
Key Points
-
Small RNAs (sRNAs) are used by plants, nematodes and arthropods in cellular defence against viruses.
-
In chordates, sRNAs were replaced with a protein-based system to inhibit viral replication.
-
microRNAs (miRNAs) are eukaryotic sRNAs that function to fine-tune host gene expression.
-
In chordates, miRNAs do not inhibit RNA viruses, and RNA viruses do not inhibit miRNAs.
-
RNA viruses can be engineered to interact with the host miRNA pathways, and miRNAs can be used to control viral tropism.
-
RNA viruses can also be used to deliver sRNAs.
Abstract
Gene silencing by small RNAs (sRNAs) occurs in all three domains of life. In recent years, our appreciation of the diverse functions of sRNAs has increased, and we have identified roles for these RNAs in cellular differentiation, fitness and pathogen defence. Interestingly, although plants, nematodes and arthropods use sRNAs to combat viral infections, chordates have replaced this defence strategy with one based exclusively on proteins. This limits chordate use of sRNAs to the silencing of genome-encoded transcripts and has resulted in viruses that do not perturb sRNA-related cellular processes. This evolutionary phenomenon provides an opportunity to exploit the pre-existing chordate sRNA pathways in order to generate a range of virus-based biological tools. Here, I discuss the relationship between sRNAs and RNA viruses, detail how microRNA expression can be harnessed to control RNA viruses and describe how RNA viruses can be designed to deliver sRNAs.
Similar content being viewed by others


Main
In all three domains of life, small RNAs (sRNAs; defined here as RNAs that are less than 60 nucleotides (nt) in length) function both in cellular defence against foreign nucleic acid and in the post-transcriptional regulation of endogenous transcripts. Through sequence-specific interactions, sRNAs can engage target transcripts, including mRNAs and pathogen-derived genomic RNAs, and influence the integrity and/or functionality of these targets. In recent years, there has been a significant expansion in our knowledge of the many functions of sRNAs, and it is now clear that sRNA-controlled genetic silencing systems are universal in biology, although their physiological uses can be unique.
The molecular biology of sRNAs demonstrates substantial species specificity, but in general sRNAs either suppress the expression of foreign genetic material or fine-tune their own expression through a process generally termed RNAi1. For instance, clustered regularly interspaced short palindromic repeat RNAs (crRNAs) are a defence system used by bacteria to restrict infection by phages; PIWI-interacting RNAs (piRNAs) are used by multicellular organisms as a germline defence against transposable elements and foreign genetic material2,3,4; and virus-derived interfering RNAs (viRNAs) constitute the antiviral innate immune system in nematodes, arthropods and plants5,6,7 (Table 1). By contrast, microRNAs (miRNAs), a class of sRNA that is conserved in eukaryotes, are similar to viRNAs in size but are thought to guide and reinforce cellular identity by post-transcriptional silencing of endogenous mRNA (Box 1) rather than provide a cellular defence against foreign nucleic acid8. miRNAs represent a unique class of sRNA and one that demonstrates significant evolutionary conservation; the genes encoding these sRNAs make up more than 2% of the genes in nematodes, arthropods and chordates and are predicted to influence more than 60% of a given host transcriptome9.
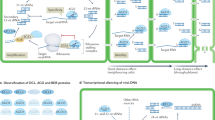
The biogenesis pathways of sRNAs do contain some common processing steps but are both complex and species specific, and a detailed discussion is beyond the scope of this Review1,10,11,12. In general, viRNAs are the product of a Dicer family protein, an RNase III endonuclease that processes double-stranded RNA (dsRNA) substrates from various origins, including viral replication intermediates and imperfect RNA hairpins that form in the viral genome12 (Fig. 1). Endonuclease substrates can derive directly from the viral genome or can be generated by a host RNA-dependent RNA polymerase (RdRp) following recognition of viral single-stranded nucleic acid13. When generated, viRNAs are loaded into a multisubunit RNA-induced silencing complex (RISC), where they mediate sequence-specific cleavage of the viral RNA within the cell. Furthermore, viRNAs can act in trans. In plants and nematodes, viRNA generation can be amplified by an RdRp, enabling systemic delivery — an activity that has been found, in some cases, to protect offspring from subsequent infection14,15,16. Interestingly, the use and systemic spread of viRNAs has also been observed in arthropods, albeit in an RdRp-independent manner; however, the mechanism remains incompletely understood17.

The response to viral infection differs dramatically when comparing chordates with nematodes, arthropods or plants. Following entry and replication, viruses commonly accumulate a number of RNA intermediates that include double-stranded RNA (dsRNA), single-stranded RNA (ssRNA) containing a 5′ triphosphate (3pRNA), or ssRNA with unusual sequence characteristics that mark it as foreign to the cell. In chordates, recognition of these pathogen-associated molecular patterns (PAMPs) occurs through pattern recognition receptors (PRRs) in the cytoplasm of the cell or through Toll-like receptors (TLRs) in endosomes. Receptor engagement results in the assembly of many adaptor protein complexes and culminates in the activation of a large kinase complex composed of four distinct inhibitor of nuclear factor-κB (NF-κB) kinase kinase (IKK kinase) family members. These kinases coordinate the activation of a number of transcription factors, including IFN regulatory factors (IRFs) and NF-κB; these factors then initiate the transcriptional induction of type I interferons (IFNs). Type I IFNs are a family of cytokines that can function in both an autocrine and paracrine manner to induce a collection of IFN-stimulated genes (ISGs), which create a cellular environment that is not amenable to further viral replication. In contrast to chordates, virus-derived RNA PAMPs in nematodes, arthropods and plants are used directly to generate virus-derived interfering RNAs (viRNAs), which can be loaded into an RNA-induced silencing complex (RISC) to mediate silencing and cleavage of viral RNA. Generation of viRNAs is mediated by an RNase III endonuclease — either a Dicer (DCR) or Dicer-like (DCL) family member — along with species-specific cofactors. In addition to primary viRNA production, nematodes and plants encode RNA-dependent RNA polymerase (RdRp), which mediates the amplification of viRNAs.
In contrast to viRNAs, miRNAs are derived from RNA polymerase II-generated transcripts that are encoded either as independent transcriptional units or in the introns of pre-mRNAs18,19,20. miRNA production begins with cleavage of the primary miRNA transcript (pri-miRNA) by the nuclear microprocessor complex, which is composed of the RNase III protein Drosha and its dsRNA-binding protein DGCR811. Cleavage of the pri-miRNA results in a 60–70 nt hairpin termed the pre-miRNA; this pre-miRNA contains a 2 nt 3′ overhang that is required for export into the cytoplasm by exportin 5 (Refs 21, 22). Cytoplasmic pre-miRNAs are recognized and processed by Dicer in a manner similar to viRNAs23,24,25,26. As with viRNAs, Dicer cleavage results in an RNA duplex in which one strand, referred to as the guide strand, is ultimately retained in the RISC27,28. This RISC-loaded guide strand is responsible for binding to target mRNAs in a sequence-dependent manner.
One noteworthy distinction between miRNA- and viRNA-mediated RISC function is the interaction of RISC with, and subsequent impact on, target RNAs. Whereas a viRNA-loaded RISC binds with perfect complementarity, effectively eliminating target expression, an miRNA-loaded RISC generally binds RNA with only partial complementarity and fine-tunes target expression, conferring less than twofold changes in protein expression29,30,31,32,33,34,35. The sequence elements that render an mRNA susceptible to the mild suppression of an miRNA are generally referred to as miRNA response elements; these elements are typically found in the ORF or 3′ UTR of the mRNA and have perfect complementarity with bases 2–8 of the miRNA guide strand, commonly referred to as the seed region of the miRNA36,37. By contrast, base pairing outside the seed region often occurs with only partial complementarity, a defining feature of the miRNA–target interaction, although a growing number of 'non-canonical' target sites are also now being described11,38,39.
Despite the similarity between the viRNA and miRNA systems, their cellular utilization is vastly disparate. Even in species with the capacity to generate both types of sRNA, viRNAs stand alone as the cellular antiviral infantry. The inability of miRNAs to function in antiviral defence is due to the fact that their sequences are fixed in the host genome and therefore their capacity to bind foreign nucleic acid is limited and easily evaded. However, the lack of viRNAs in chordates, coupled with the fact that miRNAs are not used in antiviral defence, has generated a unique opportunity for biotechnological exploitation. In this Review, I describe how the general lack of interplay between cellular miRNAs and RNA viruses permits the engineering of vectors that can deliver their own sRNAs or are controlled by the sRNAs present in the host.
Viruses and the cellular role of miRNAs
In chordates, the cellular response to viral infection begins with the recognition of viral nucleic acids. The recognition of such pathogen-associated molecular patterns (PAMPs) stimulates a signal transduction event that results in the transcriptional induction of type I interferons (IFNs) (Fig. 1). Type I IFNs are a family of cytokines that act in an autocrine and paracrine manner to induce the expression of more than 250 IFN-stimulated genes (ISGs), which function to slow the viral infection and allow time for the establishment of the adaptive immune response. ISGs slow viral infections by halting transcription, translation, cell division and the secretory pathway, and/or by inducing cell death before the virus has the opportunity to amplify.
Given their capacity to generate sRNAs, it was initially assumed that chordates were also likely to generate an RNA-mediated defence against viral infection, and that this might enhance the type I IFN response40. This was envisioned to occur in one of two ways. First, miRNAs could serve a direct antiviral function, in which case the miRNA would be responsible for silencing a viral transcript through sequence-specific binding. Second, miRNAs could function indirectly, modulating the host transcriptome to generate a less permissive environment for replication. However, despite the evolutionary continuity that these models would provide, there is little evidence to suggest that either has an important role in the physiological response to viral infection41. Although an RNA-mediated defence against viruses would seem likely, given its use in other phyla, there are three constraints that apply to miRNAs that might explain why this is not the case in the phylum Chordata.
Copy number. The first substantial constraint that would preclude most miRNAs from constituting part of the cellular defence against viruses is copy number. A conservative estimate for the minimum copy number required for an miRNA to have an impact on a host transcript is ∼100 copies per cell42,43,44,45. When one considers miRNAs acting directly on the virus, this estimate probably changes by an order of magnitude owing to the sheer number of viral mRNAs and/or genomes that would require silencing in an infected cell. This is compounded by the fact that, in any given cell type, the most abundant 20–30 miRNAs represent more than 90% of all the miRNAs in that cell, as shown by deep sequencing efforts, meaning that few miRNAs would be expressed at the concentrations required to be effective viral silencers46,47,48. Furthermore, additional studies have revealed that infection by an RNA virus does not profoundly change the sRNA landscape of the host cell or lead to the induction of any host miRNAs that accumulate to more than 100 copies per cell46,47. However, as many studies rely on PCR-based quantification of miRNAs, data concerning miRNA induction are often given as a fold change rather than as copies per cell. As a result, physiologically irrelevant miRNAs are often implicated in the response to a viral infection because of misleading fold inductions; for example, a 1,000-fold induction means little if an miRNA increases from one copy per 100 cells to ten copies per cell. This problem is further complicated by the fact that non-physiological levels of an miRNA mimetic, or a corresponding antagonist, are often used to validate these findings, a technique which has been proved to nonspecifically affect the host transcriptome and sRNA landscape of the cell34,49,50. Such studies have clouded the issue of miRNAs and their role in the cellular response to viral infection, and have led to a number of reports that are inconsistent or contradictory, or have implicated miRNAs for which the expression is too low to be of consequence in the relevant cell types studied51,52,53. Although it might be difficult to accept the conclusion that host miRNAs do not contribute to the cellular response to viral infection, clearly those studying the interplay between viruses and miRNAs must be more aware of copy number before any future hypotheses are put forward.
Silencing capacity. With the exception of latent viruses, miRNAs lack the ability to silence viral transcripts directly or to modulate the transcriptome of an infected cell in the time frame of a given infection. Under ideal conditions, an miRNA suppresses a target protein by less than twofold owing to the fact that the sequence of a genome-encoded miRNA is fixed and binding is never fully complementary29,30,31,32,33,34,35. The lack of contiguous base pairing between miRNA and mRNA prevents cleavage by an miRNA-loaded RISC; thus, complete suppression by an miRNA demands a near 1:1 ratio of miRNA to mRNA, and as this ratio is normally not achieved, this accounts for the low level of repression. By contrast, viRNAs demonstrate complete complementarity, as they are generated using the viral genome as a template. Perfect complementarity permits cleavage by a viRNA-loaded RISC, and therefore results in removal of the target RNA. Furthermore, target cleavage releases the viRNA, allowing viRNA-loaded RISC to act in an enzymatic manner, so that a single viRNA can theoretically eliminate every target mRNA in the cell54. Therefore, it follows that the only means by which miRNAs could be used to directly inhibit viral infection would be in a situation in which the complementarity spanned the length of the sRNA. This would demand that the viral genome itself be used as a template for the miRNA, as is the case for viRNAs.
This idea — that perhaps chordates were capable of generating virus-derived sRNAs — was comprehensively addressed in two landmark papers that profiled sRNAs from virus-infected cells and found no evidence of such activity55,56. These findings were further validated by an independent study that looked for evidence of an sRNA-mediated defence in the mammalian response to infection with hepatitis C virus (HCV), poliovirus, dengue virus (DENV), vesicular stomatitis virus (VSV) and West Nile virus (WNV); this study also concluded that although sRNAs could be isolated at 'vanishingly rare' concentrations, they were Dicer independent and did not demonstrate silencing capacity57. It would seem, then, that any bona fide evidence of chordate sRNAs targeting viruses directly would be examples of viral piracy. This could include viral utilization of a host miRNA to maintain the balance of its own transcripts or, as is the case for HCV, to stabilize the genome and mask the 5′ end from digestion by host nucleases58,59,60,61.
Kinetics. The last constraint that would suggest miRNAs do not contribute to the cellular response to viral infection is timing. Although it is feasible that infected cells upregulate miRNAs to fine-tune their own transcriptome in an effort to slow viral replication, this seems unlikely given the lack of evidence from deep sequencing efforts. Furthermore, when one accounts for protein half-life, de novo synthesis of miRNAs would be effective at silencing only a limited number of host and/or viral targets. With the exception of chronic viral infections, the life cycle of most viruses is less than 12 hours. Following cellular detection of the virus, the pri-miRNA would require transcriptional induction, processing and subsequent RISC loading before it could silence a viral or host mRNA. Although this might all happen in the first 4–6 hours of infection, the average protein half-life in chordates under times of stress is greater than 10 hours62. Therefore, the only viable targets for which virus-induced miRNAs would be effective would be those transcripts that are being newly transcribed or encode proteins with unusually short half-lives. One notable exception to this would be cytokine-mediated induction of miRNAs. In this case, a virus-induced cytokine could induce the transcriptional induction of an miRNA to modulate the transcriptomes of distant uninfected cells in the hours following infection. In fact, such events have been described, but it remains to be determined how extensively this type of regulation is used and whether its contribution to antiviral responses compares to the contribution made by the host antiviral cytokine signalling network63,64.
Exceptions to the rule. Despite the above arguments against miRNAs serving an antiviral function in chordates, DNA viruses might prove to be the exception to the rule. Herpesviruses not only produce their own miRNAs, they also antagonize specific miRNAs of the host65,66,67,68. These observations suggest that, in the context of the herpesvirus life cycle, a small subset of miRNAs is deleterious to the virus. Perhaps even more surprising is the recent discovery that poxvirus infection leads to the degradation of all host miRNAs69. Although the herpesvirus exception might reflect the chronic nature of herpesvirus infection, why poxviruses are threatened by host miRNAs remains unclear. One possible explanation is that this is an evolutionary artefact of antagonizing viRNAs in an RNAi-enabled species, which resulted in the requirement for an sRNA-free environment. However, one cannot exclude the possibility that poxviruses, with their lengthy 3′ UTRs, are negatively affected by host miRNAs and therefore evolved a strategy to evade them. A similar phenomenon is also apparent during adenovirus infection, in which a 160 nt non-coding RNA (called VA1) has been found to interfere with miRNA export and RISC loading70,71. Clearly, future studies examining the interplay between miRNAs and these DNA-based viruses will be required to fully elucidate the nature of this apparent host–pathogen struggle.
One means of ascertaining the importance of a particular pathway in mediating an antiviral response is by determining how much material the virus commits to circumventing it. For example, there are numerous examples of plant and insect viruses that donate a substantial proportion of their genome to preventing the biogenesis, loading and/or silencing activity of viRNAs72. In this context, it is interesting to note that a number of reports have also suggested that the dsRNA-binding proteins of influenza A virus (IAV), vaccinia virus and Ebola virus antagonize miRNA-mediated silencing, providing support for the notion that sRNA-mediated antiviral defences might indeed exist in chordates73,74,75. Although these findings are provocative, it is difficult to gauge their biological relevance, given that a number of reports have since been published which demonstrate that many of these viruses can be successfully designed to be susceptible to host miRNAs76,77. A contributing factor to this apparent contradiction comes from the fact that dsRNA is both a necessary step in miRNA formation and a PAMP. This means that although these viruses have evolved a way of masking PAMP detection, when these dsRNA-binding proteins are overexpressed at very high levels there can be a detectable impact on miRNAs. The true nature of these antagonistic proteins is perhaps best exemplified by the fact that viruses lacking them are attenuated by orders of magnitude in wild-type cells but can replicate to normal levels in type I IFN-deficient cells, regardless of miRNA expression78. Taken together, the evidence suggests that the cellular response to viruses has little overlap when comparing chordates with arthropods, plants and nematodes (Fig. 1).
Harnessing miRNAs to influence viral tropism
The reliance on protein-based antiviral defences in chordates (Box 2) is interesting from a host–pathogen interaction standpoint, but it also provides an intriguing opportunity to exploit the fact that chordates encode sRNAs which are left intact in the context of viral infection. This lack of interplay provides an opportunity to engineer viruses that interact with the miRNA pathway, in an effort to generate a range of biological tools. For example, although miRNAs might not normally function in an antiviral capacity, should one engineer perfectly complementary target sites into a virus, an miRNA will essentially be transformed into a viRNA (Fig. 2). Although copy number is still an issue, if one exploits an abundant miRNA, this strategy can be used to completely inhibit viral replication in cells expressing that miRNA79. Furthermore, because miRNAs, like mRNAs, can be expressed in a cell- or even species-specific manner, one can design viruses to behave differently depending on the site of replication, effectively dictating viral tropism. One of the first examples of exploiting an miRNA in this way incorporated target sequences for miR-142 into a lentivirus vector80. As miR-142 is a haematopoietic cell-specific miRNA, this design restricted expression of the lentivirus transgene in this particular cell lineage. A similar approach incorporated miRNA target sites into a retroviral vector as a reporter for miRNA activity81.

The presence of small RNAs (sRNAs) in chordates, in the form of microRNAs (miRNAs), gives researchers the ability to influence the tissue tropism of viruses. This can be accomplished by grafting one or more perfectly complementary miRNA target sites into a virus of interest. The resulting engineered virus will be attenuated, but only in the presence of its cognate miRNA. RNA polymerase II induces the transcription of a primary miRNA transcript (pri-miRNA) in a cell-specific manner (step 1). The pri-miRNA is processed by the microprocessor complex (composed of Drosha and DGCR8) to yield a precursor miRNA (pre-miRNA), which is exported to the cytoplasm (steps 2,3). The pre-miRNA is then further cleaved by Dicer and loaded into the RNA-induced silencing complex (RISC) (step 4). Should the virus encode targets for this cell-specific miRNA, entry into this cell would rapidly result in silencing of the viral genes and significant attenuation of the viral infection (step 5). By contrast, when the same virus enters a cell in which this pri-miRNA is not transcriptionally induced, the virus can replicate to endogenous levels and demonstrates no attenuation.
The concept of exploiting host miRNAs to control viral replication was quickly adopted by the scientific community and has since been applied to other lentiviruses by exploiting tissue-specific miRNAs to prevent translation of the viral genetic cassette in neurons and developing thymocytes82,83. miRNA-mediated targeting of viruses also expanded beyond gene therapy vectors and was found to be a powerful means of generating safe and effective live-attenuated vaccines. The first demonstration of this approach relied on the addition of target sites for the ubiquitously expressed miRNA let-7a or the neuron-specific miR-124 into two variable positions flanking the poliovirus capsid ORF79. The resulting viruses demonstrated replication levels that were inversely proportional to their cognate miRNA levels79. Furthermore, these miRNA-controlled viruses showed no evidence of pathogenesis, but their limited replication made them ideal vaccine candidates79. This same technology was also successfully applied to flaviviruses84,85. In addition, this concept was subsequently adapted for the generation of a live-attenuated IAV vaccine, but in this example, miRNA targeting was used to both attenuate the virus and address a manufacturing issue77. By exploiting a ubiquitous miRNA that was absent in the allantoic membrane of chicken eggs, a vaccine was produced that, although broadly attenuated in mammals, grew to wild-type levels in fertilized eggs. Last, in addition to gene therapy and vaccine design, harnessing of miRNAs to control viral replication has gained significant traction with researchers working with oncolytic viruses, as a means of improving safety by limiting viral tropism86,87,88.
One additional application of using miRNAs to control viral tropism is as a biological tool to determine the contribution or necessity of a certain cell type during viral infection. For example, DENV can replicate in a wide range of cell types, and its ability to establish a systemic infection could be explained by replication in endothelial cells, by piggy-backing on circulating cells or by direct infection of and amplification in haematopoietic cells. To address this question, a DENV strain was engineered to encode four haematopoietic cell-specific miR-142-targeting sites in the 3′ UTR of the virus. Although the addition of this non-coding RNA had little impact on the virus in the absence of miR-142, it completely blocked spread of the infection in vivo, suggesting that replication in haematopoietic cells is required for viral dissemination89. A similar targeting approach was also used to block IAV replication in antigen-presenting cells (APCs)76. In this example, the aim of the study was to determine the source of viral antigens that contribute to the adaptive immune response. Again exploiting the haematopoietic cell-specific nature of miR-142, IAV was designed in such a way that replication was unaffected in the lung epithelium but abolished in APCs. On the basis of the results of this study, we now know that during IAV infection, APCs engulf non-haematopoietic cells and/or cell debris and thus obtain enough viral components to generate a successful adaptive immune response76.
Escape from engineered miRNA targeting. Given the success in using miRNAs to control the properties of a virus, there is hope that this work will translate into improved therapeutic vectors for gene therapy, oncolytics and/or vaccine designs of the future. However, one common concern for this technology remains the possibility of escape mutants. Although this is less of an issue in gene therapy, for which the viral vector is not replication competent, live-attenuated viral vaccine vectors and oncolytic viruses would certainly have the potential to escape selective pressure. Fortunately, given that the targets are always multimerized and designed to be perfectly complementary to a particular miRNA, viral escape seems to be less of an issue than one would have predicted. For example, in the aforementioned study that exploited the ubiquitous miRNA let-7a to target poliovirus, the selective pressure was so great that no viral escape mutants could be isolated79. Interestingly, in contrast to this ubiquitous targeting, when the neuron-specific miRNA miR-124 was exploited, thus allowing poliovirus to replicate in some cell types, single-nucleotide mismatches were observed in the target sequences of some viral offspring, but these viruses still demonstrated substantial silencing — probably as a result of the complementarity remaining. These findings were recapitulated when miRNA targeting was applied to IAV, coxsackievirus and VSV77,86,87. However, in the case of DENV, escape of miRNA-mediated silencing did occur but was the result of a complete excision of the target area rather than through single-nucleotide mutations89. Taken together, these data suggest that providing one incorporates miRNA targets to minimize the possibility of excision (for example, incorporating multiple targets into the genome at different locations), stable sRNA-mediated silencing of a virus can be achieved.
Off-target effects of miRNA exploitation. Controlling the tropism or replication potential of a virus by harnessing host miRNA expression also has the capacity to influence the transcriptome of the host cell indirectly. This can occur through competitive hybridization to the host miRNA, making the miRNA unavailable to carry out its endogenous silencing role90. Target-mediated saturation of sRNAs has been clearly demonstrated, and the existence of perfect targets has even been associated with tailing and degradation of host miRNAs91. In fact, the capacity of viral vectors to 'soak up' the host miRNAs can be used to discern the endogenous role of miRNAs through loss-of-function assays in vivo92. Furthermore, the recent description of competing endogenous RNAs would suggest that the introduction of even small amounts of miRNA targets could have profound effects on the levels of host transcripts93. Although this phenomenon might prohibit the use of this technology with persistent viral vectors (such as lentiviruses and adeno-associated viruses (AAVs)), its use with acute RNA-based viruses or vectors should be less problematic; nonetheless, future studies directly addressing this possible complication will be needed.
In all, the capacity to control RNA viruses through the exploitation of miRNAs has the potential to transform vector-based therapeutics. Some concerns and possible complications still need to be addressed before these vectors can become a clinical reality, but the technology itself is something that requires further exploration and expansion. Taken together, the research carried out thus far in this field suggests that miRNA-mediated targeting, when executed correctly and in the proper context, is a viable strategy to improve vector design and/or vaccine development.
Virus-mediated delivery of miRNAs
The discovery that viRNA-like mimetics, in the form of small interfering RNAs (siRNAs), function in mammalian cells ushered in a new era of therapeutic potential94. The ability to silence a transcript in a sequence-specific manner was rapidly adopted as a tool to study protein function and became an attractive platform for future therapeutics (Box 3). However, despite the promise of RNAi, in vivo delivery of siRNAs to target cells has remained a substantial barrier that has prevented the field from meeting its high expectations. Vector-free delivery of siRNAs is fraught with intrinsic issues, including poor solubility of siRNAs, their poor stability, and their inability to cross membranes and enter the cytoplasm95.
An attractive solution to these issues is the use of viruses, or virus-like vectors, to deliver siRNAs in vivo (Fig. 3). Viruses have evolved over countless generations to become experts at cell entry and the subsequent production of foreign RNA, including miRNAs55,56,96,97. Viral vectors not only provide a solution to siRNA solubility, stability and entry, but also, in many cases, do not even perturb the natural sRNA landscape of the cell, unlike the direct administration of siRNAs98. Furthermore, unlike direct administration of duplex RNA, viral vectors can deliver siRNAs with a pri-miRNA-like or pre-miRNA-like structure. A pre-miRNA-like structure means that the siRNA is a Dicer substrate and does not require the microprocessor. This short hairpin RNA (shRNA) can demonstrate complete complementarity across the stem of the hairpin or can mimic a pre-miRNA and contain a number of mismatched bulges. Alternatively, delivery of a pri-miRNA generates a microprocessor substrate and is generally believed to be the least toxic delivery route because this method provides the most basic substrate to the sRNA machinery, thus ensuring that a surplus of duplex sRNAs does not accumulate98.

Viruses, regardless of their site of replication or genomic composition, can be engineered to produce robust levels of small RNAs (sRNAs). Arguably, the most effective means of delivering sRNAs is through the production of an artificial microRNA (amiRNA). a | A schematic of the neuronal microRNA (miRNA) miR-124. The primary mir-124 transcript (pri-mir-124) generates the guide sRNA shown in red, which binds to endogenous host transcripts, such as the polypyrimidine tract-binding protein 2 mRNA (PTBP2), with only partial complementarity, resulting in a modest repression of protein levels as a result of translational repression followed by RNA destabilization150. b | Schematic of an amiRNA. Designed to have the same 5′ and 3′ flanking sequences as the hairpin pre-miRNA for miR-124, as well as to maintain the overall loop sequence and structure of the hairpin, this amiRNA has an altered sequence composition in the stem. As a result, processing of this pre-amiRNA generates an sRNA with the desired sequence (blue) to engage a specific target with perfect complementarity. Given this flexibility, pri-amiRNAs can be incorporated into a viral vector and used to deliver the sRNA to a cell type of interest. c | A pri-amiRNA that is produced by the engineered virus is processed using the sRNA machinery of the host cell: the pri-amiRNA is cleaved by a cytoplasmic or nuclear microprocessor complex, generating a pre-miRNA that is further cut by Dicer and then loaded into the RNA-induced silencing complex (RISC) to target mRNA in a sequence-specific manner.
Although delivery of an miRNA has value, the enthusiasm for therapeutic sRNAs is largely due to the capacity to use these sRNAs to target any transcript in a sequence-specific manner. So, how does one deliver a specific siRNA using the miRNA machinery? The answer is artificial miRNAs (amiRNAs)99. Designed to maintain the overall structure of a particular endogenous pri-miRNA, an amiRNA has sequence-specific changes in the stem of the hairpin that tailor the sRNA to a specific host transcript but still allow the sRNA to be loaded onto the RISC29,99 (Fig. 3). Use of amiRNAs has been so successful that whole-genome libraries have now been generated and are commonly used in genetic screens, as their activities are comparable to the application of synthetic siRNAs29,100.
Constraints associated with viral delivery of sRNAs. Virus-mediated delivery of siRNAs was first described in 2002 in a study which demonstrated that an adenovirus vector could deliver functional shRNAs in both the liver and the brain101. Similar approaches using retroviruses and lentiviruses were used in the following year102,103,104. However, even as the field was gaining momentum, reports were published about a range of siRNA-mediated toxicities associated with these vectors105,106,107,108. Two distinct molecular processes were identified as the underlying causes of these adverse effects. The first is the generation of PAMPS, which trigger the cell to respond as if it has been infected by a replication-competent virus106,107,108. This effect is dependent on the vector design. In some examples, the siRNA is generated from a DNA-dependent RNA polymerase I (pol I) or pol III promoter, which leads to a primary transcript containing a 5′ triphosphate; this motif has been implicated in triggering antiviral defences and the production of pro-inflammatory cytokines109,110,111. A second factor that can result in an unwanted cellular response seems to be the shRNA itself107. The hairpin represents foreign dsRNA in the cytoplasm, and dsRNA has also been implicated in the induction of the cellular response to infection112. However, this attribute alone does not account for all of the constructs that induce the expression of pro-inflammatory cytokines. Use of exogenous siRNAs or amiRNAs with perfect complementarity in the hairpin stem would generate a 21–23 nt dsRNA fragment, and this, although short, might still be recognized by a range of pattern recognition receptors (PRRs)107,112. Pre-amiRNAs, which contain bulges in the dsRNA stem, should not be substrates for PRRs, but these too have been implicated in cytokine induction, suggesting that additional factors account for this toxicity. These factors might include improper duplex formation resulting in a lack of the 2 nt 3′ overhang that is found in natural miRNAs, and/or the inclusion of particular sequences that are recognized by the cell as foreign113,114. Taken together, the possibility of generating PAMPS from a particular siRNA precursor demands that a targeting RNA of interest be characterized for its possible pro-inflammatory properties in cell culture before it is assessed in any silencing studies.
Another possible source of siRNA-mediated toxicity is saturation of the miRNA biogenesis machinery105,115,116. This problem has been associated with the transfection of siRNAs, presumably because this can yield more RISC substrate in the cell than there are available RISC complexes98,117. However, even vector- or virus-mediated production of shRNAs in the nucleus can result in a bottleneck in the sRNA export pathway71,116,118,119. Toxicity from shRNAs produced by recombinant AAV has been observed in mouse models owing to saturation of the host machinery105,120,121. In addition to RISC saturation, AAV-mediated synthesis of shRNA also creates a bottleneck at the level of nuclear export — curiously, a phenotype comparable to that seen during adenovirus VA1 expression71,115,116. Although this bottleneck can be alleviated by reducing the activity of the shRNA promoter and so diminishing the amount of shRNA synthesized122, generating a desired siRNA in the context of an amiRNA also holds significant potential.
Taking these factors into account, it would seem that it is possible to develop a safe vector that is capable of delivering a specific siRNA which is free of cellular toxicity. Although such a delivery system might finally resolve the last remaining barrier to making sRNA therapeutics a clinical reality, the social stigma and manufacturing challenges of using such vectors would also need to be addressed. Furthermore, a single vector is unlikely to be amenable for every clinical application in which sRNAs could be therapeutically useful. Vector constraints could be specific, such as being unable to target the tissue of interest, or nonspecific, pertaining to indirect characteristics that might preclude the clinical use of a particular vector. For example, adenovirus vectors are well suited for manufacturing and delivery of sRNAs (as long as the VA1 gene is deleted), but adenovirus vectors can be complicated by their sero-prevalence in the population and an inherent cellular toxicity that is associated with particle entry123,124. Alternatively, integrating retrovirus- and lentivirus-based vectors are also effective at delivering functional sRNAs with potential long-term expression101,102,103. However, given the propensity of these viruses to integrate at transcriptional start sites125 or into introns of transcriptionally active genes126, clinical use of these vectors requires exceptional caution. Perhaps the most widely used method of clinical delivery is based on AAV vectors127. These vectors can be engineered to deliver genetic material to a wide range of organs, including the liver, lung and brain128,129,130. However, given the shRNA-mediated saturation constraints105,116,121, future work that combines amiRNAs and AAVs will be required.
RNA viruses as delivery vectors. Although the success of AAV-mediated delivery of amiRNAs could be sufficient to make sRNA therapeutics a clinical reality, it is noteworthy that a new subset of vectors capable of mediating transient sRNA delivery in vivo has also recently been described46. Although it is clear that viral production of miRNAs is largely limited to nuclear DNA viruses41, recent studies have demonstrated that RNA viruses, of both nuclear and cytoplasmic origin, could be engineered to produce functional miRNAs131,132,133,134. Combined, these studies demonstrate that encoding an miRNA does not result in substantial genomic cleavage (which could have occurred when the miRNA sequence folded into a hairpin within the genome and was processed by the host machinery) or self-targeting, disproving the hypothesis that had been suggested to explain the lack of endogenous RNA virus-encoded miRNAs41. In fact, the capacity of an RNA virus to naturally encode an miRNA was recently characterized in WNV and bovine leukaemia virus (BLV)96,135. For WNV, a virus-encoded miRNA was reported to derive from a subgenomic 3′ RNA, thus minimizing genomic cleavage, although why this was not detected in earlier studies remains unclear57. By contrast, BLV-mediated synthesis of an miRNA in the absence of self-attenuation was presumably accomplished by masking the sRNA precursor from the host machinery through extensive RNA folding in much the same way that HIV has been demonstrated to resist siRNA targeting136.
Perhaps even more surprising than the lack of viral attenuation was the fact that nuclear localization of the pri-miRNA was not necessary for hairpin processing46,132,133. An in-depth analysis aimed at determining the mechanism of cytoplasmic processing for pri-miRNAs suggests that viral infection results in the formation of a non-nuclear microprocessor with the capacity to generate Dicer and RISC substrates133. Regardless of the biological significance of cytoplasmic pri-miRNA processing, it is clear that this activity can be harnessed as a new vector platform for sRNA delivery. Although these vectors would provide only transient expression of their sRNA cargo, their ease of genetic production and lack of nuclear trafficking requirements give them unique potential for a range of applications. Furthermore, when one considers that these vectors could additionally be controlled by cell-specific miRNAs, RNA virus-inspired vectors might represent the most elegant platform to mediate transient delivery of an sRNA to a particular tissue of interest.
Another exciting application for this biological activity is to study the importance of host–pathogen interactions in vivo by endowing a virus with the capacity to silence a transcript of interest. Whole-genome amiRNA libraries can be used to determine the identity of host restriction factors for viral replication, pathogenesis, tropism or any other testable viral attribute. Perhaps the most clinically relevant use of this knowledge is in the enhancement of viral oncolytic vectors. A number of ongoing clinical trials are investigating the use of RNA viruses that preferentially replicate in transformed cells137. The activity of these viruses, which include VSV and Sindbis virus, could be further enhanced by enabling them to eliminate oncogene expression. Alternatively, one could improve the therapeutic window of these vectors or of live-attenuated viral vaccines by incorporating hairpins that silence pro-inflammatory cytokines. Transient delivery of miRNAs could also be used in the reprogramming of cells, in which changes in cell fate would not require sustained expression of the vector138,139.
Closing remarks
The use of sRNAs as an immune response to control viral pathogenesis is limited to plants, bacteria, nematodes and arthropods; in chordates, sRNAs are not used as an antiviral defence mechanism, and the sRNA biogenesis machinery can therefore be exploited for a range of vector-based therapeutics. For example, grafting miRNA target sites into a vector or virus of interest can be used to control expression in a species- and/or cell-specific manner. In addition, vectors and/or viruses can be engineered to generate amiRNAs, which themselves can be designed to target a particular gene of interest. These two concepts, used together or independently, can generate vectors in which the tropism is uniquely defined to ensure the safe and precise delivery of a particular siRNA. These advancements could breathe new life into a field that once promised a bounty of innovative medical treatments before the realization that delivery posed a seemingly insurmountable barrier. This solution will, however, require significant research support to generate clinical-grade vectors that are both safe and effective and that can overcome the social stigma associated with using virus-based products as therapeutics. If science and society can progress to this point, exploiting the host sRNA machinery with engineered viruses undoubtedly has the capacity to usher in a new era of molecular tools and change the future of therapeutics.
References
Wiedenheft, B., Sternberg, S. H. & Doudna, J. A. RNA-guided genetic silencing systems in bacteria and archaea. Nature 482, 331–338 (2012).
Shirayama, M. et al. piRNAs initiate an epigenetic memory of nonself RNA in the C. elegans germline. Cell 150, 65–77 (2012).
Lee, H. C. et al. C. elegans piRNAs mediate the genome-wide surveillance of germline transcripts. Cell 150, 78–87 (2012).
Ashe, A. et al. piRNAs can trigger a multigenerational epigenetic memory in the germline of C. elegans. Cell 150, 88–99 (2012).
Marraffini, L. A. & Sontheimer, E. J. CRISPR interference: RNA-directed adaptive immunity in bacteria and archaea. Nature Rev. Genet. 11, 181–190 (2010).
Siomi, M. C., Sato, K., Pezic, D. & Aravin, A. A. PIWI-interacting small RNAs: the vanguard of genome defence. Nature Rev. Mol. Cell Biol. 12, 246–258 (2011).
Ding, S. W. RNA-based antiviral immunity. Nature Rev. Immunol. 10, 632–644 (2010).
Ebert, M. S. & Sharp, P. A. Roles for MicroRNAs in conferring robustness to biological processes. Cell 149, 515–524 (2012).
Friedman, R. C., Farh, K. K., Burge, C. B. & Bartel, D. P. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res. 19, 92–105 (2009).
Ishizu, H., Siomi, H. & Siomi, M. C. Biology of PIWI-interacting RNAs: new insights into biogenesis and function inside and outside of germlines. Genes Dev. 26, 2361–2373 (2012).
Bartel, D. P. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 116, 281–297 (2004). A comprehensive review discussing the biology of miRNAs.
Ding, S. W. & Voinnet, O. Antiviral immunity directed by small RNAs. Cell 130, 413–426 (2007). A thorough review discussing the use of sRNAs in plants, nematodes and arthropods.
Brodersen, P. & Voinnet, O. The diversity of RNA silencing pathways in plants. Trends Genet. 22, 268–280 (2006).
Vaistij, F. E., Jones, L. & Baulcombe, D. C. Spreading of RNA targeting and DNA methylation in RNA silencing requires transcription of the target gene and a putative RNA-dependent RNA polymerase. Plant Cell 14, 857–867 (2002).
Sijen, T. et al. On the role of RNA amplification in dsRNA-triggered gene silencing. Cell 107, 465–476 (2001).
Rechavi, O., Minevich, G. & Hobert, O. Transgenerational inheritance of an acquired small RNA-based antiviral response in C. elegans. Cell 147, 1248–1256 (2011).
Saleh, M. C. et al. Antiviral immunity in Drosophila requires systemic RNA interference spread. Nature 458, 346–350 (2009). The first evidence that siRNAs can be secreted and function throughout arthropods despite the absence of an RdRp.
Lee, R. C. & Ambros, V. An extensive class of small RNAs in Caenorhabditis elegans. Science 294, 862–864 (2001).
Lau, N. C., Lim, L. P., Weinstein, E. G. & Bartel, D. P. An abundant class of tiny RNAs with probable regulatory roles in Caenorhabditis elegans. Science 294, 858–862 (2001).
Lagos-Quintana, M., Rauhut, R., Lendeckel, W. & Tuschl, T. Identification of novel genes coding for small expressed RNAs. Science 294, 853–858 (2001).
Lund, E., Guttinger, S., Calado, A., Dahlberg, J. E. & Kutay, U. Nuclear export of microRNA precursors. Science 303, 95–98 (2004).
Yi, R., Qin, Y., Macara, I. G. & Cullen, B. R. Exportin-5 mediates the nuclear export of pre-microRNAs and short hairpin RNAs. Genes Dev. 17, 3011–3016 (2003).
Bernstein, E., Caudy, A. A., Hammond, S. M. & Hannon, G. J. Role for a bidentate ribonuclease in the initiation step of RNA interference. Nature 409, 363–366 (2001).
Grishok, A. et al. Genes and mechanisms related to RNA interference regulate expression of the small temporal RNAs that control C. elegans developmental timing. Cell 106, 23–34 (2001).
Ketting, R. F. et al. Dicer functions in RNA interference and in synthesis of small RNA involved in developmental timing in C. elegans. Genes Dev. 15, 2654–2659 (2001).
Hutvagner, G. et al. A cellular function for the RNA-interference enzyme Dicer in the maturation of the let-7 small temporal RNA. Science 293, 834–838 (2001).
Schwarz, D. S. et al. Asymmetry in the assembly of the RNAi enzyme complex. Cell 115, 199–208 (2003).
Khvorova, A., Reynolds, A. & Jayasena, S. D. Functional siRNAs and miRNAs exhibit strand bias. Cell 115, 209–216 (2003).
Zeng, Y., Wagner, E. J. & Cullen, B. R. Both natural and designed micro RNAs can inhibit the expression of cognate mRNAs when expressed in human cells. Mol. Cell 9, 1327–1333 (2002). The first rigorous demonstration that miRNAs can be artificially designed and elicit silencing.
Baek, D. et al. The impact of microRNAs on protein output. Nature 455, 64–71 (2008).
Selbach, M. et al. Widespread changes in protein synthesis induced by microRNAs. Nature 455, 58–63 (2008).
Grimson, A. et al. MicroRNA targeting specificity in mammals: determinants beyond seed pairing. Mol. Cell 27, 91–105 (2007).
Giraldez, A. J. et al. Zebrafish miR-430 promotes deadenylation and clearance of maternal mRNAs. Science 312, 75–79 (2006).
Lim, L. P. et al. Microarray analysis shows that some microRNAs downregulate large numbers of target mRNAs. Nature 433, 769–773 (2005).
Krutzfeldt, J. et al. Silencing of microRNAs in vivo with 'antagomirs'. Nature 438, 685–689 (2005).
Lewis, B. P., Burge, C. B. & Bartel, D. P. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell 120, 15–20 (2005).
Bartel, D. P. MicroRNAs: target recognition and regulatory functions. Cell 136, 215–233 (2009).
Shin, C. et al. Expanding the microRNA targeting code: functional sites with centered pairing. Mol. Cell 38, 789–802 (2010).
Chi, S. W., Hannon, G. J. & Darnell, R. B. An alternative mode of microRNA target recognition. Nature Struct. Mol. Biol. 19, 321–327 (2012).
Haasnoot, J., Westerhout, E. M. & Berkhout, B. RNA interference against viruses: strike and counterstrike. Nature Biotech. 25, 1435–1443 (2007).
Cullen, B. R. Viruses and microRNAs: RISCy interactions with serious consequences. Genes Dev. 25, 1881–1894 (2011). A discussion of the physiologogical interplay between viruses and miRNAs.
Schmitter, D. et al. Effects of Dicer and Argonaute down-regulation on mRNA levels in human HEK293 cells. Nucleic Acids Res. 34, 4801–4815 (2006).
Landthaler, M. et al. Molecular characterization of human Argonaute-containing ribonucleoprotein complexes and their bound target mRNAs. RNA 14, 2580–2596 (2008).
Brown, B. D. et al. Endogenous microRNA can be broadly exploited to regulate transgene expression according to tissue, lineage and differentiation state. Nature Biotech. 25, 1457–1467 (2007).
Mullokandov, G. et al. High-throughput assessment of microRNA activity and function using microRNA sensor and decoy libraries. Nature Methods 9, 840–846 (2012).
Langlois, R. A., Shapiro, J. S., Pham, A. M. & tenOever, B. R. In vivo delivery of cytoplasmic RNA virus-derived miRNAs. Mol. Ther. 20, 367–375 (2011).
Perez, J. T. et al. Influenza A virus-generated small RNAs regulate the switch from transcription to replication. Proc. Natl Acad. Sci. USA 107, 11525–11530 (2010).
Landgraf, P. et al. A mammalian microRNA expression atlas based on small RNA library sequencing. Cell 129, 1401–1414 (2007).
Jackson, A. L. et al. Expression profiling reveals off-target gene regulation by RNAi. Nature Biotech. 21, 635–637 (2003).
Jackson, A. L. & Linsley, P. S. Recognizing and avoiding siRNA off-target effects for target identification and therapeutic application. Nature Rev. Drug Discov. 9, 57–67 (2010).
Pedersen, I. M. et al. Interferon modulation of cellular microRNAs as an antiviral mechanism. Nature 449, 919–922 (2007).
Otsuka, M. et al. Hypersusceptibility to vesicular stomatitis virus infection in Dicer1-deficient mice is due to impaired miR24 and miR93 expression. Immunity 27, 123–134 (2007).
Buggele, W. A., Johnson, K. E. & Horvath, C. M. Influenza A virus infection of human respiratory cells induces primary microRNA expression. J. Biol. Chem. 287, 31027–31040 (2012).
Haley, B. & Zamore, P. D. Kinetic analysis of the RNAi enzyme complex. Nature Struct. Mol. Biol. 11, 599–606 (2004).
Pfeffer, S. et al. Identification of microRNAs of the herpesvirus family. Nature Methods 2, 269–276 (2005).
Pfeffer, S. et al. Identification of virus-encoded microRNAs. Science 304, 734–736 (2004).
Parameswaran, P. et al. Six RNA viruses and forty-one hosts: viral small RNAs and modulation of small RNA repertoires in vertebrate and invertebrate systems. PLoS Pathog. 666, e1000764 (2010).
Jopling, C. L., Norman, K. L. & Sarnow, P. Positive and negative modulation of viral and cellular mRNAs by liver-specific microRNA miR-122. Cold Spring Harb. Symp. Quant. Biol. 71, 369–376 (2006).
Shimakami, T. et al. Stabilization of hepatitis C virus RNA by an Ago2–miR-122 complex. Proc. Natl Acad. Sci. USA 109, 941–946 (2012).
Jopling, C. L., Schutz, S. & Sarnow, P. Position-dependent function for a tandem microRNA miR-122-binding site located in the hepatitis C virus RNA genome. Cell Host Microbe 4, 77–85 (2008).
Machlin, E. S., Sarnow, P. & Sagan, S. M. Masking the 5′ terminal nucleotides of the hepatitis C virus genome by an unconventional microRNA-target RNA complex. Proc. Natl Acad. Sci. USA 108, 3193–3198 (2011).
Eden, E. et al. Proteome half-life dynamics in living human cells. Science 331, 764–768 (2011).
O'Connell, R. M., Taganov, K. D., Boldin, M. P., Cheng, G. & Baltimore, D. MicroRNA-155 is induced during the macrophage inflammatory response. Proc. Natl Acad. Sci. USA 104, 1604–1609 (2007).
Peng, X. et al. Integrative deep sequencing of the mouse lung transcriptome reveals differential expression of diverse classes of small RNAs in response to respiratory virus infection. mBio 2, e00198–11 (2011).
Libri, V. et al. Murine cytomegalovirus encodes a miR-27 inhibitor disguised as a target. Proc. Natl Acad. Sci. USA 109, 279–284 (2012).
Cazalla, D., Yario, T. & Steitz, J. A. Down-regulation of a host microRNA by a Herpesvirus saimiri noncoding RNA. Science 328, 1563–1566 (2010).
Marcinowski, L. et al. Degradation of cellular mir-27 by a novel, highly abundant viral transcript is important for efficient virus replication in vivo. PLoS Pathog. 866, e1002510 (2012).
Cullen, B. R. Herpesvirus microRNAs: phenotypes and functions. Curr. Opin. Virol. 1, 211–215 (2011).
Backes, S. et al. Degradation of host microRNAs by poxvirus poly(A) polymerase reveals terminal RNA methylation as a protective antiviral mechanism. Cell Host Microbe 12, 200–210 (2012).
Bennasser, Y. et al. Competition for XPO5 binding between Dicer mRNA, pre-miRNA and viral RNA regulates human Dicer levels. Nature Struct. Mol. Biol. 18, 323–327 (2011).
Lu, S. & Cullen, B. R. Adenovirus VA1 noncoding RNA can inhibit small interfering RNA and MicroRNA biogenesis. J. Virol. 78, 12868–12876 (2004).
Voinnet, O., Pinto, Y. M. & Baulcombe, D. C. Suppression of gene silencing: a general strategy used by diverse DNA and RNA viruses of plants. Proc. Natl Acad. Sci. USA 96, 14147–14152 (1999).
Li, W. X. et al. Interferon antagonist proteins of influenza and vaccinia viruses are suppressors of RNA silencing. Proc. Natl. Acad. Sci. USA 101, 1350–1355 (2004).
Delgadillo, M. O., Saenz, P., Salvador, B., Garcia, J. A. & Simon-Mateo, C. Human influenza virus NS1 protein enhances viral pathogenicity and acts as an RNA silencing suppressor in plants. J. Gen. Virol. 85, 993–999 (2004).
Bucher, E., Hemmes, H., de Haan, P., Goldbach, R. & Prins, M. The influenza A virus NS1 protein binds small interfering RNAs and suppresses RNA silencing in plants. J. Gen. Virol. 85, 983–991 (2004).
Langlois, R. A., Varble, A., Chua, M. A., Garcia-Sastre, A. & tenOever, B. R. Hematopoietic-specific targeting of influenza A virus reveals replication requirements for induction of antiviral immune responses. Proc. Natl Acad. Sci. USA 109, 12117–12122 (2012).
Perez, J. T. et al. MicroRNA-mediated species-specific attenuation of influenza A virus. Nature Biotech. 27, 572–576 (2009).
Garcia-Sastre, A. & Biron, C. A. Type 1 interferons and the virus-host relationship: a lesson in détente. Science 312, 879–882 (2006).
Barnes, D., Kunitomi, M., Vignuzzi, M., Saksela, K. & Andino, R. Harnessing endogenous miRNAs to control virus tissue tropism as a strategy for developing attenuated virus vaccines. Cell Host Microbe 4, 239–248 (2008).
Brown, B. D., Venneri, M. A., Zingale, A., Sergi Sergi, L. & Naldini, L. Endogenous microRNA regulation suppresses transgene expression in hematopoietic lineages and enables stable gene transfer. Nature Med. 12, 585–591 (2006).
Gottwein, E., Cai, X. & Cullen, B. R. A novel assay for viral microRNA function identifies a single nucleotide polymorphism that affects Drosha processing. J. Virol. 80, 5321–5326 (2006).
Colin, A. et al. Engineered lentiviral vector targeting astrocytes in vivo. Glia 57, 667–679 (2009).
Papapetrou, E. P., Kovalovsky, D., Beloeil, L., Sant'angelo, D. & Sadelain, M. Harnessing endogenous miR-181a to segregate transgenic antigen receptor expression in developing versus post-thymic T cells in murine hematopoietic chimeras. J. Clin. Invest. 119, 157–168 (2009).
Heiss, B. L., Maximova, O. A., Thach, D. C., Speicher, J. M. & Pletnev, A. G. MicroRNA targeting of neurotropic flavivirus: effective control of virus escape and reversion to neurovirulent phenotype. J. Virol. 86, 5647–5659 (2012).
Heiss, B. L., Maximova, O. A. & Pletnev, A. G. Insertion of microRNA targets into the flavivirus genome alters its highly neurovirulent phenotype. J. Virol. 85, 1464–1472 (2010).
Kelly, E. J., Nace, R., Barber, G. N. & Russell, S. J. Attenuation of vesicular stomatitis virus encephalitis through microRNA targeting. J. Virol. 84, 1550–1562 (2009).
Edge, R. E. et al. A let-7 microRNA-sensitive vesicular stomatitis virus demonstrates tumor-specific replication. Mol. Ther. 16, 1437–1443 (2008).
Leber, M. F. et al. MicroRNA-sensitive oncolytic measles viruses for cancer-specific vector tropism. Mol. Ther. 19, 1097–1106 (2011).
Pham, A. M., Langlois, R. A. & tenOever, B. R. Replication in cells of hematopoietic origin is necessary for Dengue virus dissemination. PLoS Pathog. 866, e1002465 (2012).
Gentner, B. et al. Stable knockdown of microRNA in vivo by lentiviral vectors. Nature Methods 6, 63–66 (2009).
Ameres, S. L. et al. Target RNA-directed trimming and tailing of small silencing RNAs. Science 328, 1534–1539 (2010).
Xie, J. et al. Long-term, efficient inhibition of microRNA function in mice using rAAV vectors. Nature Methods 9, 403–409 (2012).
Salmena, L., Poliseno, L., Tay, Y., Kats, L. & Pandolfi, P. P. A ceRNA hypothesis: the Rosetta Stone of a hidden RNA language? Cell 146, 353–358 (2011).
Elbashir, S. M. et al. Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature 411, 494–498 (2001).
Whitehead, K. A., Langer, R. & Anderson, D. G. Knocking down barriers: advances in siRNA delivery. Nature Rev. Drug Discov. 8, 129–138 (2009).
Kincaid, R. P., Burke, J. M. & Sullivan, C. S. RNA virus microRNA that mimics a B-cell oncomiR. Proc. Natl Acad. Sci. USA 109, 3077–3082 (2012).
Seo, G. J., Chen, C. J. & Sullivan, C. S. Merkel cell polyomavirus encodes a microRNA with the ability to autoregulate viral gene expression. Virology 383, 183–187 (2009).
Castanotto, D. et al. Combinatorial delivery of small interfering RNAs reduces RNAi efficacy by selective incorporation into RISC. Nucleic Acids Res. 35, 5154–5164 (2007).
Chang, K., Elledge, S. J. & Hannon, G. J. Lessons from nature: microRNA-based shRNA libraries. Nature Methods 3, 707–714 (2006). A overview of how miRNAs can be redesigned to express an sRNA of interest.
Silva, J. M. et al. Second-generation shRNA libraries covering the mouse and human genomes. Nature Genet. 37, 1281–1288 (2005).
Xia, H., Mao, Q., Paulson, H. L. & Davidson, B. L. siRNA-mediated gene silencing in vitro and in vivo. Nature Biotech. 20, 1006–1010 (2002).
Lee, M. T., Coburn, G. A., McClure, M. O. & Cullen, B. R. Inhibition of human immunodeficiency virus type 1 replication in primary macrophages by using Tat- or CCR5-specific small interfering RNAs expressed from a lentivirus vector. J. Virol. 77, 11964–11972 (2003).
Stewart, S. A. et al. Lentivirus-delivered stable gene silencing by RNAi in primary cells. RNA 9, 493–501 (2003).
Castanotto, D. & Rossi, J. J. The promises and pitfalls of RNA-interference-based therapeutics. Nature 457, 426–433 (2009).
Grimm, D. et al. Fatality in mice due to oversaturation of cellular microRNA/short hairpin RNA pathways. Nature 441, 537–541 (2006). The first evidence that overproduction of sRNAs can result in significant toxicity owing to saturation of the host machinery.
Kleinman, M. E. et al. Sequence- and target-independent angiogenesis suppression by siRNA via TLR3. Nature 452, 591–597 (2008).
Sledz, C. A., Holko, M., de Veer, M. J., Silverman, R. H. & Williams, B. R. Activation of the interferon system by short-interfering RNAs. Nature Cell Biol. 5, 834–839 (2003).
Bridge, A. J., Pebernard, S., Ducraux, A., Nicoulaz, A. L. & Iggo, R. Induction of an interferon response by RNAi vectors in mammalian cells. Nature Genet. 34, 263–264 (2003).
Kim, D. H. et al. Interferon induction by siRNAs and ssRNAs synthesized by phage polymerase. Nature Biotech. 22, 321–325 (2004).
Hornung, V. et al. 5′-triphosphate RNA is the ligand for RIG-I. Science 314, 994–997 (2006).
Pichlmair, A. et al. RIG-I-mediated antiviral responses to single-stranded RNA bearing 5′-phosphates. Science 314, 997–1001 (2006).
Kato, H. et al. Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature 441, 101–105 (2006).
Marques, J. T. et al. A structural basis for discriminating between self and nonself double-stranded RNAs in mammalian cells. Nature Biotech. 24, 559–565 (2006).
Judge, A. D. et al. Sequence-dependent stimulation of the mammalian innate immune response by synthetic siRNA. Nature Biotech. 23, 457–462 (2005).
Yi, R., Doehle, B. P., Qin, Y., Macara, I. G. & Cullen, B. R. Overexpression of exportin 5 enhances RNA interference mediated by short hairpin RNAs and microRNAs. RNA 11, 220–226 (2005).
Grimm, D. et al. Argonaute proteins are key determinants of RNAi efficacy, toxicity, and persistence in the adult mouse liver. J. Clin. Invest. 120, 3106–3119 (2010).
Khan, A. A. et al. Transfection of small RNAs globally perturbs gene regulation by endogenous microRNAs. Nature Biotech. 27, 549–555 (2009).
Koller, E. et al. Competition for RISC binding predicts in vitro potency of siRNA. Nucleic Acids Res. 34, 4467–4476 (2006).
Vickers, T. A., Lima, W. F., Nichols, J. G. & Crooke, S. T. Reduced levels of Ago2 expression result in increased siRNA competition in mammalian cells. Nucleic Acids Res. 35, 6598–6610 (2007).
Beer, S. et al. Low-level shRNA cytotoxicity can contribute to MYC-induced hepatocellular carcinoma in adult mice. Mol. Ther. 18, 161–170 (2010).
McBride, J. L. et al. Artificial miRNAs mitigate shRNA-mediated toxicity in the brain: implications for the therapeutic development of RNAi. Proc. Natl Acad. Sci. USA 105, 5868–5873 (2008).
Boudreau, R. L., Martins, I. & Davidson, B. L. Artificial microRNAs as siRNA shuttles: improved safety as compared to shRNAs in vitro and in vivo. Mol. Ther. 17, 169–175 (2009).
Schnell, M. A. et al. Activation of innate immunity in nonhuman primates following intraportal administration of adenoviral vectors. Mol. Ther. 3, 708–722 (2001).
Narvaiza, I. et al. Effect of adenovirus-mediated RNA interference on endogenous microRNAs in a mouse model of multidrug resistance protein 2 gene silencing. J. Virol. 80, 12236–12247 (2006).
Wu, X., Li, Y., Crise, B. & Burgess, S. M. Transcription start regions in the human genome are favored targets for MLV integration. Science 300, 1749–1751 (2003).
Montini, E. et al. Hematopoietic stem cell gene transfer in a tumor-prone mouse model uncovers low genotoxicity of lentiviral vector integration. Nature Biotech. 24, 687–696 (2006).
Grimm, D. Small silencing RNAs: state-of-the-art. Adv. Drug Deliv. Rev. 61, 672–703 (2009).
Nakai, H. et al. Adeno-associated viral vector-mediated gene transfer of human blood coagulation factor IX into mouse liver. Blood 91, 4600–4607 (1998).
Flotte, T. R. et al. Stable in vivo expression of the cystic fibrosis transmembrane conductance regulator with an adeno-associated virus vector. Proc. Natl Acad. Sci. USA 90, 10613–10617 (1993).
Kaplitt, M. G. et al. Long-term gene expression and phenotypic correction using adeno-associated virus vectors in the mammalian brain. Nature Genet. 8, 148–154 (1994).
Rouha, H., Thurner, C. & Mandl, C. W. Functional microRNA generated from a cytoplasmic RNA virus. Nucleic Acids Res. 38, 8328–8337 (2010).
Shapiro, J. S., Varble, A., Pham, A. M. & tenOever, B. R. Noncanonical cytoplasmic processing of viral microRNAs. RNA 16, 2068–2074 (2010). This and reference 131 demonstrate that pri-miRNAs can be generated by engineered cytoplasmic viruses.
Shapiro, J. S., Langlois, R. A., Pham, A. M. & tenOever, B. R. Evidence for a cytoplasmic microprocessor of pri-miRNAs. RNA 18, 1338–1346 (2012). The first article to address the mechanism enabling cytoplasmic synthesis of pri-miRNAs.
Varble, A. et al. Engineered RNA viral synthesis of microRNAs. Proc. Natl Acad. Sci. USA 107, 11519–11524 (2010).
Hussain, M. et al. West Nile virus encodes a microRNA-like small RNA in the 3′ untranslated region which up-regulates GATA4 mRNA and facilitates virus replication in mosquito cells. Nucleic Acids Res. 40, 2210–2223 (2012).
Westerhout, E. M., Ooms, M., Vink, M., Das, A. T. & Berkhout, B. HIV-1 can escape from RNA interference by evolving an alternative structure in its RNA genome. Nucleic Acids Res. 33, 796–804 (2005).
Russell, S. J., Peng, K. W. & Bell, J. C. Oncolytic virotherapy. Nature Biotech. 30, 658–670 (2012). A comprehensive review discussing the use of lytic viruses to treat cancer.
Judson, R. L., Babiarz, J. E., Venere, M. & Blelloch, R. Embryonic stem cell-specific microRNAs promote induced pluripotency. Nature Biotech. 27, 459–461 (2009).
Anokye-Danso, F. et al. Highly efficient miRNA-mediated reprogramming of mouse and human somatic cells to pluripotency. Cell Stem Cell 8, 376–388 (2011).
Farh, K. K. et al. The widespread impact of mammalian microRNAs on mRNA repression and evolution. Science 310, 1817–1821 (2005).
Stark, A., Brennecke, J., Bushati, N., Russell, R. B. & Cohen, S. M. Animal microRNAs confer robustness to gene expression and have a significant impact on 3′UTR evolution. Cell 123, 1133–1146 (2005).
Tsang, J., Zhu, J. & van Oudenaarden, A. MicroRNA-mediated feedback and feedforward loops are recurrent network motifs in mammals. Mol. Cell 26, 753–767 (2007).
Shkumatava, A., Stark, A., Sive, H. & Bartel, D. P. Coherent but overlapping expression of microRNAs and their targets during vertebrate development. Genes Dev. 23, 466–481 (2009).
Cerutti, H. & Casas-Mollano, J. A. On the origin and functions of RNA-mediated silencing: from protists to man. Curr. Genet. 50, 81–99 (2006).
Kato, H. et al. Length-dependent recognition of double-stranded ribonucleic acids by retinoic acid-inducible gene-I and melanoma differentiation-associated gene 5. J. Exp. Med. 205, 1601–1610 (2008).
Long, D. et al. Potent effect of target structure on microRNA function. Nature Struct. Mol. Biol. 14, 287–294 (2007).
Jackson, A. L. et al. Widespread siRNA “off-target” transcript silencing mediated by seed region sequence complementarity. RNA 12, 1179–1187 (2006).
Pusch, O. et al. Nucleotide sequence homology requirements of HIV-1-specific short hairpin RNA. Nucleic Acids Res. 31, 6444–6449 (2003).
Fellmann, C. et al. Functional identification of optimized RNAi triggers using a massively parallel sensor assay. Mol. Cell 41, 733–746 (2011).
Makeyev, E. V., Zhang, J., Carrasco, M. A. & Maniatis, T. The microRNA miR-124 promotes neuronal differentiation by triggering brain-specific alternative pre-mRNA splicing. Mol. Cell 27, 435–448 (2007).
Acknowledgements
The author thanks the members of his laboratory, both past and present, for excellent discussions on the issues addressed in this Review and for help with the illustrations, and apologizes to those whose relevant work could not be cited here. The author is indebted to the Pew Charitable Trust and to Burroughs Wellcome for their continued support to freely explore these areas of research.
Author information
Authors and Affiliations
Ethics declarations
Competing interests
The author declares no competing financial interests.
Related links
Glossary
- RNAi
-
An activity by which a small RNA negatively impacts the functionality of an mRNA.
- Clustered regularly interspaced short palindromic repeat RNAs
-
(crRNAs). 57-nucleotide RNAs that are used to target phages in a sequence-specific manner.
- PIWI-interacting RNAs
-
(piRNAs). RNAs that are ∼30 nucleotides long and are responsible for detecting foreign DNA and repressing transposable elements.
- Virus-derived interfering RNAs
-
(viRNAs). RNAs that are ∼21 nucleotides in length, processed from a viral genomic source by the host and used to silence viral replication.
- MicroRNAs
-
(miRNAs). Host-derived small RNAs that are generated by endonucleases to fine-tune mRNA and protein levels.
- RNA-dependent RNA polymerase
-
A polymerase that generates nascent RNA from an RNA template. This polymerase is commonly used by RNA viruses for genome replication and is also used in plants and nematodes for the amplification of virus-derived interfering RNAs.
- RNA-induced silencing complex
-
(RISC). A complex of RNA-binding proteins and an Argonaute protein that, together, are responsible for small-RNA-mediated silencing of target RNAs.
- Pathogen-associated molecular patterns
-
Structures that are common among a group of pathogens and are constrained from rapid evolutionary change. These structures are used in cellular recognition of the pathogen.
- Small interfering RNA
-
(siRNA). RNA that is capable of RNA-induced silencing complex (RISC)-mediated mRNA silencing and binds its target with perfect complementarity.
- Short hairpin RNA
-
(shRNA). An ∼60 nucleotide stem–loop RNA that can serve as a substrate for Dicer.
- Artificial miRNAs
-
Small RNAs that maintain the secondary structure and sequence of a specific microRNA but encode changes in the stem of the hairpin, resulting in a distinct small RNA.
- Pattern recognition receptors
-
Cellular proteins that are responsible for the recognition of pathogen-associated molecular patterns.
Rights and permissions
About this article
Cite this article
tenOever, B. RNA viruses and the host microRNA machinery. Nat Rev Microbiol 11, 169–180 (2013). https://doi.org/10.1038/nrmicro2971
Published:
Issue Date:
DOI: https://doi.org/10.1038/nrmicro2971
This article is cited by
-
Exosomal transmission of viruses, a two-edged biological sword
Cell Communication and Signaling (2023)
-
The impact of non-synonymous mutations on miRNA binding sites within the SARS-CoV-2 NSP3 and NSP4 genes
Scientific Reports (2023)
-
Virally programmed extracellular vesicles sensitize cancer cells to oncolytic virus and small molecule therapy
Nature Communications (2022)
-
microRNAs in viral acute respiratory infections: immune regulation, biomarkers, therapy, and vaccines
ExRNA (2019)
-
Expression analysis of lung miRNAs responding to ovine VM virus infection by RNA-seq
BMC Genomics (2019)
