminal noncatalytic region of NCR1, but not in the NCR2 or protein kinase (PK) domain (Fig. 4b). Sanger sequencing was performed to confirm the DSTYK driver mutation identified by whole exome sequencing analysis. As shown in Fig. 4c, the analyses revealed two types of chromosomes in the IC and IS tumors and matched blood—one carrying the DSTYK WT (IC tumor and blood) and the other one carrying both the DSTYK (M296I) mutant and DSTYK WT alleles (IS tumor). Real-time PCR was also conducted to further confirm the expression of DSTYK in IC and IS tumors. As shown in Fig. 4d, DSTYK exhibited similar gene expression in the IC and IS tumor. Consistent with genomic DNA sequencing analysis, the DSTYK (M296I) mutant allele was only observed in mRNA from the IS tumor (Fig. 4e). Interestingly, the DSTYK mutant allele in the IS tumor exhibited a higher allele fraction in mRNA than that in genomic DNA.

a FATHMM prediction on the potential pathogenicity of nonsynonymous somatic mutations. b The M296I mutation is located in the NCR1 domain of DSTYK gene. c Validation of the DSTYK M296I mutation with Sanger sequencing on genomic DNA isolated from the IC tumor, IS tumor, and matched blood. d Real-time PCR analysis of DSTYK mRNA expression in the IC and IS tumors. e Validation of the DSTYK M296I mutant with Sanger sequencing on mRNA isolated from IC or IS tumors
Establishment of the HPC3 cell line from the IC tumor
We managed to establish an SFT/HPC cell line, called HPC3, from the IC lesion. After plating, spindle-shaped HPC3 cells grew as adherent monolayers (Fig. 5a). Similar to the IC tumor, the immunofluorescence staining of HPC3 cells cultivated on coverslips revealed CD34 expression and nuclear positivity for STAT6 (Fig. 5b). HPC3 cells exhibited anchorage-independent spheroid growth in soft agar (Fig. 5c). The growth curve of different passages of HPC3 cells (P2, P4, and P8) revealed similar proliferation patterns (Fig. 5d). The mutational profiles of the IC tumor and HPC3 cells were similar with no presence of the DSTYK (M296I) mutation in genomic DNA (Fig. 5e). To confirm the derivation of the HPC3 cell line from the IC tumor, we performed microsatellite short tandem repeat (STR) analysis of 16 microsatellite loci. They showed identical results in the IC tumor and HPC3 cell line, suggesting that the HPC3 cell line originated from the IC tumor (Table S4).

a Establishment of the IC tumor-derived SFT/HPC cell line, HPC3. After plating, spindle-shaped HPC3 cells (passage 4) grew in adherent monolayers. Bars: 20 μm. b Immunofluorescence staining of HPC3 cells cultivated on coverslips revealed CD34 expression and nuclear positivity for STAT6. Bars: 10 μm. c HPC3 cells exhibited anchorage-independent spheroid growth in soft agar. Bars: 200 μm. d Grow curve of HPC3 at different passages (Passage 2, 4, 8) as measured by a CCK8 assay. e Comparison of somatic mutational profiles of the IC tumor and HPC3
DSTYK mutation promotes cell migration and invasion in HPC3 cells
We further investigated the involvement of the DSTYK mutation in maintaining malignant features of metastatic tumor cells, especially in terms of their roles in cell proliferation and mobility. HPC3 cells constitutively expressing the GFP control or DSTYK WT or M296I mutant were constructed based on a lentiviral expression system (Fig. 6a). We next examined the effect of the expression of the DSTYK mutation on the cell growth of HPC3 cells by CCK-8 assay, and the results showed that the DSTYK mutant transduced-HPC3 cells did not grow faster than those transduced with GFP or the DSTYK WT (P > 0.05; Fig. 6b). The metastatic process involves cell scattering, motility, extracellular matrix (ECM) degradation, migration, and invasion through the basement membranes [27]. To examine the effect of the DSTYK mutation on tumor cell migration in vitro, we performed a wound healing assay. A significantly higher migration speed was observed in the cells expressing the DSTYK mutant allele as compared to the HPC3 control, GFP control, and DSTYK WT cells (one-way ANOVA, P < 0.05, Fig. 6c). A transwell invasion assay was also performed in order to examine the effects of the DSTYK mutation on cell invasion. As shown in Fig. 6d, constitutive expression of the DSTYK mutation promoted the invasion of HPC3 cells, as compared with HPC3 control, GFP control, and DSTYK WT (one-way ANOVA, P < 0.05). These results illustrated that the DSTYK mutation might be an important regulator of cell migration and invasion in HPC3 cells.

a Western blot analysis of the expression of DSTYK in HPC3 control cells and HPC3 cells expressing GFP, DSTYK WT, or M296I mutant. β-actin served as the loading control. b HPC cells were seeded at a concentration of 1 × 104 cells/200 μl/well into 96-well plates, and allowed to attach overnight. After 5 days in culture, cell viability was analyzed by a CCK8 assay. c The wound healing assay using HPC3 cells. The migration of the HPC3 cells to the wound was measured at 0 and 24 h post scratch. The representative fields were photographed. The relative healing rates were quantified by measuring the gap sizes. Three different areas in each assay were chosen to measure the distances of migrating cells to the origin of the wound. Bars: 50 μm. *P < 0.05, M296I versus HPC3, GFP, and WT. d Transwell invasion assays were performed in HPC3 cells with the following groups: control, GFP, WT, and M296I. A crystal violet assay (OD570) was conducted to evaluate cell migration and invasion capability. Bars: 50 μm. All of the experiments were performed three times, and data are presented as the mean ± S.D. values. *P < 0.05, M296I versus HPC3, GFP, and WT
DSTYK mutation activates ERK1/2 signaling in HPC3 cells
DSTYK acts as a positive regulator of ERK phosphorylation in human embryonic kidney cells [28]. Our preliminary findings led to our hypothesis that the influence of the DSTYK mutation on cell migration and invasion might result from its potential function in activating ERK1/2 signaling in HPC3 cells. The protein level of phosphorylated ERK1/2 (pT202/pY204) was indeed higher in the IS tumor than that in the IC tumor (Fig. 7a). Constitutive expression of the DSTYK WT or mutant greatly increased p-ERK1/2 levels compared to the GFP control, with more ERK1/2 activation observed in the mutant group than in the WT group (Fig. 7b). AZD6244 is a potent and selective MEK/ERK inhibitor that is highly active in both in vitro and in vivo tumor models [29]. Treatment with AZD6244 markedly reduced p-ERK1/2 levels in HPC3 cells expressing the DSTYK WT or M296I mutant (Fig. 7c). A greater inhibition of migration following AZD6244 treatment was observed in the mutant group than in the GFP control or WT group (P < 0.05, Fig. 7d). As shown in Fig. 7e, the transwell invasion of HPC3 cells expressing the DSTYK mutation was markedly attenuated following AZD6244 treatment (P < 0.05, Fig. 7f).

a Western blot analysis of the protein levels of DSTYK, p-ERK1/2, and ERK1/2 in the IC and IS tumor lysates. β-actin served as the loading control. b Western blot analysis of the protein levels of p-ERK1/2 and ERK1/2 in HPC3 control cells and HPC3 cells expressing GFP, DSTYK WT, or M296I mutant. β-actin served as the loading control. c AZD6244 treatment attenuated p-ERK1/2 protein levels in HPC3 cells expressing GFP, DSTYK WT, or M296I mutant. HPC3 cells were treated with solvent (DMSO) or AZD6244 (100 nM) for 24 h, and the cell lysates were collected for western blot analysis. β-actin served as the loading control. d A wound healing assay with HPC3 cells treated with AZD6244. *P < 0.05, solvent control (DMSO) versus AZD6244 in the WT or M296I group. e Transwell invasion assay with HPC3 cells treated with AZD6244. All of the experiments were performed three times, and data are presented as the mean ± SD values. *P < 0.05, solvent control (DMSO) versus AZD6244 in the WT or M296I group
DSTYK mutation enhances MMP2/9 expression via ERK1/2 signaling
The gelatinase subgroup of MMPs consists of two highly homologous members, MMP2 and MMP9. These enzymes have been shown to be frequently overexpressed in carcinomas, and there is considerable evidence pointing to the involvement of these enzymes in malignant progression of tumors and invasion and metastasis [30, 31]. Higher expression of MMP2/9 was actually observed in the IS tumor than that in IC tumor (Fig. 8a). Further, HPC3 cells expressing the DSTYK mutation exhibited higher MMP2/9 expression than those expressing the GFP control or DSTYK WT (Fig. 8b). Consistent with these western blot results, ELISA analysis on culture supernatants showed that HPC3 cells expressing the DSTYK mutation released more MMP2/9 into the culture supernatant than those expressing the GFP control or WT (Fig. 8c). In order to confirm, the hypothesis that the DSTYK mutation might enhance MMP2/9 expression via ERK1/2 signaling, we treated the HPC3 cells with AZD6244. As shown in Fig. 8d, AZD6244 treatment greatly reduced MMP2/9 protein levels in HPC3 cells expressing the DSTYK WT or mutant. Consistently, MMP2/9 levels in the culture supernatants from the DSTYK WT and mutant group decreased following exposure to AZD6244. A considerable reduction in the secretion of MMP2/9 in the mutant group after AZD6244 treatment was observed compared to the GFP control or WT group (Fig. 8e).

a Western blot analysis of the protein levels of MMP2/9 in the IC and IS tumor lysates. β-actin served as the loading control. b Western blot analysis of the protein levels of MMP2/9 in HPC3 control cells and HPC3 cells expressing GFP, DSTYK WT, or M296I mutant. β-actin served as the loading control. c ELISA analysis of the levels of MMP2/9 in HPC3 culture supernatant. *P < 0.05, M296I versus GFP and WT. d AZD6244 treatment reduced MMP2/9 protein levels in HPC3 cells expressing GFP, DSTYK WT, or M296I mutant. HPC3 cells were treated with AZD6244 (100 nM) or solvent control (DMSO) for 24 h, and cell lysates were collected for analysis via western blot. β-actin served as the loading control. e AZD6244 treatment attenuated the secretion of MMP2/9 in HPC3 cells expressing GFP, DSTYK WT, or M296I mutant. HPC3 cells were treated with DMSO or AZD6244 (100 nM) for 24 h, and the culture supernatant was collected for ELISA analysis. *P < 0.05, solvent control (DMSO) versus AZD6244 in the WT or M296I group
Discussion
The extracranial metastatic tendency for IC SFT/HPC has been well documented, with rates ranging from 20 to 55% [3,4,5]. The common sites for metastasis include the lung, bone, and liver. Although there have been some reports of IC SFT/HPC cases with spinal metastasis, intradural extramedullary spinal metastasis of IC HPC rarely occurs in clinical practice [6]. Possible routes of IS metastasis could be explained by drop metastases of the original IC SFT/HPC through the cerebrospinal fluid (CSF) into the spinal canal. Until now, only three cases of IC SFT/HPC dissemination into the intradural extramedullary space of the spinal canal have been reported [6, 32, 33]. Although all of these cases were female, clear predisposing factors for IS metastatic SFT/HPC have not been identified. In this study, we presented a male case of IC SFT/HPC with intradural extramedullary metastasis, suggesting that sex and gender might not be a determining factor for IS metastasis. The mechanisms or predisposing factors leading to drop metastasis in different IC pathologies besides SFT/HPC have been investigated. Inoue et al. [34] reported that expression of CD133 may be a putative prognostic biomarker to predict IC dissemination of primary spinal cord astrocytoma. In oligodendroglioma, the proximity to cisterns and ventricles facilitated the spread through the CSF [35]. These reports highlight the rare clinical scenario of the tumor disseminating in the CSF and developing intradural drop metastases. The spinal seeding usually represents a negative event leading to a short survival after diagnosis (9 months in our case). Therefore, long-term follow-up, including MRI scans of the whole spine, might be warranted in IC SFT/HPC patients.
A number of next-generation sequencing studies that compared gene mutations in cancer primaries and metastases have been published in recent years. It is now well accepted that human cancers have a substantial level of intratumoral heterogeneity across multiple human cancer types [36]. Intratumoral genetic heterogeneity is the manifestation of a driving force in cancer genomes for the emergence of new subclones that have metastatic potential [37,38,39]. Zhai et al. [40] identified the spatial organization of intratumoral heterogeneity and evolutionary trajectories of metastases in hepatocellular carcinoma. However, the extent of genetic intratumoral heterogeneity in IC SFT/HPC and its IS metastatic lesions, as well as the subclonal architectures specifically associated with the development of distant metastasis, are largely unknown. In our study, we observed a morphological discrepancy between IC SFT/HPC and its IS metastasis (i.e., nuclei morphology and CD34 marker expression), suggesting their intratumoral clonality might be different. Consistently, our subclonal structural analysis revealed a differential subclonal composition in IC tumor and IS metastasis. Nevertheless, similar to the study by Xu et al. [41], we showed that metastatic tumors harbor unique subclones that are nonexistent in the matched primary tumors. Yet, how the subclonal diversity of tumors changes during metastasis still remains poorly understood in SFT/HPC during progression. Considering that the median time to distant metastasis is eight years for IC SFT/HPC, the metastasis-specific subclone might come from the pre-existing extremely rare subclone in the IC lesion, or acquire or evolve during the long-term metastatic process. Therefore, our findings might reflect the possibility of intratumoral clonal selection or evolution during the process of SFT/HPC dissemination.
Currently, only few studies have identified histological, molecular, or genetic features that are associated with SFT/HPC metastasis. Recently, Kinshow et al. [42] showed that tumor invasiveness was associated with metastasis and decreased survival in IC HPC. Kamamoto et al. [43] indicated that programmed cell death ligand-1 expression could be associated with extracranial metastasis in IC HPC. In this study, we identified a novel DSTYK mutation (M296I) as a potential driver of IS metastasis in one HPC case. DSTYK encodes a dual serine–threonine and tyrosine protein kinase, which is widely expressed in human tissues, including brain, spinal cord, pineal, gland, and skin. DSTYK is composed of two novel N-terminal non-catalytic regions (NCR1: 1-330aa; NCR2: 331-640aa) that are connected to a composite, but highly conserved C-terminal PK domain [44]. DSTYK was thought to be a regulator of apoptosis (inducer or suppressor) in some cell models [44, 45]. Recent studies have indicated that DSTYK could be a critical regulator of genitourinary development, as heterozygous mutations in DSTYK were identified as a frequent cause of autosomal dominant congenital anomalies of the kidney and urinary tract [29, 46, 47]. These loss-of-function mutations (R29Q, D200G, V210-Q218 Del) mostly occurred in NCR1 rather than PK domain, suggesting that the NCR1 domain might be crucial in regulating the catalytic activity of the PK domain. Our cellular function analysis based on a HPC3 cell model showed that the DSTYK M296I mutation further promoted cell migration and transwell invasion of HPC3 cells compared to the HP3 cells with the WT. Given the fact that the DSTYK M296I mutation was also located in the NCR1 domain, we proposed the DSTYK M296I mutation to be an activating mutation. However, the downstream signaling activated by the DSTYK mutation during metastasis still awaits further investigation.
ERK1/2 signaling plays a critical role in the progression and metastasis of cancer cells [48, 49]. Recent studies revealed that DSTYK could serve as a positive regulator of ERK1/2 phosphorylation downstream of fibroblast growth factor stimulation in HEK293T cells [29]. However, it still remains unclear whether this DSTYK mutation could aberrantly activate ERK1/2 to promote metastasis in HPC3 cells. Indeed, we observed a markedly higher level of p-ERK1/2 in the IS tumor than in IC tumor, which prompted us to look into the role of the DSTYK mutation in activating ERK1/2 signaling. We showed that constitutive expression of the DSTYK M296I mutation led to hyperactivated ERK1/2 signaling compared to the WT and GFP control, confirming the gain-of-function role of the M296I mutant. Treatment with the MEK/ERK inhibitor, AZD6244, attenuated p-ERK1/2 levels and suppressed the mutant-driven cell migration and transwell invasion in HPC3 cells. These results suggested that the effect of the DSTYK mutation on cell migration and invasion might be mediated by ERK1/2 signaling. MMPs are a family of proteolytic enzymes that degrade multiple components of the extracellular matrix. A large body of experimental and clinical evidence has implicated MMPs (especially MMP2/9) in tumor invasion, angiogenesis, and metastasis [50, 51]. We showed that the DSTYK mutation upregulated the expression of MMP2/9 compared to WT or GFP control, suggesting that HPC cells harboring the DSTYK mutation might be highly invasive and prone to metastasize. Furthermore, the high expression of MMP2/9 induced by the DSTYK mutation could be inhibited by the MEK/ERK inhibitor AZD6244. Therefore, our findings might unveil the novel function of this DSTYK activating mutation in promoting tumor metastasis through ERK1/2 and MMP2/9 signaling in this SFT/HPC case.
Aggressive tumor cell behavior is associated with a high risk of extracranial metastasis, and, as such, it is important to recognize this rare form of disease progression to determine appropriate treatments for SFT/HPC. Our study revealed the potential role of a DSTYK mutation in the regulation of IS metastasis of SFT/HPC, which might provide new biological insights into this rare disease.
Data availability
All of the data generated or analyzed during this study are included within this published article or within the supplementary materials.
References
Zhang GJ, Zhang LW, Li D, Wu Z, Zhang JT. Analysis of prognostic factors, survival rates, and treatment in anaplastic hemangiopericytoma. World Neurosurg. 2017;104:795–801.
Smith K, Talukder A, Kruse EJ. Intracranial hemangiopericytoma: a rare but aggressive tumor. Am Surg. 2015;81:314–5.
Louis DN, Perry A, Reifenberger G, von Deimling A, Figarella-Branger D, Cavenee WK, et al. The 2016 World Health Organization classification of tumors of the central nervous system: a summary. Acta Neuropathol. 2016;131:803–20.
Patel AR, Flores BC, Ban VS, Hatanpaa KJ, Mickey BE, Barnett SL. Intracranial hemangiopericytomas: recurrence, metastasis, and radiotherapy. J Neurol Surg B Skull Base. 2017;78:324–30.
Wei G, Kang X, Liu X, Tang X, Li Q, Han J, et al. Intracranial meningeal hemangiopericytoma: recurrences at the initial and distant intracranial sites and extraneural metastases to multiple organs. Mol Clin Oncol. 2015;3:770–4.
Ali HS, Endo T, Endo H, Murakami K, Tominaga T. Intraspinal dissemination of intracranial hemangiopericytoma: case report and literature review. Surg Neurol Int. 2016;7(Suppl 40):S1016–20.
Liu Q, Zhang C, Yuan J, Fu J, Wu M, Su J, et al. PTK7 regulates Id1 expression in CD44-high glioma cells. Neuro Oncol. 2015;17:505–15.
Li H, Durbin R. Fast and accurate long-read alignment with Burrows–Wheeler transform. Bioinformatics. 2010;26:589–95.
DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet. 2011;43:491–8.
Cibulskis K, Lawrence MS, Carter SL, Sivachenko A, Jaffe D, Sougnez C, et al. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat Biotechnol. 2013;31:213–9.
Kim S, Scheffler K, Halpern AL, Bekritsky MA, Noh E, Källberg M, et al. Strelka2: fast and accurate calling of germline and somatic variants. Nat Methods. 2018;15:591–4.
Liu Y, Zhang J, Li L, Yin G, Zhang J, Zheng S, et al. Genomic heterogeneity of multiple synchronous lung cancer. Nat Commun. 2016;7:13200.
Talevich E, Shain AH, Botton T, Bastian BC. CNVkit: genome-wide copy number detection and visualization from targeted DNA sequencing. PLoS Comput Biol. 2016;12:e1004873.
Shihab HA, Gough J, Cooper DN, Day INM, Gaunt TR. Predicting the functional consequences of cancer-associated amino acid substitutions. Bioinformatics. 2013;29:1504–10.
Kim D, Langmead B, Salzberg SL. HISAT: a fast spliced aligner with low memory requirements. Nat Methods. 2015;12:357–60.
Pertea M, Kim D, Pertea GM, Leek JT, Salzberg SL. Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat Protoc. 2016;11:1650–67.
Davidson NM, Majewski IJ, Oshlack A. JAFFA: high sensitivity transcriptome-focused fusion gene detection. Genome Med. 2015;7:43.
Robinson DR, Wu YM, Kalyana-Sundaram S, Cao X, Lonigro RJ, Sung YS, et al. Identification of recurrent NAB2-STAT6 gene fusions in solitary fibrous tumor by integrative sequencing. Nat Genet. 2013;45:180–5.
Hu X, Chen M, Li Y, Wang Y, Wen S, Jun F. Overexpression of ID1 promotes tumor progression in penile squamous cell carcinoma. Oncol Rep. 2019;41:1091–100.
Boeva V, Popova T, Bleakley K, Chiche P, Cappo J, Schleiermacher G, et al. Control-FREEC: a tool for assessing copy number and allelic content using next-generation sequencing data. Bioinformatics. 2012;28:423–5.
Carter SL, Cibulskis K, Helman E, McKenna A, Shen H, Zack T, et al. Absolute quantification of somatic DNA alterations in human cancer. Nat Biotechnol. 2012;30:413–21.
Roth A, Khattra J, Yap D, Wan A, Laks E, Biele J, et al. PyClone: statistical inference of clonal population structure in cancer. Nat Methods. 2014;11:396–8.
Fagerstedt KW, Salonen T, Zhao F, Kytölä S, Böhling T, Andersson LC. Establishment of a spontaneously transformed cell line (JU-PI) from a myxoinflammatory fibroblastic sarcoma. Tumour Biol. 2018;40:1010428318777936.
Yang Y, Huang JQ, Zhang X, Shen LF. MiR-129-2 functions as a tumor suppressor in glioma cells by targeting HMGB1 and is down-regulated by DNA methylation. Mol Cell Biochem. 2015;404:229–39.
Zhang J, Fu J, Pan Y, Zhang X, Shen L. Silencing of miR-1247 by DNA methylation promoted non-small-cell lung cancer cell invasion and migration by effects of STMN1. Onco Targets Ther. 2016;9:7297–307.
Shan N, Shen L, Wang J, He D, Duan C. MiR-153 inhibits migration and invasion of human non-small-cell lung cancer by targeting ADAM19. Biochem Biophys Res Commun. 2015;456:385–91.
Jun F, Hong J, Liu Q, Guo Y, Liao Y, Huang J, et al. Epithelial membrane protein 3 regulates TGF-β signaling activation in CD44-high glioblastoma. Oncotarget. 2017;8:14343–58.
Hecht I, Natan S, Zaritsky A, Levine H, Tsarfaty I, Ben-Jacob E. The motility-proliferation-metabolism interplay during metastatic invasion. Sci Rep. 2015;5:13538.
Sanna-Cherchi S, Sampogna RV, Papeta N, Burgess KE, Nees SN, Perry BJ, et al. Mutations in DSTYK and dominant urinary tract malformations. N Engl J Med. 2013;369:621–9.
Davies BR, Logie A, McKay JS, Martin P, Steele S, Jenkins R, et al. AZD6244 (ARRY-142886), a potent inhibitor of mitogen-activated protein kinase/extracellular signal-regulated kinase kinase 1/2 kinases: mechanism of action in vivo, pharmacokinetic/pharmacodynamic relationship, and potential for combination in preclinical models. Mol Cancer Ther. 2007;6:2209–19.
Singh D, Srivastava SK, Chaudhuri TK, Upadhyay G. Multifaceted role of matrix metalloproteinases (MMPs). Front Mol Biosci. 2015;2:19.
Nishio K, Motozawa K, Omagari D, Gojoubori T, Ikeda T, Asano M, et al. Comparison of MMP2 and MMP9 expression levels between primary and metastatic regions of oral squamous cell carcinoma. J Oral Sci. 2016;58:59–65.
Hayenga HN, Bishop AJ, Wardak Z, Sen C, Mickey B. Intraspinal dissemination and local recurrence of an intracranial hemangiopericytoma: case report. World Neurosurg. 2019;123:68–75.
Das A, Singh PK, Suri V, Sable MN, Sharma BS. Spinal hemangiopericytoma: an institutional experience and review of literature. Eur Spine J. 2015;24(Suppl 4):S606–13.
Taniura S, Taniguchi M, Mizutani T, Takahashi H. Metastatic hemangiopericytoma to the cauda equina: a case report. Spine J. 2007;7:371–3.
Inoue T, Endo T, Nakamura T, Shibahara I, Endo H, Tominaga T. Expression of CD133 as a putative prognostic biomarker to predict intracranial dissemination of primary spinal cord astrocytoma. World Neurosurg. 2018;110:e715–26.
Elefante A, Peca C, Del Basso De Caro ML, Russo C, Formicola F, Mariniello G, et al. Symptomatic spinal cord metastasis from cerebral oligodendroglioma. Neurol Sci. 2012;33:609–13.
Turtoi A, Blomme A, Castronovo V. Intratumoral heterogeneity and consequences for targeted therapies. Bull Cancer. 2015;102:17–23.
Büttner J, Jöhrens K, Klauschen F, Hummel M, Lenze D, Saeger W, et al. Intratumoral morphological heterogeneity can be an indicator of genetic heterogeneity in colorectal cancer. Exp Mol Pathol. 2018;104:76–81.
Gundem G, Van Loo P, Kremeyer B, Alexandrov LB, Tubio JMC, Papaemmanuil E, et al. The evolutionary history of lethal metastatic prostate cancer. Nature. 2015;520:353–7.
Kim TM, Jung SH, An CH, Lee SH, Baek IP, Kim MS, et al. Subclonal genomic architectures of primary and metastatic colorectal cancer based on intratumoral genetic heterogeneity. Clin Cancer Res. 2015;21:4461–72.
Zhai W, Lim TK, Zhang T, Phang ST, Tiang Z, Guan P, et al. The spatial organization of intra-tumour heterogeneity and evolutionary trajectories of metastases in hepatocellular carcinoma. Nat Commun. 2017;8:4565.
Xu H, Zhu X, Bao H, Wh Shek T, Huang Z, Wang Y, et al. Genetic and clonal dissection of osteosarcoma progression and lung metastasis. Int J Cancer. 2018;143:1134–42.
Kamamoto D, Ohara K, Kitamura Y, Yoshida K, Kawakami Y, Sasaki H. Association between programmed cell death ligand-1 expression and extracranial metastasis in intracranial solitary fibrous tumor/hemangiopericytoma. J Neurooncol. 2018;139:251–9.
Kinslow CJ, Rajpara RS, Wu CC, Bruce SS, Canoll PD, Wang SH, et al. Invasiveness is associated with metastasis and decreased survival in hemangiopericytoma of the central nervous system. J Neurooncol. 2017;133:409–17.
Peng J, Dong W, Chen Y, Mo R, Cheng JF, Hui CC, et al. Dusty protein kinases: primary structure, gene evolution, tissue specific expression and unique features of the catalytic domain. Biochim Biophys Acta. 2006;1759:562–72.
Zha J, Zhou Q, Xu LG, Chen D, Li L, Zhai Z, et al. RIP5 is a RIP-homologous inducer of cell death. Biochem Biophys Res Commun. 2004;319:298–303.
Lee JYW, Hsu CK, Michael M, Nanda A, Liu L, McMillan JR, et al. Large intragenic deletion in DSTYK underlies autosomal-recessive complicated spastic paraparesis, SPG23. Am J Hum Genet. 2017;100:364–70.
Payton S. Genetics: DSTYK gene linked to urinary tract defects. Nat Rev Urol. 2013;10:492.
Winer A, Adams S, Mignatti P. Matrix metalloproteinase inhibitors in cancer therapy: turning past failures into future successes. Mol Cancer Ther. 2018;17:1147–55.
Zhong Y, Lu YT, Sun Y, Shi ZH, Li NG, Tang YP, et al. Recent opportunities in matrix metalloproteinase inhibitor drug design for cancer. Exp Opin Drug Discov. 2018;13:75–87.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Tang, G., Yang, Y., Shang, L. et al. A DSTYK mutation activates ERK1/2 signaling to promote intraspinal dissemination in a case of solitary fibrous tumor/hemangiopericytoma. Lab Invest 99, 1501–1514 (2019). https://doi.org/10.1038/s41374-019-0274-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41374-019-0274-1
