
Abstract
Here we report the generation of a small focused library of 12 diversely functionalized dihydropyrimidine (DHPM) derivatives via one-pot three-component Biginelli cyclocondensation of β-ketoesters, aldehydes and (thio)ureas. By applying controlled microwave heating under sealed vessel conditions using a fully automated microwave instrument including a gripper and liquid handler, the sequential synthesis of DHPMs can be performed in a shorter reaction time (10–20 min per one DHPM derivative) compared to conventional heating methods, which normally require several hours of reflux heating. The solid products either crystallize directly upon cooling or can be precipitated upon addition of water, requiring only filtration for isolation. In this way, the DHPM derivatives are obtained in high purity and no further purification by recrystallization or chromatography is necessary. This can be ascribed to the microwave heating technology where less side-product formation is often seen. The preparation of this 12-membered DHPM library can be carried out within ∼9 h.
Similar content being viewed by others
Introduction
The generation of diverse combinatorial libraries has been shown to be a valuable tool for lead compound identification in the drug discovery process1,2. However, lead compound optimization and traditional medicinal chemistry remain the bottlenecks in this high-throughput discipline. As speed is a critical factor in the field of drug discovery and medicinal chemistry, increasing interest has focused toward technologies and concepts that allow a more rapid synthesis of such compound libraries. One such high-speed technology is microwave-assisted organic synthesis, which has attracted a substantial amount of attention in the past few years and is nowadays a popular and convenient tool for performing organic reactions in a high-speed fashion3,4,5,6,7,8. In particular, the use of dedicated microwave reactors (see Fig. 1) that enable the rapid and safe heating of reaction mixtures in sealed vessels under controlled conditions with on-line temperature and pressure monitoring has greatly increased the general acceptance of this method. The main advantages of microwave-assisted heating over conventional oil-bath heating are the significant rate enhancements (from hours to minutes and even seconds), reduced side reactions and therefore improved product yields and purities. Many reaction parameters such as reaction temperature and time, variations in solvents, additives and catalysts or the molar ratios of the substrates can be evaluated in a few hours to optimize the desired chemistry. Owing to these facts, it is not surprising that the drug discovery and medicinal chemistry communities and many pharmaceutical companies are heavily using this high-speed technology for both lead optimization and lead generation9,10,11,12,13,14,15.

Automated single-mode microwave instrument with gripper and liquid handler (Emrys Liberator, Biotage AB).
For the preparation of compound libraries, two different high-throughput techniques can be applied using microwave technology, the automated sequential or parallel approach. The concept of automated sequential microwave-assisted library synthesis is a very attractive tool if small focused libraries containing ca 20–100 compounds need to be produced. By using a dedicated single-mode microwave instrument with integrated robotic vial-handling and liquid dispensing (as shown in Fig. 1), library synthesis can become as efficient as a parallel approach under conventional heating16,17. As in this technique each individual vessel is irradiated separately, a better control over the reaction parameters is ensured, but also fast iterations in protocol development and individual rapid optimization of reaction conditions. If larger compound libraries (>200 compounds) need to be generated, the sequential approach can become impractical since the time-saving aspect of microwave synthesis is diminished by having to irradiate each reaction mixture individually. Parallel synthesis can be performed in multimode instruments using either dedicated multivessel rotor systems or deep-well microtiter plates that allow a higher throughput3,18,19,20. One limitation of the parallel mode is that all reaction vessels are exposed to the same reaction conditions and therefore the same amount of identical solvent has to be used in all the vessels employed for the library synthesis.
Multicomponent reactions (MCRs), like the Biginelli three-component condensation for the generation of multifunctionalized DHPM scaffolds, constitute one of the most efficient approaches toward rapid library production and thus identification of potentially biological active compounds21. In combination with microwave heating, MCR strategies are highly attractive to the drug discovery and medicinal chemistry communities owing to the increased diversity and efficiency that can be obtained in very short times compared to conventional linear-type strategies. The Biginelli protocol is particularly attractive from the drug discovery point of view, as the resulting DHPM scaffold covers a wide range of biological targets, which has led to the generation of a number of lead compounds based on that structural core22. In several recent publications, DHPM derivatives with novel potent biological activities have been reported (see Fig. 2). For example, compound 1 showed potent cardiotonic activity, whereas 2a and 2b proved to be β-adrenergic receptor antagonists23. DHPM derivative 3 displayed significant antispasmodic and vasodilator activity24 and scaffold 4 potent antihypertensive activity25. DHPM 5 was identified as calcitonin mimetic when administered subcutaneously26 and the boron-containing compound 6 has potential as agent against the breast cancer cell line MCF7 (see ref. 27). Cyclopentyl ester 7 proved to be a potent inhibitor of the fatty acid transporter FATP4 (the S-enantiomer showed to be ∼100 times more potent than the R-enantiomer)28. The analogs 8a and 8b both exhibited a strong in vitro antioxidant activity29 and derivative 9 plays a role in the activation of chaperones that block Aβ aggregation and thus might have a favorable therapeutic effect on neurodegenerative diseases like the Alzheimer disease30,31.

Recently published DHPM derivatives with potent biological activities (for a comprehensive review, see ref. 22)
A direct and simple method for the synthesis of DHPMs is the above-mentioned Biginelli MCR, a one-pot cyclocondensation of a β-ketoester, aldehyde and (thio)urea under acidic conditions, which was first reported by Biginelli in 1893 (see Fig. 3)32,33. As it is known that conventional heating methods for this reaction normally require several hours of reflux heating and furthermore deliver the DHPM products in rather moderate yields, we wanted to take advantage of direct and rapid microwave dielectric heating under sealed vessel conditions to shorten the reaction time and to obtain the desired DHPM derivatives in high yield and purity.

Biginelli MCR.
Based on our previous reports on the microwave-assisted generation of DHPM libraries applying an automated sequential microwave instrument (see Fig. 1)34,35, we report here an updated protocol for the synthesis of a small focused 12-membered library of DHPM scaffolds utilizing the same instrument with all available automation features (see Table 1). For the library synthesis, an Emrys Liberator instrument (Biotage AB), especially designed for library production (see Fig. 1), with incorporated EmrysWorkflowManager software was employed. In addition to the built-in gripper that moves vials in and out of the cavity, this software allows liquid dispensing using the liquid handler. Furthermore, all the weights/volumes needed for each experiment are calculated automatically (see PROCEDURE for programming). Alternatively, a SWAVE microwave synthesizer (Chemspeed Technologies), which is a fully automated system incorporating the Biotage Initiator microwave reactor in combination with automated capping/decapping, automated addition of liquid and solid reagents, transfer of the vials to the microwave cavity, intermediate addition of reagents for multistep synthesis and work-up features, can be employed for the DHPM library synthesis.
Structural diversity in the DHPM scaffolds was generated by employing seven individual CH-acidic carbonyl compounds (10), ten different aldehydes (11) and two ureas/thioureas (12). A combination of all these building blocks would lead to a library of 140 individual DHPMs (13). In the present protocol, a detailed procedure for the synthesis of a set of 12 representative DHPM analogs is given. After some reoptimization of our previously published protocols34,35, we discovered that the use of 1.5 equivalents of the CH-acidic compounds in combination with 10 mol% of Ytterbium(III) trifluoromethanesulfonate hydrate (Yb(OTf)3) as a Lewis acid catalyst at 120 °C produced the best results. Under these conditions, a 10 min reaction time was employed in cases where urea was used as a building block, and a 20 min time for thiourea. As we have reported recently, acetonitrile (MeCN) proved to be the best solvent for 2-thio-DHPM derivatives36, whereas a 3:1 mixture of acetic acid (AcOH)/ethanol (EtOH) was superior for 2-oxo-DHPM products. In some cases, pure EtOH gave somewhat higher yields compared to the AcOH/EtOH 3:1 mixture (see Table 1).
Utilizing the automation features of the microwave instrument (see Fig. 1), including a gripper that moves the vials in and out of the cavity and a liquid handler for dispensing liquid building blocks or stock solutions in the vials before irradiation, the synthesis of all 12 derivatives can be performed within 3.5 h, which implies an average processing time of ∼17 min (including dispensing, moving the vial in and out of the cavity and cooling to 50 °C). Owing to the benefits of microwave heating, further purification steps such as recrystallization or column chromatography were not required. The solid and crystalline library products were obtained in an average isolated yield of 67% and very high purity (>95% according to HPLC at 215 nm) after filtration and appropriate washing protocols (see Table 1). Here, four different HPLC methods for quality control have been employed, depending on the nature of the DHPM product (see HPLC setup and Table 1). The standard method utilized was method B, which involves an initial concentration of 35% of eluent B (see HPLC setup and Fig. 4). If the DHPM products are more polar, like 13e and 13f, the concentration had to be decreased to 20% to obtain a meaningful HPLC chromatogram (see Fig. 5). If thiourea is applied, the maximum absorption is shifted to a higher wavelength in comparison to the 2-oxo-DHPMs, therefore 305 nm is set for the second wavelength (method D, see HPLC setup).

HPLC of DHPM derivative 13a using HPLC method B (linear increase from 35% solution B to 100% solution B in 9 min, hold at 100% solution B for 1 min. The UV detector is set at 215 and 283 nm).

HPLC of DHPM derivative 13f using HPLC method A (linear increase from 20% solution B to 100% solution B in 9 min, hold at 100% solution B for 1 min. The UV detector is set at 215 and 283 nm).
This automated protocol for the generation of a diverse set of DHPM scaffolds described herein can also be employed utilizing different dedicated microwave instrumentation without a liquid handler or automation (e.g., Biotage Initiator or CEM Discover/Explorer platforms). In this case, all the building blocks have to be filled into the vial manually. For the irradiation process itself, the same time/temperature parameters can be applied as given in Table 1. The same is true for the generation of DHPM scaffolds, which are not described in this procedure; as a starting point, the same reaction conditions can be employed. If the yields are not satisfactory, a change in the ratio of the reagents or solvent from AcOH/EtOH 3:1 to pure EtOH or MeCN could help to obtain the DHPM products in higher yields. Additionally, other catalysts than Yb(OTf)3 like LaCl3 for 2-thio-DHPMs35,37 or TMSCl (trimethylsilyl chloride)38,39,40,41 can be superior, as well as prolonged reaction times or slightly higher temperatures (note that above 140 °C, more by-products are obtained owing to decomposition of urea). If acid-sensitive aldehydes such as furane-2-carbaldehyde are considered, the reaction works better when EtOH is used as a solvent at a slightly lower reaction temperature of 100 °C (ref. 34). N-Substituted (thio)ureas are known to be troublesome building blocks, resulting in lower yields of DHPM products. Especially the work-up is not as facile as presented in this protocol since the corresponding DHPM derivatives generally do not precipitate upon cooling and need column chromatography or extraction for purification.
If a microwave-assisted parallel approach is considered, it has to be ensured that identical solvents and filling volumes are used in each vessel to achieve similar temperatures. For the DHPM library presented here, this means that different reaction conditions have to be incorporated for some derivatives. If AcOH/EtOH 3:1 is employed as a solvent for all reactions at 120 °C for 10 min, scaffolds 13e and 13f are obtained in somewhat lower yields (59% versus 67% for 13e and 47% versus 53% for 13f).
Materials
Reagents
-
2-Chlorobenzaldehyde (Aldrich, cat. no. 124974)
-
2-Thiophenecarboxaldehyde (Aldrich, cat. no. T32409)
-
4-Fluorobenzaldehyde (Aldrich, cat. no. 128376)
-
o-Tolualdehyde (Fluka, cat. no. 89830)
-
3-Nitrobenzaldehyde (Aldrich, cat. no. N10845)
-
Acetaldehyde (Merck, cat. no. 800004)
-
m-Tolualdehyde (Fluka, cat. no. 89840)
-
2,3-Dichlorobenzaldehyde (Aldrich, cat. no. 254835)
-
Benzaldehyde (Acros, cat. no. 1052200)
-
3-Hydroxybenzaldehyde (Aldrich, cat. no. H190808)
-
Ethyl acetoacetate (Acros, cat. no. 1179700)
-
Methyl acetoacetate (Aldrich, cat. no. 537365)
-
Isopropyl acetoacetate (Fluka, cat. no. 00485)
-
Acetylacetone (Fluka, cat. no. 00909)
-
Methyl 4-methoxyacetoacetate (Aldrich, cat. no. 281506)
-
Methyl 3-oxo-pentanoate (Acros, cat. no. 243160250)
-
Acetoacetanilide (Fluka, cat. no. 00440)
-
Urea (Riedel de Häen, cat. no. 33247)
-
Thiourea (Acros, cat. no. 138915)
-
Yb(OTf)3 (Aldrich, cat. no. 405329)
-
MeCN (HPLC grade; Fisher Scientific, cat. no. A/0627/17)
-
AcOH (Acros, cat. no. 1240400)
-
EtOH (Merck, cat. no. 9MAU111052)
Equipment
-
Microwave process vials (0.5–2 ml, cat. no. 352016; 2–5 ml, cat. no. 351521; Biotage)
Critical
Only dedicated microwave process vials as supplied by Biotage designed to withhold elevated pressures (20 bar) and temperatures must be used. Note that the instrument vendor suggests single use for the microwave vials.
-
Solvent bottle (50 ml; Biotage)
-
Magnetic stir bar (1 cm for 2–5 ml vials; Biotage)
-
Teflon septum (Biotage)
-
Aluminum crimp (Biotage)
-
Microwave reactor (Emrys Liberator; Biotage; see Fig. 1)
-
Erlenmeyer flask
-
Disposable glass pipettes
-
Micropipettes and tips (Finnpipette; Thermo)
-
Fritted funnels (alternatively Büchner funnels + filter)
-
HPLC (Shimadzu LC-20), which includes an LC-20AD pump, a SIL-20A autosampler, a diode array detector (SPD-M20A), a column oven (CTO-20A) and a degasser (DGU-20A5)
-
HPLC column: Pathfinder AS100 reversed-phase analytical column (150 × 4.6 mm, particle size 5 μm)
Equipment setup
-
HPLC setup The separations were carried out at 25 °C using a mobile phase from (A) 0.1% trifluoroacetic acid (TFA) in 90:10 water/MeCN and (B) 0.1% TFA in MeCN (all solvents were HPLC grade, Acros; TFA was analytical reagent grade, Aldrich). The following gradients were applied at a flow rate of 0.5 ml min−1.
-
Method A: linear increase from 20% solution B to 100% solution B in 9 min, hold at 100% solution B for 1 min. The UV detector is set at 215 and 283 nm.
-
Method B: linear increase from 35% solution B to 100% solution B in 9 min, hold at 100% solution B for 1 min. The UV detector is set at 215 and 283 nm.
-
Method C: linear increase from 40% solution B to 100% solution B in 9 min, hold at 100% solution B for 1 min. The UV detector is set at 215 and 283 nm.
-
Method D: linear increase from 35% solution B to 100% solution B in 9 min, hold at 100% solution B for 1 min. The UV detector is set at 215 and 305 nm.
Procedure
Program library using EmrysWorkflowManager
-
1
Open the EmrysWorkflowManager for the programming of the library and start a new experiment.
-
2
Start adding the information about the chemicals that are necessary for the library by pressing the “New Chemical” button (red-labeled button, see Fig. 6).
Figure 6 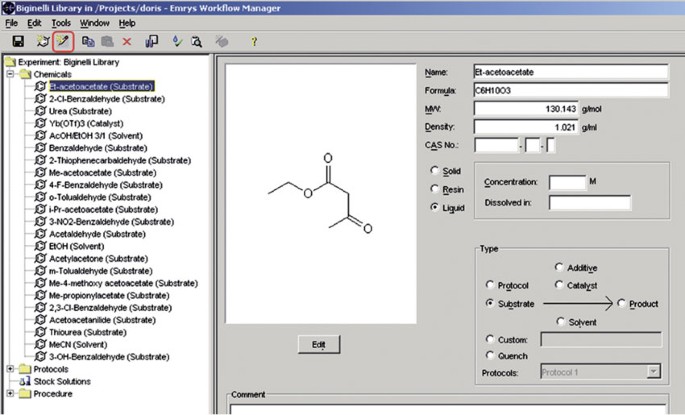
Add chemicals for the library production.
-
3
Press the “Edit” button (an ISIS-Draw window opens) and draw the structure of the first building block. By closing the ISIS-Draw window, one returns to the “Chemical” window.
-
4
Name the building block. The molecular weight is calculated by the instrument. If the building block is a liquid, enter the density. Define whether it is a solid or liquid, substrate, catalyst or solvent.
-
5
Repeat Steps 2–4 to enter all the building blocks, solvents and catalysts that are necessary for the reactions.
-
6
Start creating the protocols that are necessary for the library production by pressing the “Protocols” button (red-labeled button, see Fig. 7).
Figure 7 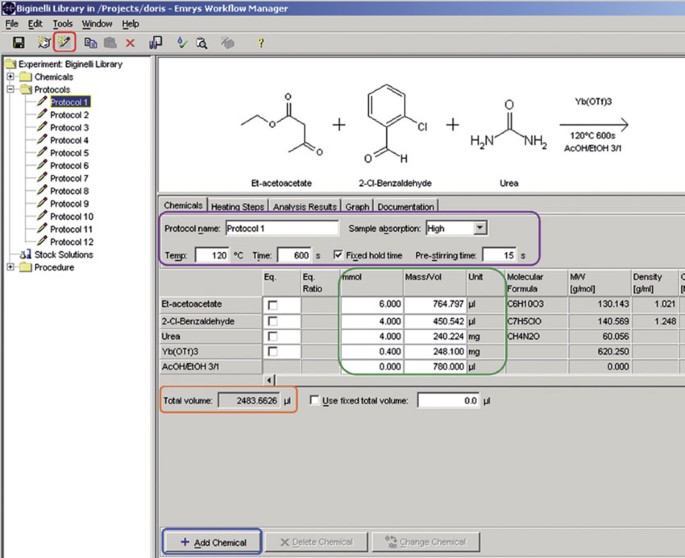
Creating protocols for the library production.
-
7
By pressing the “Add Chemicals” button (blue labeled, Fig. 7), add all the building blocks, catalysts and solvents that are needed for the first reaction (protocol).
-
8
Enter the amount (in mmol) of all the reagents: 4 mmol of aldehydes 11, 4 mmol of urea/thiourea 12, 6 mmol (1.5 equiv.) of CH-acidic compound 10, 0.4 mmol (10 mol%) of Yb(OTf)3; the instrument calculates automatically the corresponding amounts (green-labeled box, Fig. 7).
-
9
Enter the appropriate amount of solvent so that a total reaction volume of ∼2.5 ml (orange-labeled box, Fig. 7) is reached.
Critical Step
Ensure that the minimum filling volume for the corresponding vial type is reached. In the current procedure, the 2–5 ml Biotage microwave vials are used, which require a 2.0 ml minimum filling volume. If the filling volume is lower, the temperature measurement by the outside IR sensor will not be correct.
-
10
Choose “High” for the sample absorption and enter 120 °C for the temperature, 600 s (1,200 s when thiourea is used) for the reaction time and 15 s for pre-stirring; activate the “Fixed hold time” box (purple-labeled box, Fig. 7).
-
11
Follow Steps 6–10 for creating all the protocols for the 12 reactions.
-
12
By opening the “Procedure” folder, a list of all chemicals in the corresponding amounts for all reactions (protocols) opens. Define the size of the vials in which the liquid reagents should be stored and the dispensing type (here, combinatorial is chosen; see Fig. 8).
Figure 8 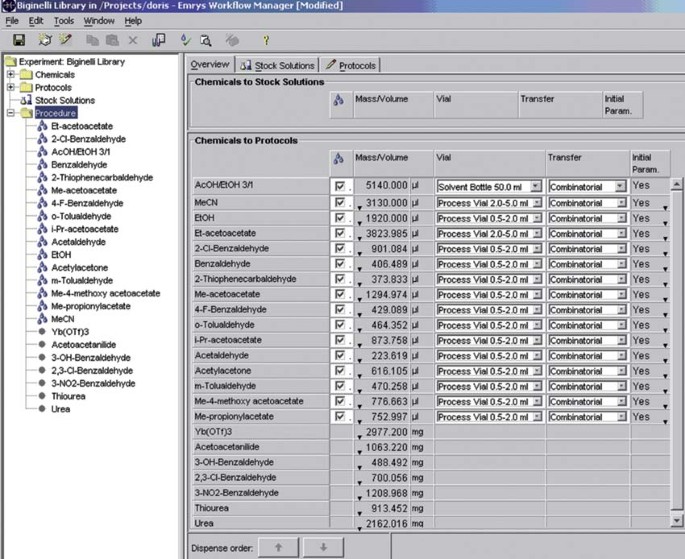
Dispensing procedure.
-
13
Save the planned experiment, open the Emrys Liberator software and upload the experiment.
-
14
Place a stir bar in each of the 12 microwave vials that are going to be irradiated in the microwave.
-
15
Add 240 mg (4 mmol) urea or 304 mg (4 mmol) thiourea, and 248 mg (10 mol%) Yb(OTf)3 into the corresponding 2–5 ml microwave vials. If other building blocks are solids, add them to the vial as well.
-
16
For the liquid dispensing procedure, fill all the liquid reagents into the appropriate microwave storage vials according to the “Procedure” instruction (see Fig. 8).
-
17
Fit a Teflon septum into the aluminum crimp and crimp all the vials.
Critical Step
The cap on the microwave vial has to be even and tight; otherwise, leakage of the reagents or solvent can occur under microwave irradiation.
-
18
Place all the microwave vials in the proper positions in the rack of the microwave instrument according to the image that appears on the screen (see Fig. 9).
Figure 9: Loading of the microwave vials onto the rack. 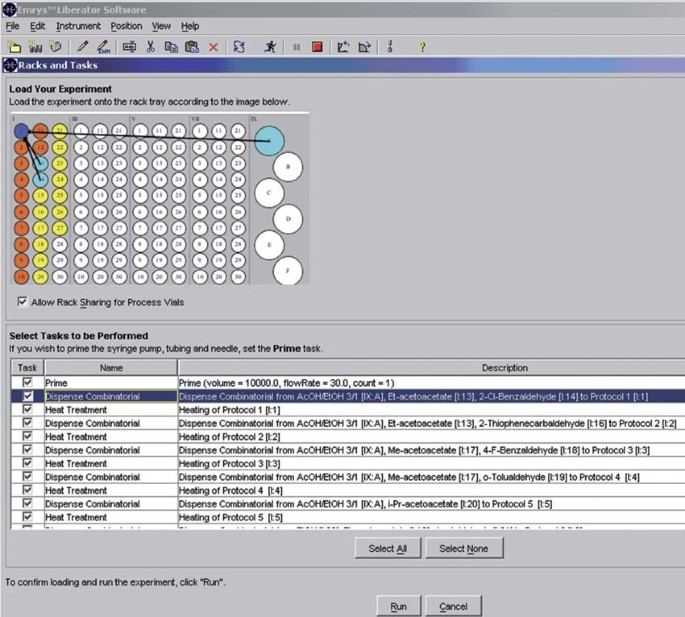
For each experiment (protocol), the procedure steps can be controlled. In the “Description” the dispensing sequence, the corresponding rack–vial position (in parentheses) of the employed chemicals and the positions of the reaction microwave vials (in parentheses), which will be irradiated (heating protocol), are provided. If the dispensing protocol is highlighted, the rack positions of the employed chemicals are highlighted in light-blue and arrows indicate in which vial they will be dispensed, which allow easier visualization of each protocol.
-
19
Run the experiment.
Caution
The solvent is heated well above its boiling point, so all necessary precautions should be taken when performing such experiments. Vessels designed to withhold elevated temperatures must be used. After completion of an experiment, the vessel must be allowed to cool to a temperature below the boiling point of the solvent before removal from the cavity and opening to the atmosphere.
-
20
After cooling to 50 °C via gas jet cooling (compressed air, 5 bar), the vials are moved out of the cavity by the gripper and placed back to the rack. In case of overpressure in the vial (∼2–3 bar), remove it by piercing the septum with a needle and open the vials.
Caution
Because of safety issues, the piercing of the septum has to be performed in the hood.
-
21
Purify the product by either filtration (A), in the case of already precipitated product, or by precipitation and filtration (B), if no product has precipitated after the cooling step.
-
A
Filtration
-
i
Place the vials in the fridge for ∼ 2 h.
Pause point
Can be left overnight.
-
ii
Filter the precipitate through a fritted funnel and wash it with 10–15 ml of a mixture of cold H2O/EtOH 2:1 in the case of 13a–f or with 15 ml of cold water in the case of 13k (see Table 1).
-
i
-
B
Precipitation and filtration
-
i
Fill an Erlenmeyer flask with ∼20 g of crushed ice.
-
ii
Transfer the reaction mixture from the vial to the Erlenmeyer flask filled with crushed ice using a glass pipette.
-
iii
Rinse the microwave vial with an additional 700–1,050 μl of the corresponding solvent and transfer it with a glass pipette to the Erlenmeyer flask filled with crushed ice.
-
iv
Let the reaction mixture stir vigorously for 2 h on a magnetic stirrer.
Pause point
Can be left overnight.
-
v
Filter the precipitates through a fritted funnel and wash it with 20 ml of a mixture of cold H2O/EtOH 1:1 in the case of 13g–i, ∼12 ml of a mixture of cold H2O/MeCN 1:2 in the case of 13j or 15 ml of cold water in the case of 13l (see Table 1).
-
i
-
A
-
22
Dry the product in the drying oven at 50 °C overnight.
-
23
Check the purity by HPLC using one of the HPLC methods described in HPLC setup.




Troubleshooting
Troubleshooting advice can be found in Table 2.
Timing
Steps 1–13: 40 min
Steps 14–17: 1 h
Step 18: 10 min
Steps 19 and 20: 3 h 30 min
Step 21A for six DHPM products: Steps (i) and (ii): ∼3 h
Step 21B for six DHPM products: Steps (i)–(iii): 15 min
Step (iv): 2 h
Step (v): 1 h
Step 22: 15 h
Anticipated results
The synthesized DHPM scaffolds together with the corresponding yield, purity and HPLC retention time can be found in Table 1.
13a: mp 210–212 °C
1H NMR (360 MHz, DMSO-d6) δ 0.99 (t, J = 7.1 Hz, 3H), 2.29 (s, 3H), 3.89 (q, J = 7.1, 2H), 5.62 (d, J = 2.7 Hz, 1H), 7.24–7.31 (m, 3H), 7.40 (d, J = 7.5 Hz, 1H), 7.70 (br s, 1H), 9.26 (br s, 1H).
13b: mp 214–217 °C
1H NMR (360 MHz, DMSO-d6) δ 1.16 (t, J = 7.1 Hz, 3H), 2.21 (s, 3H), 4.06 (q, J = 7.1, 2H), 5.41 (d, J = 3.5 Hz, 1H), 6.88–6.95 (m, 2H), 7.34 + 7.36 (dd, J = 0.9 + 5.0 Hz, 1H), 7.90 (br s, 1H), 9.31 (br s, 1H).
13c: mp 188–190 °C
1H NMR (360 MHz, DMSO-d6) δ 2.25 (s, 3H), 3.52 (s, 3H), 5.14 (d, J = 3.2 Hz, 1H), 7.14 (t, J = 8.8 Hz, 2H), 7.23–7.24 (m, 2H), 7.77 (br s, 1H), 9.25 (br s, 1H).
13d: mp 235–237 °C
1H NMR (360 MHz, DMSO-d6) δ 2.29 (s, 3H), 2.41 (s, 3H), 3.45 (s, 3H), 5.39 (d, J = 2.7 Hz, 1H), 7.11–7.17 (m, 4H), 7.63 (br s, 1H), 9.18 (br s, 1H).
13e: mp 194 °C
1H NMR (360 MHz, DMSO-d6) δ 1.09 (d, J = 6.3 Hz, 3H), 1.19 (t, J = 7.1 Hz, 3H), 2.15 (s, 3H), 4.02–4.14 (m, 3H), 7.20 (br s, 1H), 8.98 (br s, 1H).
13f: mp 250–252 °C
1H NMR (360 MHz, DMSO-d6) δ 2.09 (s, 3H), 2.28 (s, 6H), 5.21 (d, J = 3.3 Hz, 1H), 7.01–7.07 (m, 3H), 7.20 (t, J = 7.8 Hz, 1H), 7.77 (br s, 1H), 9.14 (br s, 1H).
13g: mp 199–201 °C
1H NMR (360 MHz, DMSO-d6) δ 0.98 (d, J = 6.2 Hz, 3H), 1.17 (d, J = 6.2 Hz, 3H), 2.26 (s, 3H), 4.78–4.85 (m, 1H), 5.28 (d, J = 3.0 Hz, 1H), 7.64–7.71 (m, 2H), 7.88 (br s, 1H), 8.07–8.15 (m, 2H), 9.34 (br s, 1H).
13h: mp 182–184 °C
1H NMR (360 MHz, DMSO-d6) δ 3.31 (s, 3H), 3.57 (s, 3H), 4.45–4.58 (m, 2H), 5.34 (d, J = 3.3 Hz, 1H), 7.64–7.72 (m, 2H), 8.00 (br s, 1H), 8.10–8.16 (m, 2H), 8.89 (br s, 1H).
13i: mp 212–213 °C
1H NMR (360 MHz, DMSO-d6) δ 1.15 (t, J = 7.3 Hz, 3H), 2.69 (d, J = 7.3 Hz, 2H), 3.45 (s, 3H), 5.66 (d, J = 3.1 Hz, 1H), 7.25 and 7.27 (dd, J = 1.4 and 7.8 Hz, 1H), 7.36 (t, J = 7.9 Hz, 1H), 7.54 and 7.56 (dd, J = 1.4 and 7.9 Hz, 1H), 7.77 (br s, 1H), 8.36 (br s, 1H).
13j: mp 195–197 °C
1H NMR (360 MHz, DMSO-d6) δ 2.01 (s, 3H), 5.77 (s, 1H), 7.01–7.51 (m, 9H), 9.34 (br s, 1H), 9.86 (br s, 1H), 10.05 (br s, 1H).
13k: mp 205 °C
1H NMR (360 MHz, DMSO-d6) δ 1.10 (t, J = 7.1 Hz, 3H), 2.29 (s, 3H), 4.01 (q, J = 7.0, 2H), 5.17 (d, J = 3.5 Hz, 1H), 7.20–7.37 (m, 5H), 9.65 (br s, 1H), 10.33 (br s, 1H).
13l: mp 179 °C
1H NMR (360 MHz, DMSO-d6) δ 1.12 (t, J = 7.0 Hz, 3H), 2.28 (s, 3H), 4.02 (q, J = 7.0, 2H), 5.08 (d, J = 3.3 Hz, 1H), 6.63–6.65 (m, 3H), 7.12 (t, J = 7.6 Hz, 1H), 9.44 (br s, 1H), 9.60 (br s, 1H).
References
Thompson, L.A. & Ellman, J.A. Synthesis and applications of small molecule libraries. Chem. Rev. 96, 555–600 (1996).
Dolle, R.E., Le Bourdonnec, B., Morales, G.A., Moriarty, K.J. & Salvino, J.M. Comprehensive survey of combinatorial library synthesis: 2005. J. Comb. Chem. 8, 597–635 (2006).
Kappe, C.O. & Stadler, A. Microwaves in Organic and Medicinal Chemistry (Wiley-VCH, Weinheim, 2005).
Loupy, A. (ed.) Microwaves in Organic Synthesis 2nd edn. (Wiley-VCH, Weinheim, 2006).
Kappe, C.O. & Dallinger, D. The impact of microwave synthesis on drug discovery. Nat. Rev. Drug Discov. 5, 51–64 (2006).
Lidström, P. & Tierney, J.P. (eds.) Microwave-assisted Organic Synthesis (Blackwell Publishing, Oxford, 2005).
Kappe, C.O. Controlled microwave heating in modern organic synthesis. Angew. Chem. Int. Ed. 43, 6250–6284 (2004).
Hayes, B.L. Recent advances in microwave-assisted synthesis. Aldrichim. Acta 37, 66–77 (2004).
Lindsley, C.W. et al. Allosteric Akt (PKB) inhibitors: discovery and SAR of isozyme selective inhibitors. Bioorg. Med. Chem. Lett. 15, 761–764 (2005).
Nöteberg, D., Schaal, W., Hamelink, E., Vrang, L. & Larhed, M. High-speed optimization of inhibitors of the malarial proteases plasmepsin I and II. J. Comb. Chem. 5, 456–464 (2003).
Coats, S.J. et al. Parallel methods for the preparation and SAR exploration of N-ethyl-4-[(8-alkyl-8-azabicyclo[3.2.1]-oct-3-ylidene)-aryl-methyl]-benzamides, powerful mu and delta opioid agonists. Bioorg. Med. Chem. Lett. 14, 5493–5498 (2004).
Kempson, J. et al. Fused pyrimidine based inhibitors of phosphodiesterase 7 (PDE7): synthesis and initial structure–activity relationships. Bioorg. Med. Chem. Lett. 15, 1829–1833 (2005).
Lehmann, F., Currier, E.A., Olsson, R., Hacksell, U. & Luthman, K. Isochromanone-based urotensin-II receptor agonists. Bioorg. Med. Chem. 13, 3057–3068 (2005).
Holmberg, P., Tedenborg, L., Rosqvist, S. & Johansson, A.M. Novel 3-aminochromans as potential pharmacological tools for the serotonin 5-HT7 receptor. Bioorg. Med. Chem. Lett. 15, 747–750 (2005).
Liu, J.-F. et al. Novel one-pot total syntheses of deoxyvasicinone, mackinazolinone, isaindigotone, and their derivatives promoted by microwave irradiation. Org. Lett. 7, 3363–3366 (2005).
Stadler, A. & Kappe, C.O. Automated library generation using sequential microwave-assisted chemistry. Application toward the Biginelli multicomponent condensation. J. Comb. Chem. 3, 624–630 (2001).
Larhed, M. & Hallberg, A. Microwave-assisted high-speed chemistry: a new technique in drug discovery. Drug Disc. Today 6, 406–416 (2001).
Strohmeier, G.A. & Kappe, C.O. Rapid parallel synthesis of polymer-bound enones utilizing microwave-assisted solid-phase chemistry. J. Comb. Chem. 4, 154–161 (2002).
Stadler, A. et al. Scalability of microwave-assisted organic synthesis. From single-mode to multimode parallel batch reactors. Org. Process Res. Dev. 7, 707–716 (2003).
Kremsner, J.M., Stadler, A. & Kappe, C.O. High-throughput microwave-assisted organic synthesis: moving from automated sequential to parallel library-generation formats in silicon carbide microtiter plates. J. Comb. Chem. 9, 285–291 (2007).
Zhu, J. & Bienaymé, H. (eds.) Multicomponent Reactions (Wiley-VCH, Weinheim, 2005).
Kappe, C.O. Biologically active dihydropyrimidones of the Biginelli-type. A literature survey. Eur. J. Med. Chem. 35, 1043–1052 (2000).
Sujatha, K., Shanmugam, P., Perumal, P.T., Muralidharan, D. & Rajendran, M. Synthesis and cardiac effects of 3,4-dihydropyrimidin-2-(1H)-one-5 carboxylates. Bioorg. Med. Chem. Lett. 16, 4893–4897 (2006).
Zorkun, I.S., Saraç, S., Çelebi, S. & Erol, K. Synthesis of 4-aryl-3,4-dihydropyrimidin-2(1H)-thione derivatives as potential calcium channel blockers. Bioorg. Med. Chem. 14, 8582–8589 (2006).
El-Hamouly, W.S., El-Khamry, A.-M.A. & Abbas, E.M.H. Synthesis of new 4-aryl-isoxazolo[5,4-d]pyrimidin-6-one(thione) and 4-aryl-pyrazolo[3,4-d]pyrimidin-6-one derivatives of potential antihypertensive activity. Indian J. Chem. B 45B, 2091–2098 (2006).
Matthews, J.M. et al. Discovery of a dihydropyrimidine series of molecules that selectively mimic the biological actions of calcitonin. Bioorg. Med. Chem. Lett. 14, 1155–1159 (2004).
Blacquiere, J.M. et al. Dihydropyrimidinones containing boronic acids. Can. J. Chem. 83, 2952–2059 (2005).
Blackburn, C. et al. Identification and characterization of 4-aryl-3,4-dihydropyrimidin-2(1H)-ones as inhibitors of the fatty acid transporter FATP4. Bioorg. Med. Chem. Lett. 16, 3504–3509 (2006).
Stefani, H.A. et al. Dihydropyrimidin-(2H)-ones obtained by ultrasound irradiation: a new class of potential antioxidant agents. Eur. J. Med. Chem. 41, 513–518 (2006).
Evans, C.G., Wisén, S. & Gestwicki, J.E. Heat shock proteins 70 and 90 inhibit early stages of amyloid β-(1-42) aggregation in vitro. J. Biol. Chem. 281, 33182–33191 (2006).
Fewell, S.W., Smith, C.M., Lyon, M.A., Dumitrescu, T.P., Wipf, P., Day, B.W. & Brodsky, J.L Small molecule modulators of endogenous and co-chaperone stimulated Hsp70 ATPase activity. J. Biol. Chem. 279, 51131–51140 (2004).
Biginelli, P. Aldehyde-urea derivatives of aceto- and oxaloacetic acids. Gazz. Chim. Ital. 23, 360–413 (1893).
Kappe, C.O. Recent advances in the Biginelli dihydropyrimidine synthesis. New tricks from an old dog. Acc. Chem. Res. 33, 879–888 (2000).
Stadler, A. & Kappe, C.O. Automated library generation using sequential microwave-assisted chemistry. Application toward the Biginelli multicomponent condensation. J. Comb. Chem. 3, 624–630 (2001).
Kappe, C.O. & Stadler, A. Building dihydropyrimidine libraries via microwave-assisted Biginelli multicomponent reactions. Methods Enzymol. 369, 197–223 (2003).
Dallinger, D. & Kappe, C.O. Rapid preparation of the mitotic kinesin Eg5 inhibitor monastrol using controlled microwave-assisted synthesis. Nat. Protoc. 2, 317–321 (2007).
Lu, J., Bai, Y., Wang, Z., Yang, B. & Ma, H. One-pot synthesis of 3,4-dihydropyrimidin-2(1H)-ones using lanthanum chloride as a catalyst. Tetrahedron Lett. 41, 9075–9078 (2000).
Zhu, Y., Pan, Y. & Huang, S. Trimethylsilyl chloride: a facile and efficient reagent for one-pot synthesis of 3,4-dihydropyrimidin-2(1H)-ones. Synth. Commun. 34, 3167–3174 (2004).
Matloobi, M. & Kappe, C.O. Microwave-assisted solution- and solid-phase synthesis of 2-amino-4-arylpyrimidine derivatives. J. Comb. Chem. 9, 275–284 (2007).
Prokopcová, H., Pisani, L. & Kappe, C.O. Synthesis of 5-aroyldihydropyrimidinones via Liebeskind-Srogl thiol ester-boronic acid cross-couplings. Synlett 43–46 (2007).
Pisani, L., Prokopcová, H., Kremsner, J.M. & Kappe, C.O. 5-Aroyl-3,4-dihydropyrimidin-2-one library generation via automated sequential and parallel microwave-assisted synthesis techniques. J. Comb. Chem. 9, 415–421 (2007).
Acknowledgements
We thank Biotage AB for the provision of the Emrys Liberator microwave reactor.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Rights and permissions
About this article
Cite this article
Dallinger, D., Kappe, C. Automated generation of a dihydropyrimidine compound library using microwave-assisted processing. Nat Protoc 2, 1713–1721 (2007). https://doi.org/10.1038/nprot.2007.224
Published:
Issue Date:
DOI: https://doi.org/10.1038/nprot.2007.224
This article is cited by
-
A rapid and efficient route to benzazole heterocycles
Nature Protocols (2010)
-
Synthesis of alkyl- and aryl-amino-substituted anthraquinone derivatives by microwave-assisted copper(0)-catalyzed Ullmann coupling reactions
Nature Protocols (2010)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
