
Abstract
Ingesting ethanol (EtOH) at low doses during social drinking is a common human behavior for its facilitating effects on social interactions. However, low-dose EtOH may have also detrimental effects that so far are underexplored. Here we sought to test the effects of low-dose EtOH on long-term potentiation (LTP)-like plasticity in human motor cortex. Previous cellular experiments showed that low-dose EtOH potentiates extrasynaptic GABAAR and reduces NMDAR-mediated currents, processes that would limit the expression of LTP. Paired associative transcranial magnetic stimulation (PASLTP) was employed in nine healthy subjects for induction of LTP-like plasticity, indexed by a long-term increase in motor-evoked potential input–output curves. Synaptic α1-GABAAR function was measured by saccadic peak velocity (SPV). Very low doses of EtOH (resulting in blood concentrations of <5 mM) suppressed LTP-like plasticity but did not affect SPV when compared with a placebo condition. In contrast, 1 mg of alprazolam, a classical benzodiazepine, or 10 mg of zolpidem, a non-benzodiazepine hypnotic, decreased SPV but did not significantly affect LTP-like plasticity when compared with placebo. This double dissociation of low-dose EtOH vs alprazolam/zolpidem effects is best explained by the putatively high affinity of EtOH but not alprazolam/zolpidem to extrasynaptic GABAARs and to NMDARs. Findings suggest that enhancement of extrasynaptic GABAAR-mediated tonic inhibition and/or reduction of NMDAR-mediated neurotransmission by EtOH blocks LTP-like plasticity in human cortex at very low doses that are easily reached during social drinking. Therefore, low-dose EtOH may jeopardize LTP-dependent processes, such as learning and memory formation.
Similar content being viewed by others


INTRODUCTION
Although ingestion of ethanol (EtOH) at low doses during social drinking is a common human behavior for its facilitating effects on social interactions, possible detrimental effects of low-dose EtOH remain underexplored. Cellular studies showed that low-dose EtOH (⩽30 mM) produces mainly two effects: potentiation of extrasynaptic gamma-aminobutyric acid type A receptor (GABAAR)-mediated tonic inhibition (Sundstrom-Poromaa et al, 2002; Wallner et al, 2003; Wei et al, 2004) (for review, (Olsen et al, 2007)), although this has not been unanimously supported (Borghese et al, 2006; Yamashita et al, 2006), and N-methyl-D-aspartate receptor (NMDAR) inhibition (Lovinger et al, 1989, 1990; Weitlauf and Woodward, 2008).
These effects may disrupt long-term potentiation (LTP) of synapses and LTP-dependent processes such as learning and memory formation. In slice preparations of rat motor cortex (M1), LTP induction depends on disinhibition by application of a synaptic GABAAR antagonist and can be disrupted by NMDAR blockade (Aroniadou and Keller, 1995; Castro-Alamancos et al, 1995; Fritsch et al, 2010; Hess et al, 1996). At the systems level of human M1, LTP-like plasticity, indexed by a long-term increase in motor-evoked potential (MEP) amplitude, can be induced by paired associative stimulation (PASLTP) (Cooke and Bliss, 2006; Müller-Dahlhaus et al, 2010; Stefan et al, 2002; Stefan et al, 2000; Ziemann et al, 2004). This LTP-like increase in MEP amplitude shows tight similarities to cellular LTP because it is associative, input specific, and blocked by dextromethorphan, a non-competitive NMDAR antagonist (Stefan et al, 2002; Stefan et al, 2000; Wolters et al, 2003). Furthermore, PASLTP-induced LTP-like plasticity interacts homeostatically with prior or subsequent motor learning (Elahi et al, 2013; Jung and Ziemann, 2009; Kang et al, 2011; Rosenkranz et al, 2007; Stefan et al, 2006; Ziemann et al, 2004), indicating its mechanistic importance in learning processes.
Here we sought to investigate the effects of two very low doses of EtOH (resulting in blood concentrations of <5 and <20 mM, respectively) on PASLTP-induced LTP-like plasticity in healthy volunteers, and to contrast them with those of two specific positive modulators of synaptic GABAAR-mediated inhibition (alprazolam, a classical benzodiazepine, and zolpidem, a non-benzodiazepine hypnotic with preferential α1-GABAAR affinity) (Möhler et al, 2002). We measured increases in GABAAR-mediated inhibition by slowing of saccadic peak velocity (SPV) (de Visser et al, 2003), rather than by other available techniques such as pharmacoelectroencephalography (Valle et al, 2002) or pharmacotranscranial magnetic stimulation (Paulus et al, 2008) because changes in SPV reflect changes specifically in synaptic α1-GABAAR- rather than α2- and α3-GABAAR-mediated inhibition (de Haas et al, 2009; de Haas et al, 2010) and, therefore, are expected to capture the sedative effects of alprazolam and zolpidem.
We show that low-dose EtOH abolished the PASLTP-induced LTP-like plasticity obtained in a placebo session, but had no effect on SPV. In contrast, alprazolam and zolpidem decreased SPV but exerted no significant effect on PASLTP-induced LTP-like plasticity. This provides strong evidence that LTP-like plasticity in human cortex is highly efficiently blocked by very low doses of EtOH that are easily reached during social drinking. The cellular mechanisms of this detrimental low-dose EtOH effect cannot be disentangled at the systems level but likely relate to the potentiation of tonic inhibition mediated by extrasynaptic GABAARs and/or blockade of glutamatergic neurotransmission through NMDARs. Findings have potentially significant impact at the behavioral level, as acute EtOH ingestion impairs LTP-dependent processes such as learning and memory formation (Lister et al, 1991; Lowy, 1970; Mattila et al, 1998).
METHODS
Subjects
Written informed consent was obtained prior to participation. The experiments conformed to the Declaration of Helsinki and were approved by the ethics committee of the hospital of the Goethe-University of Frankfurt am Main, Germany. All subjects completed the adult safety screen questionnaire (Keel et al, 2001). Nine healthy right-handed (Oldfield, 1971) subjects (mean (±SD) age, 26.2±4.3 years; mean (±SD) body length, 180.3±7.6 cm; mean (±SD) body weight, 80.9±15.1 kg; 6 males) were enrolled. None of the subjects had a history of neurological or psychiatric disease or was on CNS-active drugs at the time of the experiments as confirmed by comprehensive urine analysis. None of the subjects ever took alprazolam or zolpidem before, or consumed EtOH regularly. All subjects were non-smokers, as nicotine may alter PASLTP-induced plasticity (Thirugnanasambandam et al, 2011). The participating women used a hormonal contraception to avoid possible menstrual cycle-related alteration of M1 excitability and plasticity (Smith et al, 2002). Thirty subjects were screened. In a first screening step, resting motor threshold (RMT) was determined. Only those subjects with RMT⩽50% of maximum stimulator output (n=22) were retained for a second screening step (PASLTP screening) because RMT>50% of maximum stimulator output is associated with a low probability for a LTP-like response after PASLTP (Müller-Dahlhaus et al, 2008). After a second screening step, nine subjects were retained and enrolled into this study that exhibited a significant PASLTP-induced increase in MEP amplitude⩾1.2 (ratio of MEP amplitude post-PAS/pre-PAS) (Heidegger et al, 2010; Korchounov and Ziemann, 2011). Therefore, this selection excluded subjects with a long-term depression-like MEP decrease or no MEP change following PASLTP (Müller-Dahlhaus et al, 2008), as the explicit aim of this study was to study drug effects on LTP-like plasticity, a process with significant relation to motor learning (Jung and Ziemann, 2009; Kang et al, 2011; Rosenkranz et al, 2007; Ziemann et al, 2004), rather than exploring drug effects on a great variety of magnitudes and directions of PASLTP-induced plasticity in the general population.
EMG Recordings
Subjects were seated in a comfortable reclining chair with their arms and hands lying relaxed on the armrests. All transcranial magnetic stimulation (TMS) measurements were obtained by surface electromyography (EMG) from the resting abductor pollicis brevis (APB) muscle of the dominant right hand by using wafer electrodes attached to the muscle belly (active electrode) and the proximal phalanx of the index finger (reference electrode). Reproducibility of exact electrode placement within each visit was assured by marking the electrode spots after baseline measurement with a waterproof marker. The EMG raw signal was amplified and band-pass filtered (20 Hz to 2 kHz; Digitimer D360 8-channel amplifier, Digitimer, Welwyn Garden City, UK), digitized at an A/D rate of 5 kHz per channel (CED Micro 1401; Cambridge Electronic Design, Cambridge, UK) and stored in a laboratory computer for online visual display and later offline analysis using customized data collection and conditional averaging software (Spike 2 for Windows, Version 3.05, CED). All measurements were conducted during complete voluntary muscle relaxation, which was monitored audio-visually by high-gain (50 μV/Div) EMG.
Stimulation Procedures
Focal TMS of the hand area of the left primary motor cortex (M1) was performed with a figure-of-eight coil (diameter of each wing, 70 mm) and a Magstim 200 magnetic stimulator (Magstim Company, Carmarthenshire, Wales, UK) with monophasic current waveform. The optimal coil position over the hand area of the left M1 for eliciting MEPs in the right APB was determined as the site where TMS at a slightly suprathreshold intensity consistently produced the largest MEPs. This site was marked with a soft-tipped pen on the scalp in order to assure a constant placement of the coil throughout the experiment. The coil was held tangential to the scalp with the handle pointing backwards and 45 degrees away from the midline. This orientation induced a lateral-posterior to medial-anterior current in the brain, activating the corticospinal system preferentially transsynaptically via horizontal corticocortical connections (Di Lazzaro et al, 2008).
RMT was defined to the nearest 1% of maximum stimulator output (MSO) as the lowest stimulus intensity that elicited small MEPs (⩾50 μV) in at least five out of ten consecutive trials in the relaxed APB, starting determination from a slightly suprathreshold intensity (Groppa et al, 2012).
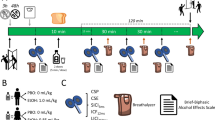
MEP input–output (IO)-curves were centered on a stimulus intensity that evoked an MEP of 1 mV peak-to-peak amplitude (SI1mV). Eight MEPs each were recorded in randomized order with stimulus intensities of 0.5, 0.7, 0.8, 0.9, 1.0 (=SI1mV), 1.1, 1.2, 1.3, and 1.5 times SI1mV (Rosenkranz et al, 2007). The peak-to-peak MEP amplitude was analyzed in the single trials and then conditional averages were calculated. MEP IO-curves were determined at baseline (time point B0, before drug intake), at B1 (90 min after tablet and 60 min after drink intake, immediately prior to PASLTP, time point B1) and 5 and 30 min after PASLTP (time points P1 and P2) (cf. time line of the experiment in Figure 1). Stimulus intensities were kept constant throughout a given experimental session.

Time line of experimental sessions. Saccadic peak velocity ((SPV), marker of sedation and α1-(gamma-aminobutyric acid type A receptor) GABAAR-mediated inhibition) and motor-evoked potential input–output curves (IO-curves), marker of corticospinal excitability) were measured at baseline (B0). Then the study drug (alprazolam tablet, zolpidem tablet, ethanol low- or high-dose drink, placebo) was administered in a double blind, double dummy design (first tablet, 30 min later drink, see also Table 1). After 60 min waiting (to reach peak plasma concentrations of study drug), SPV and IO-curve were re-tested (B1) to measure the effects of the study drug on these markers. Then, PASLTP was applied and IO-curve was re-tested 5 min later (P1) and 30 min later (P2) to investigate the long-term potentiation (LTP)-like increase in corticospinal excitability. In addition, a blood sample was taken at P1 to measure the blood concentration of the study drug.
Paired Associative Stimulation (PASLTP)
PASLTP was performed according to a protocol originally described by (Stefan et al, (2000)) and later on slightly modified by our group (Müller et al, 2007; Müller-Dahlhaus et al, 2008). It consisted of 225 pairs of electrical stimulation of the right median nerve at the wrist followed by TMS delivered at a rate of 0.25 Hz (duration: 15 min). Electrical stimulation was applied through a bipolar electrode (cathode proximal) at an intensity of three times the perceptual threshold. The intensity of TMS was adjusted to SI1mV in the resting APB when given without the preceding median nerve stimulus. To produce a long lasting LTP-like increase in MEP amplitude the interstimulus interval was equal to the individual N20-latency of the median nerve somatosensory-evoked cortical potential plus 2 ms (N20+2 ms) (Müller et al, 2007). The mean (±SD) interstimulus interval was 21.9±0.6 ms. Attention may have considerable effects on the magnitude of the PASLTP effect (Kamke et al, 2012; Stefan et al, 2004). Therefore, to control the level of attention, a randomly flashing light emitting electrode was attached to the back of the stimulated hand. Subjects were instructed to count the flashes and to report the number at the end of the PAS intervention.
Saccadic Peak Velocity Measurements
Visually guided SPV is a biomarker of sedation mediated through the α1-GABAAR (de Haas et al, 2009; de Haas et al, 2010; de Visser et al, 2003). We were interested in obtaining this marker at baseline (time point B0) and after drug intake (time point B1, cf. time line in Figure 1) to estimate the contribution of GABAAergic sedation to drug effects on LTP-like plasticity. Subjects sat in front of a screen (eyes-to-screen distance, 90 cm) and were instructed to make visually guided saccades in response to a white dot subtending an angle of view of 1° on a black screen while the head was maintained in straight position. The dot jumped at randomized intertrial intervals of 2–3 s (to prevent anticipation of the next event) horizontally from one lateral edge to the opposite edge of the screen, subtending an angle of view of 40°. Per time point 90 s were recorded, resulting on average in 36 trials. Saccade recordings were obtained by electronystagmography using surface wafer electrodes placed at the outer canthus of each eye. The electronystagmography raw signals were amplified and band-pass filtered (20 Hz to 2 kHz; Digitimer D360), digitized at an A/D rate of 5 kHz per channel (CED Micro 1401) and stored in a laboratory computer for online visual display and later offline analysis using customized data collection and conditional averaging software (Spike 2 for Windows, Version 3.05). The raw data were exported into MATLAB (version 6.1; Natick, MA). Software written in-house was used for manually setting markers of saccade onsets and offsets. SPV (in °/s) was determined by automatic identification of the maximum value between a pair of markers of saccade onset and offset using a third-order polynomial fit of the raw signal (Velazquez-Perez et al, 2004). Conditional SPV averages were calculated for left- and rightward saccades and a grand mean was finally calculated for each individual, session, and time point.
Pharmacokinetic Measurements
Pharmacokinetic measurements were performed in the Institute for Forensic Toxicology, Goethe-University Frankfurt/Main using well-established chromatographic-mass spectrometric screening and target compound analyses. At each visit, a urine sample before drug intake was screened for medical drugs and drugs of abuse to ensure that subjects were free of the study drugs or any other psychoactive substance at baseline. Blood samples were taken at time point P1 to measure the plasma levels of the study medications.
Experimental Design
We performed a randomized double blind, double dummy, placebo (PBO)-controlled five-period crossover study (Table 1). Study visits always started at 8:00 a.m. to exclude diurnal variability of PASLTP effects (Sale et al, 2007), and were separated by at least 1 week to prevent carry-over effects between sessions. Subjects were in a fastening state and received during each visit one tablet immediately after the MEP IO-curve and SPV measurements at time point B0 and one drink 30 min later (cf. Table 1 and time line in Figure 1). Study medications were: alprazolam (APZ, 1 mg), zolpidem (ZLP, 10 mg), 96% ethanol in two different dosages (resulting in blood concentrations of <5 mM and <20 mM, respectively, and hence termed EtOH<5mM, and EtOH<20mM), and PBO tablet and PBO drink (mixture of orange juice and bitter syrup to imitate ethanol content). EtOH<5mM and EtOH<20mM doses were individually calculated according to the Widmark formula to reach a peak blood ethanol concentration of 0.35‰ and 0.65‰, respectively. Male subjects received ethanol doses of 0.29 and 0.55 g/kg, and female subjects 0.25 and 0.47 g/kg in the EtOH<5mM and EtOH<20mM conditions, respectively. Due to the fast metabolism of alcohol, we expected plasma levels of ∼0.2‰ (≈4.6 mM) and 0.5‰ (≈11.6 mM) at the time of determination (time point P1, Figure 1). One blood sample in the EtOH<5mM condition could not be retrieved for analysis. Dosages of APZ and ZLP were selected according to previously shown sedative effects indexed by significant slowing of SPV (Blom et al, 1990; de Haas et al, 2010), while selection of dosages of EtOH<5mM and EtOH<20mM was based on their specific action on recombinant α4ß3δ- and α6ß3δ-GABAARs (Wallner et al, 2003). Timing of B1 and PASLTP after tablet and drink intake (cf. Figure 1) was planned according to the expected times of peak plasma concentration of APZ, ZLP, and EtOH in healthy young adults (de Haas et al, 2010; Greenblatt and Wright, 1993; Welling et al, 1977).
Statistics
Statistical testing was performed with IBM SPSS Statistics (Version 20.0.0). Drug effects on SPV were analyzed with a mixed repeated measures analysis of variance (rmANOVA) with DRUG as between-subject effect (5 levels: PBO, APZ, ZLP, EtOH<5mM, EtOH<20mM) and TIME (2 levels: B0, B1) as within-subject effect. Post hoc rmANOVAs compared the effects of each drug (APZ, ZLP, EtOH<5mM, EtOH<20mM) pairwise with the effects of PBO on SPV with DRUG as between-subject effect (2 levels: drug vs PBO) and TIME (2 levels: B0, B1) as within-subject effect. Post hoc two-tailed t-tests were performed in case of significant DRUG * TIME interactions.
The MEP data were not normally distributed according to Wilk–Shapiro testing. Therefore, a logarithmic transformation was applied to obtain a normal distribution of the MEP data (Bland and Altman, 1996). All statistical tests were performed on these transformed MEP data. The effects of DRUG on MEP IO-curve were tested in a mixed rmANOVA with DRUG as between-subject effect (5 levels: PBO, APZ, ZLP, EtOH<5mM, EtOH<20mM) and TIME (2 levels: B0, B1) and Stimulus Intensity (SI, 9 levels: 0.5 × SI1mV, 0.7 × SI1mV, 0.8 × SI1mV, 0.9 × SI1mV, 1.0 × SI1mV, 1.1 × SI1mV, 1.2 × SI1mV, 1.3 × SI1mV, 1.5 × SI1mV) as within-subject effects. Similarly, the effects of DRUG on PASLTP-induced changes in MEP IO-curve were tested by a mixed rmANOVA with DRUG (5 levels) as between-subject effect, and TIME (3 levels: B1, P1, P2) and SI (9 levels) as within-subject effects. Post hoc rmANOVAs compared the effects of each drug (APZ, ZLP, EtOH<5mM, EtOH<20mM) pairwise with the effects of PBO on PASLTP-induced changes in MEP IO-curve.
In order to obtain a single measure of the drug and PASLTP-induced changes in the MEP IO-curves, the area under logarithmically transformed MEP IO-curves (AUIOC) was calculated for each subject, time point, and experimental session. The AUIOC is a highly reliable and valid measure to characterize the excitability state of the corticospinal projection to hand muscles (Carson et al, 2013). To correlate drug blood concentrations (determined at time point P1, cf. Figure 1) and changes in SPV with drug effects on the PASLTP-induced changes in AUIOC, the following index was calculated: (AUIOC(P1)-AUIOC(B1))DRUG—(AUIOC(P1)-AUIOC(B1))PBO. This way, the PASLTP-induced change in AUIOC in any of the DRUG conditions is related to the PASLTP-induced AUIOC increase in the PBO condition. This index is referred to as normalized ΔAUIOC in this paper. Correlation analyses of the normalized ΔAUIOC with drug blood concentrations and changes in SPV (difference between time points B1–B0) were performed by linear regression.
For all rmANOVAs, Mauchly’s test was applied to test for sphericity and in case of violation of sphericity, the degrees of freedom were corrected by the Greenhouse–Geisser test. Significance was assumed when p<0.05. All data are reported as means±SEM, unless stated otherwise.
RESULTS
Experimental procedures and study drugs were generally well tolerated except for ZLP, which caused nausea and vomiting in one subject. Other common adverse events were mild to moderate sedation or dizziness, and did not limit full compliance of the subjects with the requirements of this study.
Baseline Excitability Data (RMT, SI1mV at Time Point B0)
ANOVAs did not show differences between DRUG conditions for RMT or SI1mV at time point B0 (all p>0.75, Table 2).
Plasma Concentrations of Drugs at Time Point P1
The following plasma concentrations were measured in the respective drug conditions: APZ: 9.61±0.48 ng/ml; ZLP: 98.2±37.1 ng/ml; EtOH<5mM: 0.13±0.02‰ (≈3.01±0.46 mM; range: 0.46–4.40 mM); EtOH<20mM: 0.55±0.05‰ (≈12.74±1.16 mM; range, 5.79–18.30 mM). The EtOH<5mM vs EtOH<20mM concentrations were significantly different (unpaired two-tailed t-test, t15=6.15, p<0.001).
Drug Effects on Saccadic Peak Velocity (Comparison of SPV at Time Points B1 vs B0)
In the PBO condition, there was no effect of TIME on SPV (two-tailed paired t-test, t8=0.26, p=0.80, Figure 2). The mixed rmANOVA for all drug conditions revealed significant effects of TIME (F1,40=25.62, p<0.001) and the DRUG * TIME interaction (F4,40=3.08, p=0.027) but not DRUG (F4,40=1.70, p=0.17) (Figure 2a–d). Post hoc pairwise rmANOVAs demonstrated significant DRUG * TIME interactions for APZ vs PBO (F1,16=7.40, p=0.015, Figure 2a) and ZLP vs PBO (F1,16=12.08, p=0.003, Figure 2b), but not for EtOH<5mM vs PBO (F1,16=0.21, p=0.65, Figure 2c) or EtOH<20mM vs PBO (F1,16=3.11, p=0.10, Figure 2d). The significant DRUG * TIME interactions were explained by a reduction in SPV post-drug compared with PBO (all p<0.05 in two-tailed unpaired t-tests, indicated by asterisks in Figure 2a and b). These data demonstrate that only APZ and ZLP, but not EtOH<5mM or EtOH<20mM resulted in significant SPV reduction, indicative of a sedative effect mediated by positive modulation at the α1-GABAAR by APZ and ZLP, but not by EtOH<5mM or EtOH<20mM.

Saccadic peak velocity (SPV, in °/s) at time points B0 (before drug intake) and B1 (after drug intake) in the alprazolam (APZ) (a), zolpidem (ZLP) (b), ethanol-low (EtOH<5mM) (c), and ethanol-high (EtOH<20mM) (d) condition compared with the placebo (PBO) condition (black circles in a–d). All data are means (n=9)±SEM. Asterisks indicate significant difference to PBO (p<0.05).
Drug Effects on MEP IO-Curve (Comparison of Time Points B1 vs B0)
The rmANOVA revealed no effects of DRUG (F4,40=0.98, p=0.43), or the DRUG * TIME (F4,40=0.61, p=0.66) or DRUG * TIME * SI interactions (F14.77,147.68=1.19, p=0.28) (Figure 3a–d). This is an important nil finding because it indicates that there were no significant drug effects on MEP IO-curve at time point B1 (immediately prior to PASLTP) that could have potentially confounded interpretation of the PASLTP data.

Logarithmically transformed motor-evoked potential (MEP) input–output (IO)-curves at time points B0 (before drug intake, filled symbols) and B1 (after drug intake, open symbols) as a function of stimulus intensity (in multiples of SI1mV) in the alprazolam (APZ) (a), zolpidem (ZLP) (b), ethanol-low (EtOH<5mM) (c) and ethanol-high (EtOH<20mM) (d) condition compared to the placebo (PBO) condition (black circles and lines in a–d). All data are means (n=9)±SEM. Note that drugs had no significant effect on MEP IO-curve in comparison to PBO.
Drug Effects on PASLTP-Induced Changes of MEP IO-Curve (Comparison of Time Points P1 and P2 vs B1)
At time point B1 (immediately before PASLTP, cf. Figure 1), there was no difference in MEP IO-curves between drugs, ie there were no significant effects of DRUG (F4,40=0.57, p=0.69) or DRUG * SI (F9.48,94.78=0.66, p=0.75) (Figure 4). This is an important nil finding as there were no differences in corticospinal excitability prior to intervention (PASLTP) that could have accounted for the differential drug effects (see below) on the PASLTP-induced changes in MEP IO-curves.

Logarithmically transformed motor-evoked potential (MEP) input–output (IO)-curves at time points B1 (after drug intake and before PASLTP, dark-filled symbols), P1 (5 min after PASLTP, light-filled symbols) and P2 (30 min after PASLTP, open symbols) as a function of stimulus intensity (in multiples of SI1mV) in the alprazolam (APZ) (a), zolpidem (ZLP) (b), ethanol-low (EtOH<5mM) (c) and ethanol-high (EtOH<20mM) (d) condition compared with the placebo (PBO) condition (black circles and lines in a–d). All data are means (n=9)±SEM. Note that the significant increase in MEP IO-curves after PASLTP in the PBO session was significantly suppressed in the EtOH<5mM and EtOH<20mM sessions.
In the PBO condition, the effects of TIME (F2,16=4.92, p=0.022) and the TIME * SI interaction (F5.42,43.35=2.91, p=0.021) were significant (Figure 4). Post hoc testing revealed that PASLTP resulted in increased MEP IO-curves at P1 (TIME: F1,8=6.70, p=0.032, TIME * SI interaction: F2.75,22.00=4.91, p=0.011) and P2 (TIME * SI interaction: F2.89,23.11=3.83, p=0.024).
The rmANOVA for all drug conditions revealed a significant DRUG * TIME interaction (F8,80=2.20, p=0.036), while the effects of TIME (F2,80=2.29, p=0.06), DRUG (F4,40=1.81, p=0.15), and the DRUG * TIME * SI interaction (F25.04,250.37=0.86, p=0.66) were not significant. Post hoc pairwise rmANOVAs revealed significant DRUG * TIME interactions for EtOH<5mM vs PBO (F2,32=4.25, p=0.023, Figure 4c) and EtOH<20mM vs PBO (F2,32=4.95, p=0.013, Figure 4d) but not for APZ vs PBO (F2,32=2.11, p=0.14, Figure 4a) and ZLP vs PBO (F2,32=0.59, p=0.56, Figure 4b).
These effects are explained by abolition of LTP-like effects in the EtOH<5mM and EtOH<20mM conditions but significant, although weak LTP-like effects in the APZ (at time point P2) and ZLP (at time point P1) conditions (see also Figure 5a).

(a). Areas under the logarithmically transformed motor-evoked potential (MEP) input–output curves (AUIOCs) are shown for time points B1 (before PASLTP), P1 (5 min after PASLTP) and P2 (30 min after PASLTP) for the five different drug conditions. The significant PASLTP-induced increase in AUIOC in the placebo (PBO), alprazolam (APZ) and zolpidem (ZLP) conditions (one-tailed paired t-tests, *p<0.05) was suppressed in the ethanol-low (EtOH<5mM) and ethanol-high (EtOH<20mM) conditions. (b). The normalized ΔAUIOC in the EtOH<5mM condition correlated negatively with EtOH<5mM blood concentration (r=−0.71, p<0.05), ie, higher EtOH concentrations were associated with a stronger suppression of the LTP-like plasticity obtained in the PBO condition.
The selection of PASLTP LTP-responders (see methods) may have biased the drug effects toward suppression of the LTP-like increase in MEP IO-curve obtained in the PBO condition. However, this was unlikely in the present study, as linear regression analyses of (AUIOC(P1)-AUIOC(B1))PBO vs (AUIOC(P1)-AUIOC(B1))Drug did not reveal negative correlations (all p>0.10). Only the linear regression of (AUIOC(P1)-AUIOC(B1))PBO vs (AUIOC(P1)-AUIOC(B1))EtOH<5mM revealed a non-significant trend, but toward a positive correlation (r=0.54, p=0.13), ie the strongest PASLTP LTP-responders in the PBO condition had the weakest suppressive effect in the EtOH<5mM condition. Therefore, preselection of PASLTP LTP-responders did not set a bias toward drug suppression of LTP-like plasticity in this study.
Relation of Sedation and Drug Levels to PASLTP-Induced Changes of AUIOC (Comparison of Time Points P1 vs B1)
The AUIOC group data are displayed in Figure 5a. Linear regression analyses showed that the blood concentrations of EtOH<5mM correlated negatively with the normalized ΔAUIOC (EtOH<5mM: r=−0.71, p=0.048, Figure 5b). This means that higher ethanol concentrations were associated with a stronger suppression of the LTP-like plasticity obtained in the PBO condition. In contrast, the blood concentrations of APZ, ZLP and EtOH<20mM did not correlate with the normalized ΔAUIOC (all p>0.25). Finally, changes in SPV as a biomarker of sedation mediated through the α1-GABAAR did not correlate with the normalized ΔAUIOC in any of the drug conditions (all p>0.1).
DISCUSSION
The pharmacological effects of this study showed double dissociation: enhancement of synaptic GABAAR-mediated inhibition by APZ and ZLP resulted in sedation indexed by a decrease of SPV but no significant effect on PASLTP-induced LTP-like plasticity when compared with the PBO condition. Conversely, rather low doses of EtOH led to abolition of LTP-like plasticity but not to sedation, ie SPV remained unchanged. We argue that these findings suggest detrimental effects of low doses of EtOH on mechanisms of learning and memory and support the importance of extrasynaptic GABAAR-mediated tonic inhibition and/or glutamatergic neurotransmission through NMDARs in regulating plasticity in neuronal networks of human cortex.
Drug Effects on Saccadic Peak Velocity (SPV)
SPV is an established biomarker of sedation mediated through the α1-GABAAR (de Visser et al, 2003). Accordingly, classical benzodiazepines and also ZLP decrease SPV in a sigmoid dose-dependent manner (de Haas et al, 2009; de Haas et al, 2010) while a specific agonist at the α2- and α3-GABAAR did not induce sedation or a decrease in SPV (de Haas et al, 2009). This is consistent with a wealth of data in support of the notion that the different synaptic GABAAR subtypes mediate different functions: α1-GABAAR activation results in sedative and anticonvulsant effects, α2-GABAAR activation leads to anxiolytic and muscle relaxant effects, while the biological functions of the α3-GABAAR are as of yet unclear (Möhler, 2007; Möhler et al, 2002). EtOH at the low doses tested here had no effect on α1-GABAAR-mediated currents in acutely dissociated cells (Criswell et al, 2003) and did not affect SPV in monkeys (Fuster et al, 1985) while higher doses⩾0.6‰ (≈13.9 mM) resulted in SPV slowing (Fransson et al, 2010). Therefore, the lack of any SPV slowing in the EtOH<5mM (Figure 2c) condition provides further evidence that EtOH at very low doses has no significant action at the synaptic α1-GABAAR.
Drug Effects on MEP IO-Curve
The nil findings of this study on corticospinal excitability as tested by MEP IO-curve are largely consistent with previous studies. While the effects of APZ and ZLP on MEP IO-curve have not been tested previously, other benzodiazepines such as lorazepam or diazepam had either no effect (Ilic et al, 2002; Ziemann et al, 1996) or they produced a moderate MEP IO-curve depression (Boroojerdi et al, 2001). EtOH at a higher blood concentration (16.5±2.3 mM) than in the present study had no effect (Ziemann et al, 1995). Therefore, MEP IO-curve used for testing the PASLTP effects on corticospinal excitability was not affected per se by any of the drugs at the dosages tested in the present experiments.
Drug Effects on PASLTP-Induced LTP-Like Increase of MEP IO-Curve
EtOH but not APZ or ZLP significantly suppressed the PASLTP-induced LTP-like increase in MEP IO-curve seen in the PBO condition. This effect was obtained already at the lowest EtOH dose (the EtOH<5mM condition; blood concentration, 3.01±0.46 mM), and in this condition the suppressive effect on LTP-like plasticity correlated with the individual EtOH blood concentration. The non-significant trends toward suppression of LTP-like plasticity by APZ and ZLP are consistent with a similarly non-significant trend toward a suppressive effect of diazepam in one previous study (Heidegger et al, 2010). Of note, others have demonstrated in animal studies that diazepam can suppress neocortical LTP, but this may require high dosages ⩾5 mg/kg (Komaki et al, 2007; Trepel and Racine, 2000). Therefore, the present findings should not be interpreted as an all-or-none dissociation of suppression of LTP-like plasticity in human cortex by benzodiazepines vs EtOH, but they point to a mechanism particularly sensitive to EtOH. The present findings are not in disagreement with the lack of an effect of acute EtOH exposure on MEP increase during a 5 Hz-train of 10 TMS pulses in one previous study (Conte et al, 2008), as this MEP increase is very short-lasting (<1 s), thus reflecting short-term synaptic enhancement rather than LTP (Ziemann et al, 2008).
What are the mechanisms through which low-dose EtOH exerted its suppressive effects on LTP-like plasticity in the present study? The δ-subunit containing extrasynaptic α4ß3δ and α6ß3δ GABAARs are uniquely sensitive to EtOH and show significant increases in GABA-related currents at EtOH concentrations of 3 mM or less (Sundstrom-Poromaa et al, 2002; Wallner et al, 2003). As this was the mean blood concentration in the EtOH<5mM condition, the suppressive effects on LTP-like plasticity could have been mediated through these GABAARs. The α6ß3δ GABAAR is exclusively located on granule cells in the cerebellum (McKernan and Whiting, 1996; Nusser et al, 1998), while the α4ß3δ GABAAR is expressed with decreasing abundance in thalamus, the dentate gyrus, the striatum, and the outer layers of neocortex (Pirker et al, 2000). We cannot tell with certainty if any of these two receptor subtypes was more likely to mediate the observed EtOH effects on LTP-like plasticity. However, it was recently shown that increasing cerebellar excitability by anodal transcranial direct current stimulation or intermittent theta-burst stimulation abolished PAS-induced LTP-like plasticity (Hamada et al, 2012; Popa et al, 2013), but this suppressive effect was seen only with PAS25ms (ie with the interval between the electrical stimulus to the median nerve and TMS of the contralateral M1 equaling 25 ms) and not with PAS21.5ms (Hamada et al, 2012). The mean PAS interstimulus interval in our study was 21.9 ms. Therefore, it is very likely that we have investigated PASLTP-induced LTP-like plasticity that is not influenced by processing of sensory afferent information in the cerebellum. This indirectly supports the notion that the effect of low-dose EtOH was mediated by enhancement of tonic inhibition through the α4ß3δ GABAAR. In summary, this would be first indirect evidence that extrasynaptic GABAAR-mediated tonic inhibition has an exquisite role in regulating LTP-like plasticity in human cortex. However, these statements deserve caution, given that the systems level approach of our experiments does not permit direct insights into the cellular mechanisms. Furthermore, the high sensitivity of extrasynaptic GABAARs to low doses of EtOH has not been unanimously replicated (Borghese et al, 2006; Yamashita et al, 2006).
Of particular note are possible alterations of neurotransmission through the NMDAR by EtOH because the NMDAR is a major target of EtOH (Kumari and Ticku, 2000). EtOH at low concentrations of ⩽25 mM resulted in NMDAR inhibition in various experimental preparations (Lovinger et al, 1989, 1990; Weitlauf and Woodward, 2008), and in significant suppression of LTP (Blitzer et al, 1990; Morrisett and Swartzwelder, 1993), although other studies failed to reported significant LTP suppression by low concentrations of EtOH (Pyapali et al, 1999; Schummers et al, 1997). Taken together, NMDAR inhibition may have contributed to the suppressive effect of low-dose EtOH on LTP-like plasticity in our study, in agreement with previous studies that have demonstrated suppression of LTP-like plasticity by specific pharmacological NMDAR blockade (Stefan et al, 2002; Wankerl et al, 2010).
Finally, EtOH may exert excitability-depressant actions through a variety of other receptors such as non-NMDA glutamate receptors (Badanich et al, 2013; Frye and Fincher, 2000) or G protein-coupled inwardly rectifying potassium channels (Lewohl et al, 1999), but these effects were consistently observed only at high, intoxicating EtOH concentrations >50 mM and, therefore, are unlikely to have contributed to our findings.
Together, our findings are consistent with the notion that enhancement of tonic inhibition through the extrasynaptic α4ß3δ GABAAR has contributed to the acute suppressive effect of low-dose EtOH on LTP-like plasticity. Of note, others have shown that NMDAR-dependent hippocampal LTP can be attenuated by tonic inhibition through the α4ß3δ GABAAR (Shen et al, 2010). Tonic inhibition is mediated through extrasynaptic GABAARs that are exquisitely sensitive to low concentrations of ambient GABA (Farrant and Nusser, 2005). Increase of tonic inhibition shifts the input–output relationship of cells to the right, ie the probability of action potential generation to a given excitatory input is decreased (Mitchell and Silver, 2003). The input–output relationship of corticospinal cells in human motor cortex can be shifted to the right or left by cortex polarization through cathodal or anodal transcranial direct current stimulation, respectively (Nitsche and Paulus, 2000). A shift to the right by cathodal stimulation abolished PASLTP-induced LTP-like plasticity, while a shift to the left by anodal stimulation enhanced it (Nitsche et al, 2007). Similarly, cathodal vs anodal stimulation of rat hippocampal slices respectively suppressed or enhanced subsequent LTP induction (Ranieri et al, 2012).
PASLTP-induced LTP-like plasticity shares common mechanisms with motor skill learning (Elahi et al, 2013; Jung and Ziemann, 2009; Kang et al, 2011; Rosenkranz et al, 2007; Stefan et al, 2006; Ziemann et al, 2004) and acute EtOH ingestion has deleterious effects on memory formation and learning (Lister et al, 1991; Lowy, 1970; Mattila et al, 1998). Therefore, the present findings suggest a negative impact of EtOH on memory formation and learning at doses as low as reached by a single drink, but this will need to be tested in further experiments.
On the other hand, NMDAR-dependent LTP has also been implicated in the induction and maintenance of alcohol addiction (for reviews, (Krystal et al, 2003; Ron and Wang, 2009)). Basic experiments suggested that this is caused by facilitation rather than inhibition of LTP in the presence of high concentrations of EtOH (100 mM) and that blockade of this aberrant LTP facilitation can attenuate operant self-administration of EtOH in rats (Wang et al, 2007). TMS can be used to investigate and therapeutically interfere with this aberrant plasticity in alcohol addicts (Barr et al, 2011; Naim-Feil and Zangen, 2013).
In conclusion, very low, non-sedating doses of EtOH show deleterious effects on LTP-like plasticity at the systems level of human motor cortex, contrasting with non-significant effects on LTP-like plasticity by sedating doses of alprazolam and zolpidem. We argued that one possibility to explain this double dissociation is enhanced tonic inhibition through the extrasynaptic α4ß3δ GABAAR by low-dose EtOH. Findings may stimulate more extensive research of the physiological importance of tonic inhibition in regulating excitability and plasticity of cortical neuronal networks, and its potential role in abnormalities of these processes in neurological disorders such as epilepsy (Semyanov et al, 2004; Walker and Semyanov, 2008).
FUNDING AND DISCLOSURE
The authors declare no conflict of interest.
References
Aroniadou VA, Keller A (1995). Mechanisms of LTP induction in rat motor cortex in vitro. Cereb Cortex 5: 353–362.
Badanich KA, Mulholland PJ, Beckley JT, Trantham-Davidson H, Woodward JJ (2013). Ethanol reduces neuronal excitability of lateral orbitofrontal cortex neurons via a glycine receptor dependent mechanism. Neuropsychopharmacology 38: 1176–1188.
Barr MS, Farzan F, Wing VC, George TP, Fitzgerald PB, Daskalakis ZJ (2011). Repetitive transcranial magnetic stimulation and drug addiction. Int Rev Psychiatry 23: 454–466.
Bland JM, Altman DG (1996). Transforming data. BMJ 312: 70.
Blitzer RD, Gil O, Landau EM (1990). Long-term potentiation in rat hippocampus is inhibited by low concentrations of ethanol. Brain Res 537: 203–208.
Blom MW, Bartel PR, de Sommers K, Van der Meyden CH, Becker PJ (1990). The effects of alprazolam, quazepam and diazepam on saccadic eye movements, parameters of psychomotor function and the EEG. Fundam Clin Pharmacol 4: 653–661.
Borghese CM, Storustovu S, Ebert B, Herd MB, Belelli D, Lambert JJ et al (2006). The delta subunit of gamma-aminobutyric acid type A receptors does not confer sensitivity to low concentrations of ethanol. J Pharmacol Exp Ther 316: 1360–1368.
Boroojerdi B, Battaglia F, Muellbacher W, Cohen LG (2001). Mechanisms influencing stimulus-response properties of the human corticospinal system. Clin Neurophysiol 112: 931–937.
Carson RG, Nelson BD, Buick AR, Carroll TJ, Kennedy NC, Cann RM (2013). Characterizing changes in the excitability of corticospinal projections to proximal muscles of the upper limb. Brain Stimul 6: 760–768.
Castro-Alamancos MA, Donoghue JP, Connors BW (1995). Different forms of synaptic plasticity in somatosensory and motor areas of the neocortex. J Neurosci 15 (7 Pt 2): 5324–5333.
Conte A, Attilia ML, Gilio F, Iacovelli E, Frasca V, Bettolo CM et al (2008). Acute and chronic effects of ethanol on cortical excitability. Clin Neurophysiol 119: 667–674.
Cooke SF, Bliss TV (2006). Plasticity in the human central nervous system. Brain 129 (Pt 7): 1659–1673.
Criswell HE, Ming Z, Griffith BL, Breese GR (2003). Comparison of effect of ethanol on N-methyl-D-aspartate- and GABA-gated currents from acutely dissociated neurons: absence of regional differences in sensitivity to ethanol. J Pharmac Exp Ther 304: 192–199.
de Haas SL, Franson KL, Schmitt JA, Cohen AF, Fau JB, Dubruc C et al (2009). The pharmacokinetic and pharmacodynamic effects of SL65.1498, a GABA-A alpha2,3 selective agonist, in comparison with lorazepam in healthy volunteers. J Psychopharmacol 23: 625–632.
de Haas SL, Schoemaker RC, van Gerven JM, Hoever P, Cohen AF, Dingemanse J (2010). Pharmacokinetics, pharmacodynamics and the pharmacokinetic/ pharmacodynamic relationship of zolpidem in healthy subjects. J Psychopharmacol 24: 1619–1629.
de Visser SJ, van der Post JP, de Waal PP, Cornet F, Cohen AF, van Gerven JM (2003). Biomarkers for the effects of benzodiazepines in healthy volunteers. Br J Clin Pharmacol 55: 39–50.
Di Lazzaro V, Ziemann U, Lemon RN (2008). State of the art: physiology of transcranial motor cortex stimulation. Brain Stimul 1: 345–362.
Elahi B, Hutchison WD, Daskalakis ZJ, Gunraj C, Chen R (2013). Dose response curve of associative plasticity in human motor cortex and interactions with motor learning. J Neurophysiol.
Farrant M, Nusser Z (2005). Variations on an inhibitory theme: phasic and tonic activation of GABA(A) receptors. Nat Rev Neurosci 6: 215–229.
Fransson PA, Modig F, Patel M, Gomez S, Magnusson M (2010). Oculomotor deficits caused by 0.06% and 0.10% blood alcohol concentrations and relationship to subjective perception of drunkenness. Clin Neurophysiol 121: 2134–2142.
Fritsch B, Reis J, Martinowich K, Schambra HM, Ji Y, Cohen LG et al (2010). Direct current stimulation promotes BDNF-dependent synaptic plasticity: potential implications for motor learning. Neuron 66: 198–204.
Frye GD, Fincher A (2000). Sustained ethanol inhibition of native AMPA receptors on medial septum/diagonal band (MS/DB) neurons. Br J Pharmacol 129: 87–94.
Fuster JM, Willey TJ, Riley DM (1985). Effects of ethanol on eye movements in the monkey. Alcohol 2: 611–616.
Greenblatt DJ, Wright CE (1993). Clinical pharmacokinetics of alprazolam. Therapeutic implications. Clin Pharmacokinet 24: 453–471.
Groppa S, Oliviero A, Eisen A, Quartarone A, Cohen LG, Mall V et al (2012). A practical guide to diagnostic transcranial magnetic stimulation: report of an IFCN committee. Clin Neurophysiol 123: 858–882.
Hamada M, Strigaro G, Murase N, Sadnicka A, Galea JM, Edwards MJ et al (2012). Cerebellar modulation of human associative plasticity. J Physiol 590: 2365–2374.
Heidegger T, Krakow K, Ziemann U (2010). Effects of antiepileptic drugs on associative LTP-like plasticity in human motor cortex. Eu j Neurosci 32: 1215–1222.
Hess G, Aizenman CD, Donoghue JP (1996). Conditions for the induction of long-term potentiation in layer II/III horizontal connections of the rat motor cortex. J Neurophysiol 75: 1765–1778.
Ilic TV, Meintzschel F, Cleff U, Ruge D, Kessler KR, Ziemann U (2002). Short-interval paired-pulse inhibition and facilitation of human motor cortex: the dimension of stimulus intensity. J Physiol 545.1: 153–167.
Jung P, Ziemann U (2009). Homeostatic and non-homeostatic modulation of learning in human motor cortex. J Neurosci 29: 5597–5604.
Kamke MR, Hall MG, Lye HF, Sale MV, Fenlon LR, Carroll TJ et al (2012). Visual attentional load influences plasticity in the human motor cortex. J Neurosci 32: 7001–7008.
Kang J-S, Terranova C, Hilker R, Quartarone A, Ziemann U (2011). Deficient homeostatic regulation of practice-dependent plasticity in writer’s cramp. Cereb Cortex 21: 1203–1212.
Keel JC, Smith MJ, Wassermann EM (2001). A safety screening questionnaire for transcranial magnetic stimulation [letter]. Clin Neurophysiol 112: 720.
Komaki A, Shahidi S, Lashgari R, Haghparast A, Malakouti SM, Noorbakhsh SM (2007). Effects of GABAergic inhibition on neocortical long-term potentiation in the chronically prepared rat. Neurosci Lett 422: 181–186.
Korchounov A, Ziemann U (2011). Neuromodulatory neurotransmitters influence LTP-like plasticity in human cortex: a Pharmaco-TMS Study. Neuropsychopharmacol 36: 1894–1902.
Krystal JH, Petrakis IL, Mason G, Trevisan L, D'Souza DC (2003). N-methyl-D-aspartate glutamate receptors and alcoholism: reward, dependence, treatment, and vulnerability. Pharmacol Ther 99: 79–94.
Kumari M, Ticku MK (2000). Regulation of NMDA receptors by ethanol. Prog Drug Res 54: 152–189.
Lewohl JM, Wilson WR, Mayfield RD, Brozowski SJ, Morrisett RA, Harris RA (1999). G-protein-coupled inwardly rectifying potassium channels are targets of alcohol action. Nat Neurosci 2: 1084–1090.
Lister RG, Gorenstein C, Fisher-Flowers D, Weingartner HJ, Eckardt MJ (1991). Dissociation of the acute effects of alcohol on implicit and explicit memory processes. Neuropsychologia 29: 1205–1212.
Lovinger DM, White G, Weight FF (1989). Ethanol inhibits NMDA-activated ion current in hippocampal neurons. Science 243: 1721–1724.
Lovinger DM, White G, Weight FF (1990). NMDA receptor-mediated synaptic excitation selectively inhibited by ethanol in hippocampal slice from adult rat. J Neurosci 10: 1372–1379.
Lowy R (1970). Toxicology of single doses of ethyl alcohol. Int Encycl Pharmacol Ther 20: 277–299.
Mattila MJ, Vanakoski J, Kalska H, Seppala T (1998). Effects of alcohol, zolpidem, and some other sedatives and hypnotics on human performance and memory. Pharmacol Biochem Behav 59: 917–923.
McKernan RM, Whiting PJ (1996). Which GABAA-receptor subtypes really occur in the brain? Trends Neurosci 19: 139–143.
Mitchell SJ, Silver RA (2003). Shunting inhibition modulates neuronal gain during synaptic excitation. Neuron 38: 433–445.
Möhler H (2007). Molecular regulation of cognitive functions and developmental plasticity: impact of GABA(A) receptors. J Neurochem 102: 1–12.
Möhler H, Fritschy JM, Rudolph U (2002). A new benzodiazepine pharmacology. J Pharmacol Exp Ther 300: 2–8.
Morrisett RA, Swartzwelder HS (1993). Attenuation of hippocampal long-term potentiation by ethanol: a patch-clamp analysis of glutamatergic and GABAergic mechanisms. J Neurosci 13: 2264–2272.
Müller JFM, Orekhov Y, Liu Y, Ziemann U (2007). Homeostatic plasticity in human motor cortex demonstrated by two consecutive sessions of paired associative stimulation. Eur J Neurosci 25: 3461–3468.
Müller-Dahlhaus F, Ziemann U, Classen J (2010). Plasticity resembling spike-timing dependent synaptic plasticity: the evidence in human cortex. Front Syn Neurosci 2: 1–11.
Müller-Dahlhaus JF, Orekhov Y, Liu Y, Ziemann U (2008). Interindividual variability and age-dependency of motor cortical plasticity induced by paired associative stimulation. Exp Brain Res 187: 467–475.
Naim-Feil J, Zangen A (2013). Addiction. Handb Clin Neurol 116: 613–630.
Nitsche MA, Paulus W (2000). Excitability changes induced in the human motor cortex by weak transcranial direct current stimulation. J Physiol 527 (Pt 3): 633–639.
Nitsche MA, Roth A, Kuo M-F, Fischer AK, Liebetanz D, Lang N et al (2007). Timing-dependent modulation of associative plasticity by general network excitability in the human motor cortex. J Neurosci 27: 3807–3812.
Nusser Z, Sieghart W, Somogyi P (1998). Segregation of different GABAA receptors to synaptic and extrasynaptic membranes of cerebellar granule cells. J Neurosci 18: 1693–1703.
Oldfield RC (1971). The assessment and analysis of handedness: the Edinburgh inventory. Neuropsychologia 9: 97–113.
Olsen RW, Hanchar HJ, Meera P, Wallner M (2007). GABAA receptor subtypes: the ‘one glass of wine’ receptors. Alcohol 41: 201–209.
Paulus W, Classen J, Cohen LG, Large CH, Di Lazzaro V, Nitsche M et al (2008). State of the art: pharmacologic effects on cortical excitability measures tested by transcranial magnetic stimulation. Brain Stimul 1: 151–163.
Pirker S, Schwarzer C, Wieselthaler A, Sieghart W, Sperk G (2000). GABA(A) receptors: immunocytochemical distribution of 13 subunits in the adult rat brain. Neuroscience 101: 815–850.
Popa T, Velayudhan B, Hubsch C, Pradeep S, Roze E, Vidailhet M et al (2013). Cerebellar processing of sensory inputs primes motor cortex plasticity. Cereb Cortex 23: 305–314.
Pyapali GK, Turner DA, Wilson WA, Swartzwelder HS (1999). Age and dose-dependent effects of ethanol on the induction of hippocampal long-term potentiation. Alcohol 19: 107–111.
Ranieri F, Podda MV, Riccardi E, Frisullo G, Dileone M, Profice P et al (2012). Modulation of LTP at rat hippocampal Ca3-Ca1 synapses by direct current stimulation. J Neurophysiol 107: 1868–1880.
Ron D, Wang J (2009) The NMDA receptor and alcohol addiction In: Van Dongen AM (eds). Biology of the NMDA Receptor: Boca Raton (FL).
Rosenkranz K, Kacar A, Rothwell JC (2007). Differential modulation of motor cortical plasticity and excitability in early and late phases of human motor learning. J Neurosci 27: 12058–12066.
Sale MV, Ridding MC, Nordstrom MA (2007). Factors influencing the magnitude and reproducibility of corticomotor excitability changes induced by paired associative stimulation. Exp Brain Res 181: 615–626.
Schummers J, Bentz S, Browning MD (1997). Ethanol's inhibition of LTP may not be mediated solely via direct effects on the NMDA receptor. Alcohol Clin Exp Res 21: 404–408.
Semyanov A, Walker MC, Kullmann DM, Silver RA (2004). Tonically active GABA A receptors: modulating gain and maintaining the tone. Trends Neurosci 27: 262–269.
Shen H, Sabaliauskas N, Sherpa A, Fenton AA, Stelzer A, Aoki C et al (2010). A critical role for alpha4betadelta GABAA receptors in shaping learning deficits at puberty in mice. Science 327: 1515–1518.
Smith MJ, Adams LF, Schmidt PJ, Rubinow DR, Wassermann EM (2002). Effects of ovarian hormones on human cortical excitability. Ann Neurol 51: 599–603.
Stefan K, Kunesch E, Benecke R, Cohen LG, Classen J (2002). Mechanisms of enhancement of human motor cortex excitability induced by interventional paired associative stimulation. J Physiol 543 (Pt 2): 699–708.
Stefan K, Kunesch E, Cohen LG, Benecke R, Classen J (2000). Induction of plasticity in the human motor cortex by paired associative stimulation. Brain 123 (Pt 3): 572–584.
Stefan K, Wycislo M, Classen J (2004). Modulation of associative human motor cortical plasticity by attention. J Neurophysiol 92: 66–72.
Stefan K, Wycislo M, Gentner R, Schramm A, Naumann M, Reiners K et al (2006). Temporary occlusion of associative motor cortical plasticity by prior dynamic motor training. Cereb Cortex 16: 376–385.
Sundstrom-Poromaa I, Smith DH, Gong QH, Sabado TN, Li X, Light A et al (2002). Hormonally regulated alpha(4)beta(2)delta GABA(A) receptors are a target for alcohol. Nat Neurosci 5: 721–722.
Thirugnanasambandam N, Grundey J, Adam K, Drees A, Skwirba AC, Lang N et al (2011). Nicotinergic impact on focal and non-focal neuroplasticity induced by non-invasive brain stimulation in non-smoking humans. Neuropsychopharmacology 36: 879–886.
Trepel C, Racine RJ (2000). GABAergic modulation of neocortical long-term potentiation in the freely moving rat. Synapse 35: 120–128.
Valle M, Barbanoj MJ, Jane F (2002). Pharmaco-electroencephalography and pharmacokinetic/pharmacodynamic modeling in basic research: focus on human pharmacology. Methods Find Exp Clin Pharmacol 24 (Suppl D): 129–137.
Velazquez-Perez L, Seifried C, Santos-Falcon N, Abele M, Ziemann U, Almaguer LE et al (2004). Saccade velocity is controlled by polyglutamine size in spinocerebellar ataxia 2. Ann Neurol 56: 444–447.
Walker MC, Semyanov A (2008). Regulation of excitability by extrasynaptic GABA(A) receptors. Results Probl Cell Differ 44: 29–48.
Wallner M, Hanchar HJ, Olsen RW (2003). Ethanol enhances alpha 4 beta 3 delta and alpha 6 beta 3 delta gamma-aminobutyric acid type A receptors at low concentrations known to affect humans. Proc Natl Acad Sci USA 100: 15218–15223.
Wang J, Carnicella S, Phamluong K, Jeanblanc J, Ronesi JA, Chaudhri N et al (2007). Ethanol induces long-term facilitation of NR2B-NMDA receptor activity in the dorsal striatum: implications for alcohol drinking behavior. J Neurosci 27: 3593–3602.
Wankerl K, Weise D, Gentner R, Rumpf JJ, Classen J (2010). L-type voltage-gated Ca2+ channels: a single molecular switch for long-term potentiation/long-term depression-like plasticity and activity-dependent metaplasticity in humans. J Neurosci 30: 6197–6204.
Wei W, Faria LC, Mody I (2004). Low ethanol concentrations selectively augment the tonic inhibition mediated by delta subunit-containing GABAA receptors in hippocampal neurons. J Neurosci 24: 8379–8382.
Weitlauf C, Woodward JJ (2008). Ethanol selectively attenuates NMDAR-mediated synaptic transmission in the prefrontal cortex. Alcohol Clin Exp Res 32: 690–698.
Welling PG, Lyons LL, Elliott R, Amidon GL (1977). Pharmacokinetics of alcohol following single low doses to fasted and nonfasted subjects. J Clin Pharmacol 17: 199–206.
Wolters A, Sandbrink F, Schlottmann A, Kunesch E, Stefan K, Cohen LG et al (2003). A temporally asymmetric Hebbian rule governing plasticity in the human motor cortex. J Neurophysiol 89: 2339–2345.
Yamashita M, Marszalec W, Yeh JZ, Narahashi T (2006). Effects of ethanol on tonic GABA currents in cerebellar granule cells and mammalian cells recombinantly expressing GABA(A) receptors. J Pharmacol Exp Ther 319: 431–438.
Ziemann U, Ilic TV, Pauli C, Meintzschel F, Ruge D (2004). Learning modifies subsequent induction of LTP-like and LTD-like plasticity in human motor cortex. J Neurosci 24: 1666–1672.
Ziemann U, Lönnecker S, Paulus W (1995). Inhibition of human motor cortex by ethanol. A transcranial magnetic stimulation study. Brain 118 (Pt 6): 1437–1446.
Ziemann U, Lönnecker S, Steinhoff BJ, Paulus W (1996). The effect of lorazepam on the motor cortical excitability in man. Exp Brain Res 109: 127–135.
Ziemann U, Paulus W, Nitsche MA, Pascual-Leone A, Byblow WD, Berardelli A et al (2008). Consensus: motor cortex plasticity protocols. Brain Stimul 1: 164–182.
Acknowledgements
This work was funded by the Interdisciplinary Center for Neuroscience Frankfurt (ICNF), an intramural funding program of the Goethe-University Frankfurt. This work was funded by an intramural grant from Goethe-University, Frankfurt. The authors declare that over the past three years UZ has received compensation for professional advice from CorTec GmbH Freiburg, Germany and Biogen Idec GmbH Ismaning, Germany; LK was supported by the grant ZI 542/5-1 from the German Research Foundation (DFG), and after completion of the work reported here LK was employed at EyeSense GmbH Großostheim, Germany. All of these compensations are unrelated to the research presented in this paper. Apart from this, all authors declare that, except for income received from the primary employer, no financial support or compensation has been received from any individual or corporate entity over the past three years for research or professional service and that there are no personal financial holdings that could be perceived as constituting a potential conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Lücke, C., Heidegger, T., Röhner, M. et al. Deleterious Effects of a Low Amount of Ethanol on LTP-Like Plasticity in Human Cortex. Neuropsychopharmacol 39, 1508–1518 (2014). https://doi.org/10.1038/npp.2013.350
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/npp.2013.350
Keywords
This article is cited by
-
Impairment of Neuroplasticity in the Dorsolateral Prefrontal Cortex by Alcohol
Scientific Reports (2017)
-
Low Doses of Ethanol Enhance LTD-like Plasticity in Human Motor Cortex
Neuropsychopharmacology (2015)
