
Abstract
Individuals with type 2 diabetes (T2D) display vascular insulin resistance and decreased nitric oxide production leading to vasoconstriction and atherosclerosis. Soluble factors such as pro-inflammatory molecules, and various genetic and epigenetic mechanisms have been implicated to induce insulin resistance in vascular endothelial cells. Epigenetic mechanisms such as altered promoter DNA methylation have been demonstrated in development and progression of metabolic disorders and atherosclerosis. However, underlying precise epigenetic mechanisms regulating cross talk between insulin signaling genes and inflammation in vascular cells remains to be fully understood. Human endothelial cells when (a) treated with interleukin-6 (IL-6) and insulin together, (b) pretreated with IL-6, and (c) under hyperinsulinemic conditions led to a state of vascular insulin resistance resulting in decreased Akt/eNOS activation and subsequent stabilization of STAT3 phosphorylation. IL-6 abrogated insulin effects on angiogenesis in 3D spheroid and matrigel assays. IL-6-induced insulin resistance was associated with decreased activity of DNA methyltransferase isoforms and global DNA hypomethylation, which inversely correlated with S-phase of cell cycle. CpG microarray analysis in IL-6 treated endothelial cells revealed promoters associated hypo- and hypermethylation of 199 and 98 genes respectively. Promoter DNA methylation status of genes associated with insulin signaling and angiogenesis such as RPS6KA2, PIK3R2, FOXD3, EXOC7, MAP3K8, ITPKB, EPHA6, IGF1R, and FOXC2 were validated by bisulfite DNA sequencing. Concentration and time-dependent analysis revealed that IL-6 reduced DNMT1 and DNMT3B but not DNMT3A protein levels. Our data indicate a causal link between IL-6-induced changes in global and promoter-specific DNA methylation, due to reduced DNMT1 and DNMT3B protein levels leading to altered expression of critical genes involved in insulin signaling and angiogenesis.
Similar content being viewed by others

Introduction
Diabetes is a chronic inflammatory disease characterized by an increase in pro-inflammatory cytokines, transcription factors, and surface proteins inducing insulin resistance [1]. Altered metabolic homeostasis in individuals with type 2 diabetes (T2D) has been causally related to inflammation. Acute-phase proteins such as C-reactive protein, haptoglobin, fibrinogen, plasminogen activator inhibitor, and cytokines such as interleukin (IL)-1β, IL-6, tumor necrosis factor (TNF)-α, and monocyte chemoattractant protein-1 are upregulated in T2D [2]. Such increased levels of cytokines orchestrate inter-organ communication modulating insulin sensitivity.
Increased levels of circulating IL-6 is associated with pre-disposition to T2D, insulin resistance [3], and T2D-associated vascular complications [4]. Experimental and clinical evidences have indicated increased serum IL-6 levels to alter insulin sensitivity in insulin-responsive tissues such as adipose, hepatic, and vasculature. Chronic infusion of IL-6 in mice led to decreased autophosphorylation of insulin receptor (IR) and phosphorylation of signal transducer and activator of transcription 3 (STAT3), subsequently resulting in reduced insulin sensitivity [5]. In mouse hepatocytes and HepG2 cells, IL-6 treatment decreased tyrosine phosphorylation of IR substrate-1 (IRS1) and led to 75% reduction of insulin-dependent glycogen synthesis [6]. IL-6 induced the disruption of insulin signaling upon activation of suppressor of cytokine signaling family member, SOCS-3, in hepatic tissue subcutaneously injected with IL-6 by inhibiting IR autophosphorylation [7]. Fat cells of insulin-resistant subjects expressed increased IL-6 levels, and in vitro in adipose cells (3T3-L1), IL-6 inhibited IRS1 and GLUT4 transcription, suggesting its role in impairing insulin signaling [8]. In vascular endothelial cells, IL-6 induced an imbalance in phosphorylation of IRS1Tyr612/IRS1Ser616, associated with activation of JNK and Erk kinases, which subsequently led to decreased endothelial nitric oxide synthase (eNOS) activity [9].
T2D is multifactorial complex disease. Over the years, epigenetic mechanisms such as chromatin modifications, DNA methylation, and small non-coding RNAs have been demonstrated to influence the onset, glycemic memory, progression, and overt vascular complications in T2D [10, 11]. Insulin resistance is one of the common pathological conditions of T2D, affecting liver and peripheral tissues such as skeletal muscle, adipose, and vasculature. Global DNA methylation, as studied by Alu repeat analysis in T2D subjects in monozygotic twins, demonstrated correlation of altered CpG methylation with homeostasis model assessment (Homa-IR), suggesting an association between insulin resistance and DNA methylation [12]. Methylome analysis of human pancreatic islets revealed 1649 CpG sites in 853 genes to be differentially methylated in T2D individuals compared with normal individuals [13]. Genes such as CDK1B, EXO3CL, INS and PPARGC1A (involved in insulin secretion), and MEF2A, NDUFB6, and TFAM (related to insulin resistance) were epigenetically influenced in islets analyzed from T2D individuals [13]. Dynamic modifications in DNA methylation of matrix metallopeptidase-9 promoter due to an interplay between active methylation and hydroxymethylation by DNA methyltransferase 1 (DNMT1) and Tet2 in diabetic retinopathy was observed [14]. In vitro methylation of promoter of insulin gene (INS) led to suppression of gene expression [15], and further four CpG sites in the INS promoter were significantly methylated in islets obtained from T2D subjects leading to reduced insulin levels and was inversely correlated to HbA1c levels [16]. DNA methylation of insulin-like growth factor binding protein-1 (IGFBP1) gene was higher in blood cells of T2D subjects and concomitantly led to low serum levels of IGFBP1, which was correlated with insulin resistance [17].
T2D is characterized by comorbid conditions of vascular diseases including insulin resistance and endothelial dysfunction. Metabolic insulin resistance leads to decreased insulin response resulting in depleted glucose transport via GLUT4. However, in the context of endothelial cells lacking GLUT4 expression, insulin maintains vasodilator effects facilitating the production of nitric oxide via IR/IRS/PI3K/Akt signaling axis. Hence, vascular insulin resistance manifests in reduced nitric oxide production and subsequently leads to vasoconstriction and cardiovascular complications. Pro-inflammatory cytokines such as IL-6 have been demonstrated to disrupt insulin signaling leading to vascular insulin resistance. We hypothesized that IL-6 might induce epigenetic changes and modulate gene expression associated with insulin signaling. The present study was designed to identify key epigenetic signatures and its functional relevance during IL-6-induced vascular insulin resistance.
Methods
Cell culture
Human umbilical vein endothelial cells (HUVECs) and human dermal microvascular endothelial cells (HDMECs) were isolated from the umbilical vein and foreskin, respectively, after obtaining ethical clearance from the Institutional Ethics Committee, Kasturba Medical College, MAHE, Manipal. Prior written consent was obtained from all volunteers.
For isolation of HUVEC cells, umbilical veins were flushed with phosphate-buffered saline (PBS), followed by collagenase Type I (Sigma Chemicals, St. Louis, USA) treatment for 30 min. Cells were dislodged and cultured in gelatin (0.5%)-coated tissue culture plates. For HDMEC isolation, foreskin tissue was minced and digested with dispase-II (0.5 U/ml) (Sigma Chemicals) overnight and further treated with collagenase Type IA for 2 h. Endothelial cells were sorted using endothelial-specific surface antigen CD31 by magnetic beads (Miltenyi Biotec, Germany). HUVEC and HDMEC cells were cultured in endothelial cell growth medium with 2% Supplements (PromoCell GmbH, Heidelberg, Germany). Isolated cells were validated with cell-specific markers such as CD31 by flow cytometry and VE-Cadherin by immunocytochemistry. Both HUVEC and HDMEC cells were used at passage 3–7, with occasional screening for expression of CD31 and VE-cadherin. Experiments were carried out in tissue culture surfaces precoated with 0.5% gelatin.
Three-dimensional spheroid culture
Three-dimensional (3D) spheroid culture was prepared from endothelial cells using hanging drop method as described earlier [18]. Briefly, spheroids were prepared from endothelial cells (1000 cells/spheroid) in 20% methylcellulose and after 24 h, 50–100 spheroids were embedded in collagen gels (3 mg/ml) (Sigma Chemicals) and the culture medium was overlaid with activators as mentioned in figure legends. Spheroids were fixed with 4% paraformaldehyde for 12 h. Outgrowth of capillary-like structures from spheroids was evaluated by morphometric analysis of the number and total length of sprouts per spheroid, using ImageJ (NIH, USA) program. At least 20 spheroids from three parallel wells were analyzed for each experimental point.
Reagents and antibodies
Human recombinant insulin was purchased from Novo Nordisk Private Limited, Bangalore. Human recombinant IL-6, TNF-α, Nuclease P1, and methylcellulose were purchased from Sigma Chemicals. Collagen was purchased from SERVA Electrophoresis GmbH (Heidelberg, Germany). The following antibodies were procured from Cell Signaling Technology (Massachusetts, USA): anti-β-actin, anti-pAkt (Ser473), anti-Akt, anti-phospho eNOS (Ser177), eNOS, anti-IL-6 receptor, anti-DNMT1, anti-DNA methyltransferase 3A (DNMT3A), and anti-DNA methyltransferase 3B. Nitrocellulose membrane was purchased from Whatman plc (Little Chalfont, Chiltern, UK). Enhanced chemiluminescence (ECL) to develop western blotting images was from ThermoFisher Scientific (Massachusetts, USA). IL-6 enzyme-linked immunosorbent assay (ELISA) kit was from R&D Systems (Minneapolis, USA). DNase and calf intestinal phosphatase were purchased from New England Biolabs (Massachusetts, USA). The following nucleotide standards were purchased from MP Biomedicals (California, USA): Adenine, Guanine, Thymine, Cytosine, and 5-methylcytosine.
Neutrophil isolation
Neutrophils were isolated from peripheral blood of healthy individuals at Kasturba Hospital, Manipal. The protocol was approved by the Institutional Ethics Committee, Kasturba Medical College, Manipal. Blood cells were separated in a density gradient set up using Ficoll (GE Healthcare, Bangalore, India) and neutrophils were sedimented using 4% Dextran T500 (HiMedia, Mumbai, India) as described earlier. Purified neutrophils were resuspended in RPMI 1640 medium supplemented with 2% heat-inactivated human serum, 2 mM l-glutamine, 100 U/ml penicillin, and 100 μg/ml streptomycin (HiMedia).
Interleukin-6 ELISA assay
Neutrophils were seeded at a density of 1 × 106 /ml cells in 96-well plate. Neutrophil conditioned medium was collected after 3 and 16 h, centrifuged, and the accumulated IL-6 was measured using sandwich ELISA method according to the manufacturer’s instruction. Absorbance was read at 450 nm in an ELISA reader (TECAN, Austria).
Immunoblotting
Cells were briefly rinsed with PBS and lysed with lysis buffer containing 1% SDS in PBS along with protease inhibitor cocktail (Sigma Chemicals) and phosphatase inhibitors such as sodium orthovanadate (1 mM) and sodium fluoride (5 mM). Protein concentration was measured by Lowry’s method [19]. Equal amount of proteins was loaded on to 8% polyacrylamide gels, electrophoresed, and subsequently transferred to nitrocellulose membrane. Proteins were immunoblotted with corresponding primary antibody and detected with horseradish peroxidase-conjugated secondary antibodies and ECL. Images of immunoblots were analyzed by ImageJ (NIH).
DNA methyltransferase activity assay
Nuclear extracts were prepared from HUVEC cells as described previously [20]. Concentration of protein in nuclear extracts was measured by Lowry’s method [19]. Reaction mixture to measure DNA methyltransferase activity contained poly(dI-dC), protein extract (0.2 µg/μl), and [3H]-SAM (1.65 µCi) in a buffer containing Tris-HCl (200 mM, pH 7.6), EDTA (50 mM), dithiothreitol (10 mM), glycerol (50%), and protease inhibitor cocktail, and incubated at 37 °C for 3 h. DNMT1 activity was calculated by measuring incorporation of radiolabeled C3H3 in poly(dI-dC) by scintillation counter (Perkin Elmer 2900TR, USA).
Cell cycle analysis
HUVEC cells were seeded in six-well plates at the density of 100,000 cells/ml and treated with IL-6 as indicated in figure legends, and subsequently fixed with ethanol, washed with PBS and treated with RNAse (0.5 µg/ml). The cells were stained with propidium iodide (1 µg) and analyzed by flow cytometer (BD FACS Calibur, BD Biosciences, USA). Ten thousand events were collected from each sample. Data acquisition and cell cycle distribution was carried out using the BD CellQuest software (BD Biosciences).
Global DNA methylation analysis
DNA was isolated from endothelial cells by phenol-chloroform method. Initially, DNA was digested with DNase for 16 h at the concentration of 1 U/μg and further DNA fragments were converted into single strands by treating with Nuclease P1 (0.2 U/μl) overnight. Phosphate group was removed from nucleosides by treating with calf intestine phosphatase (0.5 U/μl) for 6 h. 5-Methylcytosine levels were measured by running the digested nucleotides along with standards in reverse phase high-performance liquid chromatography (HPLC) (Waters Alliance 2695, MA, USA) by using 0.1 M potassium hydrogen phosphate and methanol (9:1) in C18 column (Phenomenox, CA, USA). Waters 2487 dual λ-absorbance detector was used to detect the eluted nucleotides. The intensity graphs of eluted nucleotides were plotted using Empower Waters software (version 6.10).
Differential methylation hybridization
Differential methylation hybridization (DMH) was performed as described previously [21, 22]. Briefly, 2 μg of genomic DNA was subjected to digestion by MseI, which restricts genomic DNA into small fragments, but retains GC-rich CpG island fragments. The digested fragments were ligated with H-24 and H-12 universal linkers. Methylation sensitive endonucleases, HpaII and BstUI were used for further digestion of DNA samples and fragments were amplified by using H-24 primer. DNA isolated from IL-6 treated cells was labeled with Cy5 dye (Red), whereas the control DNA was labeled with Cy3 dye (Green). Equal amounts of IL-6-treated and control DNA were hybridized to a Human 244 K CpG island array slide (Agilent Technologies, CA, USA). After hybridization, the microarray slide was scanned using Agilent p/n G2565BA scanner. Differentially methylated probes were identified by calculating M-value [M = log2 (Cy5 /Cy3)]. Intensity of individual probe was calculated by L value [L = 0.5 × log2 (Cy5 × Cy3)]. Locally weighted scatterplot smoothing (LOWESS) was used to normalize both the values.
Bisulfite DNA sequencing
Bisulfite conversion of endothelial cell DNA was achieved using the EZ DNA Methylation-Gold kit (Zymo Research, Orange, CA, USA) according to the manufacturer’s protocol. In brief, about 1.5 μg of total genomic DNA from control and IL-6-treated ECs was treated with sodium bisulfite, purified according to the manufacturer’s protocol before bisulfite-specific PCR. The resultant amplicons were gel extracted from 1.5% agarose gel and the purified PCR products were sequenced using BigDye terminator cycle sequencing kit (ABI, USA) using Genetic Analyzer 3130 (Applied Biosystems, USA). Methylation at CpG positions were analyzed using ESME software [23].
Statistics and bioinformatics analysis
All the experiments were performed in triplicates at independent occasions. Unpaired two-tailed Student’s test and analysis of variance were performed with or without post-hoc (Bonferroni’s multiple comparison test) using Graphpad Prism. Data were represented along with SD and a p-value < 0.05 was considered as a significant change. Microarray data was extracted with Feature Extraction software (Agilent) with “no background correction” and signals of the probes were normalized with LOWESS normalization. The quality of data was assessed using the box plot, M–A plot, and RG density plot using R basic packages. Circular representation of probes was plotted using Circos graph [24]. Differentially methylated genes were tested for enrichment of specific gene ontology by DAVID [25]. Pathway and literature-based gene-disease association analysis were performed using Ingenuity Pathway Analysis (Qiagen, Germany). Differentially methylated CpG probes were screened for regulatory elements for STAT3 and E26 transformation-specific (ETS) family-binding proteins by using the UCSC genome browser from ENCODE database.
Primers
Primers for bisulfite DNA were designed using MethylPrimer Express v1.0 by following the standard guidelines. Primer list is provided as Supplementary Table-S1.
Results
Interplay between IL-6 and insulin results in stabilization of STAT3 activation and impeded eNOS phosphorylation
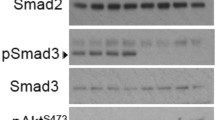
As a first step, based on dose-dependent assays (Fig S1A) and our earlier observations [18], we selected the concentrations of IL-6 and insulin showing highest STAT3Tyr705 and AktSer473 phosphorylation with 25 ng/ml and 100 nM, respectively. Treatment of endothelial cells with IL-6 and insulin, both individually and together, for 36 h did not affect cell survival (Fig S1B & Fig S1D). Further, we investigated the cooperative effects of IL-6 and insulin signaling pathways by measuring the kinetics of effector molecules associated with both the pathways in the presence of (a) IL-6, (b) insulin, and (c) IL-6 + insulin. In HUVECs, IL-6 induced robust phosphorylation of STAT3Tyr705 after 6 h of treatment (compared with basal levels) (Fig. 1a). Stimulation of endothelial cells with IL-6 resulted in a fourfold elevation of AktSer473 phosphorylation at 18 h, which increased to fivefold at 24 h before returning to basal levels at 36 h. IL-6 induced a steady increase in eNOSSer1177 phosphorylation levels at 6 h and decreased subsequently to control levels by 36 h (Fig. 1a). Insulin-activated profound phosphorylation of AktSer473 by 6 h, which was nearly eightfold higher compared with untreated control, and subsequently reduced to basal levels (Fig. 1b). In response to insulin, phosphorylation of eNOSSer1177 increased by threefold and subsequently reduced to steady state. Interestingly, IL-6 effects on the kinetics of phospho-eNOS did not mirror that of phospho-Akt, where IL-6-induced phosphorylation of eNOS was followed by phosphorylation of Akt (Fig. 1a, b). Hence, to understand the mechanism for IL-6-induced eNOS activation, we treated endothelial cells with IL-6 in the presence or absence of LY294002, a potential inhibitor of phosphatidyl inositol 3-kinase (PI3K). We observed that LY294002 inhibited IL-6-induced AktSer473 phosphorylation but not eNOSSer1177 phosphorylation. This suggested that IL-6-activated eNOS was independent of Akt phosphorylation (Fig S2). Further, we examined the influence of IL-6 and insulin together on endothelial cells. Any influence of both IL-6 and insulin on phosphorylation of AktSer473 and eNOSSer1177 was nullified when treated together (Fig. 1c). On the other hand, insulin along with IL-6 stabilized phospho-STAT3Tyr705 levels. Both IL-6 and insulin-induced expression of total STAT3 and these effects were abrogated when treated together (Fig S1F).

Influence of IL-6 and insulin alone or in combination on endothelial cells: HUVEC cells were serum deprived for 4 h and treated with IL-6 (25 ng/ml) (a) or insulin (100 nM) (b), or together (c) for indicated time periods and cells were lysed. Cell lysates were processed for immunoblotting and probed for phospho-AktSer473 and total Akt, phospho-STAT3Tyr705 and total STAT3, phospho eNOSSer117, and total eNOS and actin. Changes in phosphorylation/total effector ratios are expressed relative to respective initial time. Significant modulation in phosphorylation status of three independent experiments is denoted by asterisk, *p < 0.05, **p < 0.01, ***p < 0.001
Chronic IL-6 and hyperinsulinemic conditions lead to vascular insulin resistance
Next, we investigated the influence of insulin on endothelial cells pretreated with chronic IL-6. In response to insulin, we observed activation of AktSer473 (2-fold) and eNOSSer1177 (2.5-fold) phosphorylation and insulin effects were abrogated in cells pretreated with IL-6 (Fig. 2a). The IL-6 induced insulin resistance was dose dependent where cells treated with higher doses (25 ng/ml) did not respond to insulin as compared with lower doses (10 ng/ml) (Fig S1C). Previous studies have demonstrated hyperinsulinemia as one of the potential causes of insulin resistance [26]; we noticed that IL-6 stimulated AktSer473 and eNOSSer1177 phosphorylation (Fig. 1a). Therefore, we tested the influence of IL-6 on AktSer473 in cells pretreated with insulin. Hyperinsulinemic conditions were mimicked by treating the cells with insulin for 18 h and further stimulated with IL-6 for different time periods. We observed minimal activation (twofold) of AktSer473 by IL-6 at 6 h in insulin pretreated cells, which showed a nearly threefold increase at 12 h and subsequently returned to basal levels (Fig. 2b). This suggested chronic insulin treatment influences IL-6 effects. IL-6-induced insulin resistance was also supported by significant elevation (threefold compared with control at 36 h) in the phosphorylation of IRS1Ser636/639 (Fig. 2c), which is known to signal for proteasomal degradation of IRS1, leading to a state of insulin resistance in various cell types [27].

Effects of chronic IL-6 on insulin response in endothelial cells. a HUVECs were pretreated with or without IL-6 in serum free (SF) medium (basal ECGM + 0.5% FBS) for 36 h and followed by stimulation with insulin (100 nM) for 30 min. b HUVECs were treated with or without insulin (100 nM) in serum-deprived medium for 18 h and followed by IL-6 (25 ng/ml) treatment for indicated time points. Cell lysates from (a) and (b) were processed for immunoblotting and stained for pAkt Ser473 and total Akt. Significant differences in Phospo/total Akt ratios are denoted by *p < 0.05, **p < 0.01. c Cells were treated with IL-6 for indicated time points and lysates were processed for immunoblotting and stained for phospho and IRSSer636/639 and total IRS and actin. Experiments (a, b, c) were conducted three times independently. Spheroids from endothelial cells were prepared in methylcellulose (20%) and embedded in collagen gels in the presence or absence of IL-6 (25 ng/ml) for 8 h followed by inclusion of insulin (100 nM) for further 24 h. Data from four independent experiments is represented as percentage sprout length (d) and numbers (e) relative to control. f Representative micrographs of 3D cultures in collagen gels are provided. Sprouts are indicated by red arrows. Scale bar: 50 µm. g Cells were serum deprived for 4 h and treated with or without IL-6 (25 ng/ml) for 36 h and equal number of cells were seeded in plates precoated with matrigel for 6 h in the presence or absence of insulin (100 nM). Tube formation was assessed and represented as percentage number of tubes/field and significant differences of four independent experiments are denoted as **p < 0.01, ***p < 0.001. h Representative fields of tube formation is given. Scale bar: 200 µm
IL-6 abrogates insulin effects on angiogenesis
We examined the effect of insulin and IL-6 alone and together in in vitro angiogenesis models. Both insulin and IL-6 facilitated significant increase in number of sprouts and their length in collagen spheroid assays (Fig. 2d, e). In response to insulin, both sprout length and numbers declined in the spheroids pretreated with IL-6, indicating that insulin-resistant conditions exerted by IL-6 led to decrease in insulin-induced angiogenesis (Fig. 2f). Similar effects of insulin and IL-6 together on matrigel tube forming assays were observed, where both insulin and IL-6 individually induced significant tube formations, but failed to do so when treated together (Fig. 2g, h).
IL-6 reduces 5-methylcytosine levels in endothelial cells
Next, we conducted a series of experiments to elucidate constitutive and inducible changes in DNA methylation patterns in endothelial cells. HPLC-based analysis revealed basal levels of total methylcytosine contents to be 3.42 ± 0.1% and 3.6 ± 0.14% in HUVECs and HDMECs, respectively. Cells cultured in the presence of a DNA methyltransferase inhibitor, 5-azacytidine for 36 h, reduced methylcytosine levels by nearly 1%, yielding 2.45 ± 0.07% and 2.5 ± 0.13% in HUVECs and HDMECs, respectively (Fig. 3a). MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide)-based cytotoxicity assays revealed 1–3% cell death in 5-Azacytidine-treated cells (data not shown), suggesting cytosine methylation changes were not due to severe cytotoxic effects.

Global DNA hypomethylation in endothelial cells is associated with insulin resistance and DNA synthesis. a Genomic DNA isolated from cultures of HUVEC and HDMEC treated with or without 5-Azacytidine (3 µM) for 36 h was subjected to nucleoside isolation and processed for HPLC. Data from four independent experiments is cumulated. b Genomic DNA isolated from cultures of HUVEC treated with or without IL-6 (25 ng/ml) for 36 h was subjected to nucleoside isolation and processed for HPLC. Data from experiments conducted in quadruplicates is represented as % 5-methylcytosine. c HUVECs were treated with 5-Azacytidine (3 μM and 6 μM) for 36 h and cells were serum starved for 4 h and further treated with insulin (100 nM) for 30 min, and lysates were processed for immunoblotting. Fold change with respect to untreated control in levels of phospho-AktSer473, phospho-eNOSSer1147, and DNMT1 are shown with significant changes marked by asterisk **p < 0.01, ***p < 0.001. Representative blots from four individual experiments are shown. d Cell cycle analysis was performed in HUVEC cells treated with IL-6 (25 ng/ml) or 5-Azacytidine (3 µM) at indicated time points. Cells after treatment were fixed and stained with propidium iodide and processed for flow cytometry analysis. Percentage of cells in different phases of cell cycle and PI intensity are shown
To determine DNA methylation changes under the conditions of vascular insulin resistance, we investigated the influence of IL-6 on global DNA methylation. Neutrophils are one of the major sources of IL-6 and endothelial-neutrophil interactions are common during inflammation. Neutrophil cultures showed accumulation of significant levels of IL-6 in a time-dependent manner, which reached 1275 ± 106 pg/105 cells by 16 h of culture (Fig S3A). Subsequently, HUVECs were treated with neutrophil conditioned medium for 36 h and tested for methylcytosine content. Neutrophil conditioned medium (NCM) led to significant decrease in methylcytosine levels (3.8% vs. 2.2% in treated) (Fig S3B), indicating that inflammatory response induces genome-wide hypomethylation, which might be responsible for expression of pro-inflammatory genes. Further inclusion of neutralizing antibody of IL-6R in the presence of NCM, increased the DNMT1 protein levels (Fig S3C), suggesting that IL-6 present in NCM might be one of the cytokines responsible for modulation of DNA methylation during inflammation. Next, we examined the direct effect of IL-6 on methylcytosine content in HUVECs (Fig. 3b). Kinetic analysis suggested that IL-6 induced a transient increase in methylated cytosine levels (3.87 ± 0.02%) at 24 h and a significant decline of 0.7% at 36 h compared with control (3.15 ± 0.05 vs. 2.62 ± 0.12%). IL-6-induced demethylation of DNA was within the modifiable range of 1% that was observed with 5-Azacytidine treatment.
IL-6-induced vascular insulin resistance is paralleled with DNA hypomethylation and DNA synthesis
Our earlier experiments indicated that IL-6-induced insulin resistance was associated with kinetics of DNA hypomethylation. Hence, to examine possible inter-relationship between insulin resistance and DNA methylation, we tested the consequence of pharmacological inhibition of DNA methyltransferases on insulin response. We observed that cells cultured in the presence of 3 and 6 μM of 5-Azacytidine did not respond to insulin for AktSer473 (Fig. 3c) and eNOSSer1177 (Fig. 3d) phosphorylation, which was in parallel with degradation of DNMT1 (Fig. 3e). This suggested that vascular insulin resistance was associated with DNA hypomethylation. As DNA methylation is coupled with DNA replication, we investigated cell cycle status in IL-6 treated endothelial cells in a time-dependent manner. IL-6 stimulated DNA synthesis (12.27% vs. 10.81% of control) at 12 h, which further increased to 15% and sustained for 36 h. Although 12.09% control cells entered synthetic phase at 24 h, which further declined to 8.7% at 36 h. DNA methyltransferase inhibitor 5-Azacytidine treatment also induced and maintained DNA synthesis until 36 h though not as potent as IL-6 (Fig. 3f).
IL-6 significantly alters endothelial epigenome
Next, we performed CpG microarray analysis encompassing 237,220 probes to identify differentially methylated genes responsible for IL-6-induced alterations in gene expression patterns under insulin-resistant conditions. Global changes and localization in DNA methylation pattern has been illustrated as density graph (Fig. 4a) and circos diagram (Fig. 4b). We observed significant changes in methylation patterns across the genome. Statistical analysis of microarray data indicated a total of 4390 probes showing > 1.5-fold change compared with untreated control. These probes were further annotated based on their genomic location to their nearest genes and classified into promoter region, 3′-untranslated region, gene body, and intergenic space. Major ratio of probes with significant modulation of DNA methylation was distributed in gene body, followed by promoter regions (Fig S5A). When precisely analyzed, we found 2438 hypomethylated probes pertaining to 199 genes and 1892 hypermethylated probes associated with 98 genes. In both the cases, the probes were mostly accumulated at transcription start sites, suggesting that changes in DNA methylation of these regions might strongly affect gene transcription (Fig. 4a). Probes located in promoter regions were annotated and further screened for the presence of putative regulatory elements for endothelial cell-specific ETS family transcription factors (Fig S5B). As STAT3 is attributed in modulating angiogenesis in vascular pathologies [28], we stratified our data based on localization of STAT3-binding elements (Table 1). Among the 297 differentially methylated genes, 9 were selected for further validation based on number of probes per gene, fold-change level (Table 2), network analysis (Fig S5C, S5D), pathway analysis, gene ontology (Table 3), and literature survey.

Influence of IL-6 on endothelial methylome. Density plot shows the distance of hypomethylated (a) probes and hypermethylated sequences (b) from TSS. c Circos plot shows distribution and variation of methylation patterns of CpG probes in endothelial cells. Green dots represent hypermethylated genes and red dot represent hypomethylated genes. Significant alterations (more than 1.5-fold) of DNA methylation in response to IL-6 is represented with thicker stroke. d Graphical representation of differentially methylated CpG sites in response to IL-6. Genomic DNA extracted from untreated and IL-6-treated HUVEC were subjected to PCR after bisulfite conversion. Methylation pattern was obtained by direct DNA sequencing using bisulfite-specific primers. Data from three independent experiments are represented as percentage methylated CG in the amplicon pool. Ticks in X axis denotes CpG sites. UCSC CpG track is shown to indicate position CpG island in chromosome for each gene
Validation of differentially methylated genes was carried out by direct bisulfite DNA sequencing method (Fig. 4c). Among the hypomethylated genes, RPS6KA2, PIK3R2, FOXD3, EXOC7, and MAP3K8 were considered for validation, of which, RPS6KA2 and PIK3R2 showed maximum influence of IL-6 on promoter hypomethylation. In promoter fragment of RPS6KA2, 8/30 CpG sites and in PIK3R2, 17/35 CpG sites were significantly demethylated in response to IL-6. CpG sites of FOXD3, EXOC7, and MAP3K8 genes were moderately hypomethylated, which showed demethylation of 7/15, 3/18, and 3/28 CpG sites, respectively. For hypermethylated genes, IGF1R, ITPKB, EPHA6, and FOXC2 were validated. We observed significant hypermethylation on 8 CpG sites and 19 CpG sites in FOXC2 and EPHA6, respectively in IL-6-treated cells. ITPKB and IGF1R showed moderate hypermethylation with seven and nine CpG sites, respectively.
Further, we selected six genes and studied kinetics of CpG methylation at 12, 24, and 36 h post IL-6 treatment (Fig S6). RPS6KA2 and PIK3R2 genes showed delayed response to IL-6 for demethylation of CpG sites, which exhibited alterations only after 36 h. On the other hand, CpG sites in EXOC7 promoter region demethylated at 12 h and showed sustained hypomethylation until 36 h. Promoter of MAP3K8 exhibited varied methylation pattern over different time points but subsequently showed complete hypomethylation at 36 h. CpG sites in promoter region of ITPKB showed hypermethylation only after 36 h and methylation of CpG residues in FOXC2 promoter started at 24 h and increased at 36 h.
IL-6 modulates DNMT1 and DNMT3B but not DNMT3A protein levels
Time-dependent assays indicated that IL-6 significantly modulated DNMT1 (Fig. 5a) and DNMT3B (Fig. 5c) but not DNMT3A (Fig. 5b) protein levels. IL-6 transiently increased DNMT1 (threefold) and DNMT3B (approximately twofold) levels by 6 h, gradually started declining to basal levels at 24 h and further decreased at 36 h. However, we did not find any influence of IL-6 on DNMT3A levels. Concentration-dependent assays indicated a 50% decline in DNMT1 levels in response to 10 ng/ml IL-6 and a further 75% decrease with 25 ng/ml (Fig. 5d). However, 25 ng/ml IL-6 was required to downregulate protein levels of DNMT3B (Fig. 5f). In line with the kinetic analysis, DNMT3A levels remained unaltered in response to IL-6 (Fig. 5e). To elucidate the functional consequence of decreased DNMT1 and DNMT3B levels in response to IL-6 in endothelial cells, we measured DNMT enzyme activity by analyzing transfer of radiolabeled methyl group from S-adenosyl methionine to DNA. IL-6 treatment for 36 h significantly (p < 0.05) reduced DNMT activity in nuclear extracts of HUVECs and HMDECs (Fig. 5g). IL-6 effects on reduced DNMT1 levels were further confirmed by inclusion of neutralizing antibody against IL-6R. Influence of IL-6 (10 and 25 ng/ml) on DNMT1 levels were abolished significantly upon inclusion of IL-6R neutralizing antibody for 36 h (Fig. 5h), confirming that IL-6 has an important role in the regulation of DNMT1 expression. To investigate whether IL-6 modulates DNMT1 protein levels by effecting stabilization, we measured DNMT1 levels in the cells in the presence of MG132, a potent proteasome inhibitor. IL-6 reduced DNMT1 levels in 24 h but MG132 rescued the degradation and brought back to higher than control levels (Fig. 5i), suggesting involvement of IL-6 in maintaining stability of DNMT1. To address whether downstream effects of IL-6 on DNMT1 and DNMT3B changes were due to modulation in IL-6 receptor (IL-6R) per se, we examined expression levels of IL-6R in a time-course experiment. However, IL-6R expression levels remained unaltered in response to IL-6 treatment (Fig S4).

IL-6 modulates DNMT isoforms. Kinetics of DNMT isoform protein expression in response to IL-6 (25 ng/ml) in HUVEC cells were measured after treating for indicated time points. Cell lysates were processed for immunoblotting for the detection of DNMT1 (183 kDa) (a) and DNMT3A (120 kDa) (b), and DNMT3B (100 kDa) (c), and actin (44 kDa). Data from three independent experiments is represented as fold change of untreated control. HUVEC cells were treated with IL-6 (10 and 25 ng/ml) for 36 h and cell lysates were immunoblotted for DNMT1 (d), DNMT3A (e), and DNMT3B (f). Representative blots and graphical representation are provided. g HUVEC and HDMEC cells were treated with or without IL-6 for 36 h and nuclear extracts were tested for DNMT activity by 3H-labeled S-adenosyl methionine using poly-dI-dC (30 µg/ml) as substrate. DNMT1 activity was measured in scintillation counter and data is represented as counts per minute. *p < 0.05 indicates statistical significance. HUVEC were pretreated with or without IL-6R neutralizing antibody (1 µg/ml) (h) or MG132 (5 µM) (i) in presence of IL-6 (25 ng/ml) for 36 h. Cell lysates were processed for western analysis and probed for DNMT1 and actin. Data are represented as fold change of untreated control. Statistical significance calculated from three different experiments is denoted by *p < 0.05, **p < 0.01
Discussion
Our study was aimed at dissecting the intricate role of inflammatory mediators on insulin signaling and identifying underlying epigenetic changes in the vascular endothelium. Using 2D and 3D endothelial cell culture models, we demonstrated the role of IL-6 in manifestation of vascular insulin resistance. Global DNA methylation patterns under the influence of IL-6 indicated possible inter-relationship of impeded insulin response, DNA hypomethylation, and DNA synthesis. CpG microarray analysis revealed that IL-6 significantly altered promoter DNA methylation patterns of genes involved in insulin signaling and angiogenesis. Finally, mechanistic studies showed IL-6 facilitated modulation of DNMT1 and DNMT3B protein levels, providing a causal link for IL-6-induced differential DNA methylation and its possible role in deregulation of insulin signaling axis (Fig. 6).

Hypothetical model showing interplay between IL-6 and insulin signaling pathways as a consequence of DNMT1 and DNMT3B degradation, and altering DNA methylation changes in genes involved in insulin signaling and angiogenesis
Although IL-6 and insulin alone induced activation of AktSer473, STAT3Tyr705, eNOSSer1177 phosphorylation, and angiogenesis in vitro, the magnitude and kinetics varied between both inducers. The effects were abrogated when IL-6 and insulin were treated together, suggesting a state of insulin resistance in presence of IL-6. Our observation of IL-6 effects on insulin resistance was in agreement with earlier studies that demonstrated IL-6 treatment in endothelial cells impaired insulin effects on IRS1/Akt/eNOS activity in Erk1/2- and JNK-dependent manner [9]. Further, IL-6-induced eNOS activation was PI3K/Akt independent. Similar observations were made in bovine aortic endothelial cells, where thrombin-induced eNOS was calcium dependent and Akt independent [29]. AMPK has been shown as a potential kinase to phosphorylate eNOS in endothelial cells [30]. In another study, IL-6 impaired insulin-induced eNOS activation and nitric oxide production that were associated with increased expression of TNF-α in human aortic endothelial cells [31]. Hyperinsulinemia is also known to induce insulin resistance [26] and circulating IL-6 has been correlated with hyperinsulinemia and insulin resistance [32]. In a time-course analysis, IL-6 showed an additive effect on Akt phosphorylation in insulin pretreated cells at an early time point, followed later by complete reduction, suggesting that hyperinsulinemia along with IL-6 caused disruption in insulin signaling.
HPLC analysis showed 3.42 ± 0.1% and 3.6 ± 0.14% of basal DNA methylation levels in HUVEC and HDMEC cells, respectively. Studies have indicated the dynamics in cell type-specific DNA methylation. A microarray-based analysis of 82 human cell lines and tissues showed 3–15% of global DNA methylation, which significantly varied between normal and transformed cells [33]. Inflammatory mediators have been demonstrated as potential modulators of DNA methylation and subsequent cause for altered gene expression [34].
Epigenetic mechanisms including DNA methylation have been implicated in endothelial dysfunction in T2D [35] and development and progression of atherosclerosis [36]. Global DNA analysis of atherosclerosis plaques indicated hypomethylation [37] and was associated with decreased DNMT1 expression in human carotid plaques [38]. In our study, global DNA methylation analysis and microarray data of endothelial cells treated with IL-6 showed significant DNA hypomethylation. Endothelial cell-specific methylome analysis has been carried out with different stimulants such as high glucose [39] and shear stress [40]. In aortic endothelial cells, hyperglycemia induced significant alterations in both DNA methylation and chromatin modifications, resulting in changes in expression of genes related to pro-inflammatory pathway, endothelial dysfunction, and atherogenesis [39]. Disturbed flow upon employing carotid ligation surgery in murine model and oscillatory shear stress in cultured endothelial cells led to increased expression of DNMT1 and subsequent microarray analysis indicated hypermethylation of mechanosensitive genes involved in induction of atherosclerosis [40].
Knowledge of regulation and function of DNMT isoforms in vascular endothelial cells is sparse. In brain endothelial cells, homocysteine increased the expression of DNMT1 and DNMT3A, and simultaneously decreased DNMT3B levels and caused mitochondrial toxicity [41]. Krause et al. [42] showed the role of DNMT1 in upregulating eNOS expression in endothelial cells isolated from intrauterine growth restricted fetuses, which resulted in altered response to hypoxia. Mesenchymal stem cells differentiated into arterial endothelial cells and induced angiogenesis upon treating with DNMT inhibitor 5-aza-dc and when knocked out with DNMT1 and DNMT3A genes [43]. We observed transient increase, followed by a decline, in both DNMT1 and DNMT3B in response to IL-6, whereas DNMT3A remained unaltered. Earlier studies have demonstrated role of IL-6 in regulating DNMT1 function [44]. IL-6 induced Akt dependent phosphorylation of nuclear localization signal and subsequently increased nuclear translocation of DNMT1 [45]. In pancreatic cancer cells, IL-6-induced STAT3 recruited DNMT1 to the promoter site of SOCS3 and inhibited its transcription via DNA methylation [46].
Enhanced activity of DNMT1 was demonstrated during the S-phase of cell cycle [47]. Our analysis indicated that IL-6 increased the population of S-phase cells, which was kinetically associated with DNA hypomethylation, suggesting that reduced replication-associated DNMT1 levels may be due to activation of certain genes by DNA hypomethylation. 5-Azacytidine has been shown to induce proteasomal degradation of DNMT1 [48], which reduced its activity leading to DNA hypomethylation. Increased S-phase cell population was associated with decreased DNMT1 and DNMT3B levels upon treatment with 5-azacytidine and thus followed similar pattern of IL-6 effects, suggesting that DNA methylation changes due to IL-6 might be transmitted to next generation.
Our study provides novel insights into IL-6 biology modulating DNMT1 and DNMT3B in vascular endothelial cells and thereby regulating the expression of genes involved in insulin signaling and angiogenesis by promoter DNA methylation. Validation of the methylation signatures of associated genes and their function in T2D warrants animal and clinical models. Abnormal endothelial function due to insulin resistance is a known risk factor for the development of diabetes associated micro and macro vascular diseases. Novel therapeutic interventions that restore epigenetic changes, thereby ameliorating endothelial function, might facilitate lowering of the cardiovascular risk in diabetes mellitus.
References
Shoelson SE, Lee J, Goldfine AB. Inflammation and insulin resistance. J Clin Invest. 2006;116:1793–801.
Donath MY, Shoelson SE. Type 2 diabetes as an inflammatory disease. Nat Rev Immunol. 2011;11:98–107.
Pradhan AD, Manson JE, Rifai N, et al. C-reactive protein, interleukin 6, and risk of developing type 2 diabetes mellitus. JAMA. 2001;286:327–34.
Lowe G, Woodward M, Hillis G, et al. Circulating inflammatory markers and the risk of vascular complications and mortality in people with type 2 diabetes and cardiovascular disease or risk factors: the ADVANCE study. Diabetes. 2014;63:1115–23.
Klover PJ, Zimmers TA, Koniaris LG, et al. Chronic exposure to interleukin-6 causes hepatic insulin resistance in mice. Diabetes. 2003;52:2784–9.
Senn JJ, Klover PJ, Nowak IA, et al. Interleukin-6 induces cellular insulin resistance in hepatocytes. Diabetes. 2002;51:3391–9.
Senn JJ, Klover PJ, Nowak IA, et al. Suppressor of cytokine signaling-3 (SOCS-3), a potential mediator of interleukin-6-dependent insulin resistance in hepatocytes. J Biol Chem. 2003;278:13740–6.
Rotter V, Nagaev I, Smith U. Interleukin-6 (IL-6) induces insulin resistance in 3T3-L1 adipocytes and is, like IL-8 and tumor necrosis factor-alpha, overexpressed in human fat cells from insulin-resistant subjects. J Biol Chem. 2003;278:45777–84.
Andreozzi F, Laratta E, Procopio C, et al. Interleukin-6 impairs the insulin signaling pathway, promoting production of nitric oxide in human umbilical vein endothelial cells. Mol Cell Biol. 2007;27:2372–83.
Rönn T, Ling C. DNA methylation as a diagnostic and therapeutic target in the battle against Type 2 diabetes. Epigenomics. 2015;7:451–60.
Keating ST, Plutzky J, El-Osta A. Epigenetic changes in diabetes and cardiovascular risk. Circ Res. 2016;118:1706–22.
Zhao J, Goldberg J, Bremner JD, et al. Global DNA methylation is associated with insulin resistance: a monozygotic twin study. Diabetes. 2012;61:542–6.
Dayeh T, Volkov P, Salö S, et al. Genome-wide DNA methylation analysis of human pancreatic islets from type 2 diabetic and non-diabetic donors identifies candidate genes that influence insulin secretion. PLoS Genet. 2014;10:e1004160.
Kowluru RA, Shen Y, Mishra M. Dynamic DNA methylation of matrix metalloproteinase-9 in the development of diabetic retinopathy. Lab Investig J Tech Methods Pathol. 2016;96:1040–9.
Kuroda A, Rauch TA, Todorov I, et al. Insulin gene expression is regulated by DNA methylation. PLoS ONE. 2009;4:e6953.
Yang BT, Dayeh TA, Kirkpatrick CL, et al. Insulin promoter DNA methylation correlates negatively with insulin gene expression and positively with HbA1c levels in human pancreatic islets. Diabetologia. 2011;54:360–7.
Gu T, Gu HF, Hilding A, et al. Increased DNA methylation levels of the insulin-like growth factor binding protein 1 gene are associated with type 2 diabetes in Swedish men. Clin Epigenetics. 2013;5:21.
Philippova M, Joshi MB, Pfaff D, et al. T-cadherin attenuates insulin-dependent signalling, eNOS activation, and angiogenesis in vascular endothelial cells. Cardiovasc Res. 2012;93:498–507.
Lowry OH, Rosebrough NJ, Farr AL, et al. Protein measurement with the Folin phenol reagent. J Biol Chem. 1951;193:265–75.
Joshi MB, Ivanov D, Philippova M, et al. A requirement for thioredoxin in redox-sensitive modulation of T-cadherin expression in endothelial cells. Biochem J. 2008;416:271–80.
Yan PS, Potter D, Deatherage DE, et al. Differential methylation hybridization: profiling DNA methylation with a high-density CpG island microarray. Methods Mol Biol Clifton NJ. 2009;507:89–106.
Bhat S, Kabekkodu SP, Noronha A, et al. Biological implications and therapeutic significance of DNA methylation regulated genes in cervical cancer. Biochimie. 2016;121:298–311.
Lewin J, Schmitt AO, Adorján P, et al. Quantitative DNA methylation analysis based on four-dye trace data from direct sequencing of PCR amplificates. Bioinforma Oxf Engl. 2004;20:3005–12.
Krzywinski M, Schein J, Birol İ, et al. Circos: an information aesthetic for comparative genomics. Genome Res. 2009;19:1639–45.
Huang DW, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009;37:1–13.
Catalano KJ, Maddux BA, Szary J, et al. Insulin resistance induced by hyperinsulinemia coincides with a persistent alteration at the insulin receptor tyrosine kinase domain. PLoS ONE. 2014;9:e108693.
Ozes ON, Akca H, Mayo LD, et al. A phosphatidylinositol 3-kinase/Akt/mTOR pathway mediates and PTEN antagonizes tumor necrosis factor inhibition of insulin signaling through insulin receptor substrate-1. Proc Natl Acad Sci USA. 2001;98:4640–5.
Becerra A, Rojas M, Vallejos A, et al. Endothelial fibrosis induced by suppressed STAT3 expression mediated by signaling involving the TGF-β1/ALK5/Smad pathway. Lab Investig J Tech Methods Pathol. 2017;97:1033–46.
Thors B, Halldórsson H, Jónsdóttir G, et al. Mechanism of thrombin mediated eNOS phosphorylation in endothelial cells is dependent on ATP levels after stimulation. Biochim Biophys Acta. 2008;1783:1893–902.
Thors B, Halldórsson H, Thorgeirsson G. Thrombin and histamine stimulate endothelial nitric-oxide synthase phosphorylation at Ser1177 via an AMPK mediated pathway independent of PI3K-Akt. FEBS Lett. 2004;573:175–80.
Yuen DYC, Dwyer RM, Matthews VB, et al. Interleukin-6 attenuates insulin-mediated increases in endothelial cell signaling but augments skeletal muscle insulin action via differential effects on tumor necrosis factor-alpha expression. Diabetes. 2009;58:1086–95.
Franckhauser S, Elias I, Rotter Sopasakis V, et al. Overexpression of Il6 leads to hyperinsulinaemia, liver inflammation and reduced body weight in mice. Diabetologia. 2008;51:1306–16.
Varley KE, Gertz J, Bowling KM, et al. Dynamic DNA methylation across diverse human cell lines and tissues. Genome Res. 2013;23:555–67.
Stenvinkel P, Karimi M, Johansson S, et al. Impact of inflammation on epigenetic DNA methylation - a novel risk factor for cardiovascular disease? J Intern Med. 2007;261:488–99.
Prattichizzo F, Giuliani A, Ceka A, et al. Epigenetic mechanisms of endothelial dysfunction in type 2 diabetes. Clin Epigenetics. 2015;7:56.
Hai Z, Zuo W. Aberrant DNA methylation in the pathogenesis of atherosclerosis. Clin Chim Acta. 2016;456:69–74.
Aavik E, Lumivuori H, Leppänen O, et al. Global DNA methylation analysis of human atherosclerotic plaques reveals extensive genomic hypomethylation and reactivation at imprinted locus 14q32 involving induction of a miRNA cluster. Eur Heart J. 2015;36:993–1000.
Greißel A, Culmes M, Napieralski R, et al. Alternation of histone and DNA methylation in human atherosclerotic carotid plaques. Thromb Haemost. 2015;114:390–402.
Pirola L, Balcerczyk A, Tothill RW, et al. Genome-wide analysis distinguishes hyperglycemia regulated epigenetic signatures of primary vascular cells. Genome Res. 2011;21:1601–15.
Dunn J, Qiu H, Kim S, et al. Flow-dependent epigenetic DNA methylation regulates endothelial gene expression and atherosclerosis. J Clin Invest. 2014;124:3187–99.
Kamat PK, Kalani A, Tyagi SC, et al. Hydrogen sulfide epigenetically attenuates homocysteine-induced mitochondrial toxicity mediated through NMDA receptor in mouse brain endothelial (bEnd3) cells. J Cell Physiol. 2015;230:378–94.
Krause BJ, Costello PM, Muñoz-Urrutia E, et al. Role of DNA methyltransferase 1 on the altered eNOS expression in human umbilical endothelium from intrauterine growth restricted fetuses. Epigenetics. 2013;8:944–52.
Zhang R, Wang N, Zhang L-N, et al. Knockdown of DNMT1 and DNMT3a promotes the angiogenesis of human mesenchymal stem cells leading to arterial specific differentiation. Stem Cells Dayt Ohio. 2016;34:1273–83.
Foran E, Garrity-Park MM, Mureau C, et al. Upregulation of DNA methyltransferase–mediated gene silencing, anchorage-independent growth, and migration of colon cancer cells by interleukin-6. Mol Cancer Res. 2010;8:471–81.
Hodge DR, Cho E, Copeland TD, et al. IL-6 enhances the nuclear translocation of DNA cytosine-5-methyltransferase 1 (DNMT1) via phosphorylation of the nuclear localization sequence by the AKT kinase. Cancer Genom Proteom. 2007;4:387–98.
Huang L, Hu B, Ni J, et al. Transcriptional repression of SOCS3 mediated by IL-6/STAT3 signaling via DNMT1 promotes pancreatic cancer growth and metastasis. J Exp Clin Cancer Res. 2016;35:27.
Leonhardt H, Page AW, Weier HU, et al. A targeting sequence directs DNA methyltransferase to sites of DNA replication in mammalian nuclei. Cell. 1992;71:865–73.
Ghoshal K, Datta J, Majumder S, et al. 5-Aza-deoxycytidine induces selective degradation of DNA methyltransferase 1 by a proteasomal pathway that requires the KEN box, bromo-adjacent homology domain, and nuclear localization signal. Mol Cell Biol. 2005;25:4727–41.
Acknowledgements
We acknowledge TIFAC, Government of India, and Manipal Academy of Higher Education Manipal, India, for infrastructure. AB was supported by fellowship from Dr. TMA Pai Foundation, Manipal, and Indian Council of Medical Research, Government of India. We thank Dr Harish Rotti for technical assistance in microarray analysis and Drs Lavanya Rai and Prashant Adiga, Department of Obstetrics and Gynecology, Kasturba Medical College, Manipal, for clinical advice and selecting umbilical cords for isolation of endothelial cells. We thank Dr. T. G. Vasudevan for critically reading the manuscript and helpful advice.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Electronic supplementary material
Rights and permissions
About this article

Cite this article
Balakrishnan, A., Guruprasad, K.P., Satyamoorthy, K. et al. Interleukin-6 determines protein stabilization of DNA methyltransferases and alters DNA promoter methylation of genes associated with insulin signaling and angiogenesis. Lab Invest 98, 1143–1158 (2018). https://doi.org/10.1038/s41374-018-0079-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41374-018-0079-7
