
Abstract
Medulloblastoma (MB) is the most common malignant brain tumor in childhood. At present, there is no well-established targeted drug for majority of patients. The kinesin family member 14 (KIF14) is a novel oncogene located on chromosome 1q and is dysregulated in multiple cancers. The objectives of this study were to evaluate KIF14 expression and chromosome 1q copy number in MB, and to delineate its biological functions in MB pathogenesis. By quantitative RT-PCR and immunohistochemistry, we found KIF14 was overexpressed in MB. Increased KIF14 expression at protein level was strongly associated with shorter progression-free survival (P=0.0063) and overall survival (P=0.0083). Fluorescence in situ hybridization (FISH) analysis confirmed genomic gain of chromosome 1q in 17/93 (18.3%) of MB. Combined genetic and immunohistochemical analyses revealed that 76.5% of MB with 1q gain showed consistent overexpression of KIF14, and a tight link between chromosome 1q gain and KIF14 overexpression (P=0.03). Transient, siRNAs-mediated downregulation of KIF14 suppressed cell proliferation and induced apoptosis in two MB cell lines. Stably KIF14 knockdown by shRNAs inhibited cell viability, colony formation, migration and invasion, and tumor sphere formation in MB cells. We conclude that KIF14 is dysregulated in MB and is an adverse prognostic factor for survival. Furthermore, KIF14 is part of MB biology and is a potential therapeutic target for MB.
Similar content being viewed by others


Main
Medulloblastoma (MB) is the most common malignant brain neoplasm in childhood and represents about 20% of all pediatric central nervous system (CNS) tumors.1, 2 About 15% of MB is found in adult.3 Recent integrative genomic and transcriptome analyses have led to an international consensus that recognizes four principle molecular subgroups in MB.4 However, molecular grouping has not guided standard-of-care management as of now. The disease risk stratification based on patient age, metastatic stage at diagnosis, and extent of surgical resection has so far remained the most important factors determining outcome and clinical practice.5 Development of new therapeutic treatment for MB has remained slow. Better understanding of the molecular pathogenesis of MB may facilitate development of novel therapies for MB.
Kinesin family member 14 (KIF14) is a member of kinesin family proteins located on long arm of chromosome 1. The gene encodes 1648 amino acids, a protein containing a kinesin motor domain with ATPase activity. KIF14 has been implicated in cancer progression and is frequently overexpressed in various tumors.6, 7, 8, 9 Upregulation of KIF14 was a poor prognostic indicator for poor survival in lung and breast cancers.10, 11 It has been shown that KIF14 overexpression accelerated initiation of tumor formation.12 In contrast, siRNA-induced silencing KIF14 resulted in tumor growth inhibition and cell cycle arrest in hepatocellular carcinoma and glioma.13, 14
In the present study, we demonstrated increased KIF14 mRNA and protein abundance in MB samples. Furthermore, high KIF14 expression was significantly associated with unfavorable progression-free survival (PFS) and overall survival. We detected a strong correlation between overexpression and gain of chromosome 1q in MB. We then explored the biological effects of KIF14 in transiently and stably knockdown experiments. Our data indicated that KIF14 regulated cell proliferation, apoptosis, clonogenic potential, migration and invasion, and tumor sphere formation ability in MB. Taken together, we provided evidence that targeting KIF14 might be a novel strategy for MB treatment.
MATERIALS AND METHODS
Frozen Tissue Specimens
A sum of 32 MB tumors were collected at the time of neuro-oncology service. Frozen tissues were stored at −80 °C until use. Tumor samples were histologically reviewed by neuropathologist (Ng HK). Clinicopathological information of the patients is summarized in Supplementary Table 1. This study was approved by the Ethics Committee of Huashan Hospital, Fudan University. Six normal cerebella were purchased from Ambion (Austin, TX, USA), Clontech Laboratories (Palo Alto, CA, USA), and Biochain Institute (Hayward, CA, USA).
Formalin-Fixed Paraffin-Embedded Specimens
A total of 93 formalin-fixed paraffin-embedded (FFPE) MB samples were retrieved from Huashan Hospital, Shanghai. They were collected between January 2002 and December 2007. Clinical features of these patients were summarized in Table 1. Follow-up data were collected by telephone interviews or follow-up at the hospital clinics.
Molecular Subgroup Affiliation
Molecular subgroup affiliation was achieved by immunohistochemical evaluation of β-catenin, GAB1, Filamin A, and YAP1 described previously.15, 16 In addition, Sanger sequencing was carried out to confirm that WNT tumors detected by IHC carried CTNNB1 mutation. The primers for CTNNB1 were forward 5′-GATTTGATGGAGTTGGACATGG-3′, and reverse 5′-TGTTCTTGAGTGAAGGACTGAG-3′. PCR amplification was achieved using KAPA2G Robust HotStart Ready Mix (Kapa Biosystems, Wilmington, MA, USA). PCR reaction was run at 95 °C for 5 min, followed by 40–45 cycles of 95 °C for 15 s, 60 °C for 15 s, and 72 °C for 15 s, with a final extension of 72 °C for 5 min. Amplification products were purified using MEGAquick-spin Total Fragment DNA Purification Kit (Intron Biotechnology, Korea). Sequencing was performed using Big Dye Terminator Cycle Sequencing kit v1.1. The products were resolved in Genetic Analyzer 3130xl and analyzed by Sequencing Analysis software. Examples of wild-type and mutant tumors are shown in Supplementary Figure 1.
Cell Lines
DAOY, D283, and D341 were purchased from American Type Culture Collection (Manassas, VA, USA). ONS-76 was obtained from Japanese Cancer Research Resources Bank. D384, D425, and D458 were a generous gift from Dr Darrell Bigner (Department of Pathology, Duke University, Durham, NC, USA). DAOY was cultured in MEMα supplemented with 10% heat-activated fetal bovine serum (FBS). ONS-76 was grown in RPMI 1640 supplemented with 10% heat-inactivated FBS. Other cell lines were maintained in recommended media and housed at 37 °C in a humidified atmosphere of 5% CO2.
RNA Extraction
Total RNA was extracted by TRIzol reagent (Invitrogen, Carlsbad, CA, USA) as described by manufacturer.
Quantitative RT-PCR
Two-step RT-PCR was performed to quantify the mRNA abundance. In the first step, total RNA was reverse transcribed into cDNA with MultiScribe reverse transcriptase (Invitrogen) according to the manufacturer’s protocol. Briefly, a 20 μl-reaction mixture containing 1 μg RNA, 1 × reverse transcription buffer, 5.5 mM magnesium chloride, 2 mM deoxyribonucleoside triphosphates, 1.25 U/μl MultiScribe reverse transcriptase, 2.5 μM random hexamer, and 0.4 U/μl RNase inhibitor was prepared. The reaction mixture was incubated for 10 min at 25 °C, 30 min at 48 °C, and 5 min at 95 °C. In the second step, amplification mixture consisting of diluted cDNA, 1 × TaqMan Universal PCR Master Mix, and 1 × KIF14 TaqMan Gene Expression Assay (Hs00208408_m1), or 1 × GAPDH TaqMan Gene Expression Assay (Hs02758991_g1) (Applied Biosystems) was prepared. PCR was run under the conditions of 95 °C for 10 min, followed by 40 cycles of 95 °C for 15 s, and 60 °C for 1 min in QuantStudio 12 K Flex Real-Time PCR System (Invitrogen). Relative expression level for target genes was normalized by the Ct value of GAPDH using a 2−ΔΔCt relative quantification method. The relative KIF14 expression in MB was then calculated by comparison to the average of normal cerebella, which was set to 1.
Western Blot Analysis
Cells were washed with ice-cold phosphate-buffered saline (PBS), lysed in RIPA buffer (50 mM Tris-HCl pH 7.4, 150 mM NaCl, 1% Triton X-100, 1% sodium deoxycholate, 0.1% SDS, and 1 × protease inhibitor mix) on ice. Total protein concentration was quantified by Bradford Protein Assay (Bio-Rad, Hercules, CA, USA). A total of 50 μg of protein lysates were resolved on 8% SDS-polyacrylamide gel electrophoresis gel and electroblotted onto polyvinylidene difluoride membranes (GE Healthcare, Buckinghamshire, UK). Non-specific binding of antibodies was prevented by blocking membrane with 3% non-fat milk. Membranes were then incubated with primary polyclonal anti-KIF14 antibody (1:1000; Bethyl Laboratories Inc, Montgomery, TX, USA) or monoclonal anti-β-actin antibody (1:20000; Sigma, St Louis, MO, USA) at 4 °C overnight. Antibody-incubated membranes were washed with a series of 1 × TBST and reincubated with appropriate horseradish peroxidase-conjugated secondary antibody for 1 h at room temperature. Immunoreactive bands were visualized with Immobilon Western Chemiluminescent HRP substrate western blotting reagent (Millipore, Billerica, MA, USA).
Immunohistochemistry
FFPE tissues of 4 μm thickness were dewaxed by xylene and rehydrated in a serious of descending grades of alcohol. Antigen retrieval was achieved with EDTA buffer (pH 8.0) heated in a microwave for 20 min. The slides were then incubated with primary polyclonal anti-KIF14 antibody (A300-233A, Bethyl Laboratories, Montgomery, TX, USA) at a dilution of 1:500 dilution at 37 °C for 32 min on Ventana Benchmark XT automated immunostainer (Ventana, Oro Valley, AZ, USA). Endogenous peroxidase activity was blocked by incubation with OptiView Peroxidase Inhibitor for 5 min at room temperature. OptiView HQ Universal Linker was used as the secondary antibody. Signals were detected by 3,3′-diaminobenzidine tetrahydrochloride (DAB) and counterstained with Mayer’s hematoxylin solution.
We adopted a semi-quantitative scoring system including signal intensity and percentage of positive cells as parameters for grading IHC staining result. Intensity level were graded as 0 (no staining), 1 (weak staining), 2 (moderate staining), or 3 (strong staining). The extent of immunopositivity cells was given a score of 0 (0–<25%), 1 (25–<50%), 2 (50–<75%), or 3 (75–100%). Assessment of the specimens was made by two independent observers. An IHC score was calculated by multiplying the intensity and extent of positivity stained cells, with a minimum score of 0 and a maximum score of 9. The final score was obtained by calculating the average of scores provided by the two observers. Tumors with score between 0 and 4.5 were classified as low expression, and those with score >4.5 were considered high expression.
Fluorescence In Situ Hybridization Analysis for Chromosome 1q Gain
Dual-probe fluorescence in situ hybridization (FISH) assay was performed on FFPE sections to detect chromosome 1q gain, with locus-specific probes BAC clone RP11-92G12 (Invitrogen) on 1q32.1 served as target probe (red) and centromere 1 (Cytocell, Tarrytown, NY, USA) served as reference probe (green). Sections were dewaxed in xylene, followed by dehydration in 100% ethanol, retrieval in 1 M sodium thiocyanate at 80 °C for 10 min, and digestion by 0.04% pepsin at 37 °C. The probes were mixed human Cot-1 DNA in hybridization mixture (50% formamide, 10% dextran sulfate, 2 × SSC). After denaturation at 80 °C for 10 min, probes were hybridized onto the denatured chromosomes on sections overnight at 37 °C in oven. The sections were then washed two times in 1.5 M urea/2 × saline sodium citrate at 50 °C for 10 min. After washing, sections were incubated with Vectashield mounting medium containing 4′,6-diamidino-2-phenylindole (Vector Laboratories) and visualized under a fluorescent microscope (Carl Zeiss Microscopy, LLC, NY, USA). Hybridizing signals in at least 200 non-overlapping nuclei were counted.
We adopted the Adamson et al17 criteria for analysis of chromosome 1q gain in MB. Copy number was calculated by taking the mean ratio of red to green signals per cell, and tumors were considered to be gain when red/green ratio was >1.2.
Data Retrieval of Gene Expression and SNP Arrays
Data of expression profilings in Cho et al18 and Robinson et al19 studies were retrieved from the Gene Expression Omnibus (GSE37418) and from http://www.broadinstitute.org/pubs/medulloblastoma/cho, respectively. Data were then analyzed by Agilent GeneSpring v13 software. Data of chromosome alteration in Robinson et al19 were obtained from the Supplementary Data of the publication.
Transient Transfection of KIF14 siRNAs
Two gene-specific siRNAs, siKIF14#1 (HSS145380) and siKIF14#2 (HSS190721) were obtained from Invitrogen. SiKIF14#1 and siKIF14#2 targeted exon 25 and exon 12 of KIF14 (NM_014875.2), respectively. The Stealth siRNA Negative Control (siNegative control) showing minimal sequence identical to vertebrate transcriptome was served as internal control (Invitrogen). Transfection was achieved using Lipofectamine 2000 according to the recommended protocol (Invitrogen). In brief, cells were seeded subconfluently into culture plate a day before transfection. Prior transfection, KIF14-specific siRNAs or negative control was diluted in Opti-MEM Reduced Serum Medium (Invitrogen). Subsequent transfection was conducted according to manufacturer's instructions.
Establishment of Stable KIF14 Knockdown Clones
Two KIF14-specific MISSION short hairpin RNAs (shKIF14-816 and shKIF14-817) targeting different regions of KIF14 were purchased from Sigma (Sigma Chemical, St Louis, MO, USA). The two shRNAs were constructed with pLKO.1-puro vector as the backbone. The targeted shRNA inserts were placed next to human U6 promoter for effective generation of shRNA transcripts. The target sequence of shKIF14-816 was 5′-CCCTGTTATCTGTGTTTCATA-3′, located on exon 29; and the target sequence of shKIF14-817 was 5′-GCCCGTTTAATAGTCAACATT-3′, located on exon 11. The pLKO.1-puro empty vector was served as negative control.
To establish stably KIF14 knockdown clones, DAOY and ONS-76 cells were transfected with shKIF14-816, shKIF14-817, or pLKO.1-puro (empty vector) plasmid with Lipofectamine 2000 (Invitrogen). After 48 h post transfection, transfected cells were cultured in medium containing puromycin (Santa Cruz Biotechnology, Dallas, Texas, USA) at a final concentration of 2 μg/ml (DAOY) or 5 μg/ml (ONS-76). Single cell dilution was employed to select monoclonal cell lines and selection medium were changed every 2–3 days for 3–4 weeks when individual foci developed. Representative stable KIF14 knockdown clones and control clones were selected by quantitative RT-PCR and western blot analyses. Two clones derived from each shRNA construct were picked in DAOY and ONS-76 cells for further functional study.
Cell Proliferation Assay
Cell proliferation was determined by Vybrant MTT cell proliferation assay kit (Invitrogen). In transiently knockdown experiment, 1.0 × 103 cells were seeded into each well of a 96-well culture plate a day before transfection. Cell proliferation was determined at 24, 48, 72, and 96 h post transfection. In stable transfection study, cells were seeded in 96-well culture plate and cell proliferation was examined at the followed four consecutive days. To perform the assay, culture media were first replaced. In each culture well, 10 μl of MTT solution was added, and cells were incubated at 37 °C for 3 h. The solution was then removed and replaced with 100 μl of DMSO (MP Biomedicals, Santa Ana, CA, USA). After the insoluble crystals were completely dissolved, absorbance was recorded at 560 nm by a Glomax Multi-Detection system (Promega, Madison, WI, USA). In each experiment, three measurements were taken for each sample and the experiment was repeated three times.
Cell Viability Count
Trypan blue dye exclusion assay was employed to quantify cell viability. Cells were dissociated with trypsin, and stained with 0.4% (w/v) Trypan blue. Cell counting was achieved with a hemocytometer and a light microscope. The test was repeated three times.
Annexin V Assay
The percentage of apoptotic cells was measured with FITC Annexin V Apoptosis Detection Kit I (BD Pharmingen, San Diego, CA, USA). After 72 h of siRNA transfection, cells were collected, washed twice with cold PBS, and stained with Annexin V-FITC and propidium iodide (PI) for 15 min at room temperature in the dark. The stained cells were quantified by BD FACSCalibur flow cytometer (BD Bioscience). Early apoptotic cells were characterized by Annexin V positive and PI negative. Cells positive for Annexin V and PI represented late (end-stage) apoptotic cells.
Cell Migration Assay
Cell migration assay was examined using BD uncoated transwell inserts chamber with 8 μm pore size (BD Bioscience, Bedford, MA, USA). Stably transfected cells were trypsinized, collected, resuspended in serum-free media. Cells were then seeded into the upper chamber at a density of 2.5 × 104 cells. Culture medium containing 10% FBS as chemoattractant was filled in the lower chamber. After 22 h of incubation, non-migrated cells were removed with cotton swabs. The migrated cells were fixed by 100% methanol and stained with 2% crystal violet. The number of migrated cells was counted in five random fields per chamber at a magnification of 200 under light microscope. Three independent experiments were carried out.
Cell Invasion Assay
Cell invasion was determined by BD BioCoat Matrigel Invasion Chamber (BD Bioscience). Stably transfected cells were seeded on upper reservoir of rehydrated Matrigel-coated invasion chamber at a density of 2.5–5 × 104 cells. At 22 h post incubation, non-invading cells were removed. Invading cells passed to the lower reservoir of chamber were treated with 100% methanol and then with 2% crystal violet. The number of invaded cells was determined by counting five random fields per chamber at a magnification of 200 under light microscope. The experiment was conducted three times.
Soft Agar Colony Formation Assay
Soft agar colony formation assay was carried out to investigate anchorage-independent cell growth in stably KIF14 knockdown clones. A total of 2 × 104 cells was seeded in 0.3% sterile agarose solution containing a final concentration of 1 × MEMα and 10% FBS on top of 0.6% sterile agarose/1 × MEMα/10% FBS solution (bottom layer) in 60 mm culture plate. Plates were incubated at 37 °C for 3–4 weeks. During this period, medium was replenished as needed to prevent dehydration of the agarose. At the end of the assay, colonies were stained with 0.005% crystal violet and photographed. Colonies with >100 μm diameter were counted. The experiment was repeated three times independently.
Tumor Sphere Formation
A sum of 4.0 × 104 stably KIF14 knockdown cells were seeded in ultra-low attachment six-well culture plate (Corning, NY, USA) under serum-free condition, consisting of neurobasal medium (Invitrogen), 20 ng/ml basic fibroblast growth factor (bFGF) (Invitrogen), 20 ng/ml epidermal growth factor (EGF) (Invitrogen), 1 × B-27 (Invitrogen), and 1 × l-glutamine (Invitrogen). After incubation at 37 °C for 7 days, pictures were taken under a microscope and the tumor spheres formed with diameter >50 μm were counted. The number of tumor spheres represented total count from four different wells. The experiments were repeated three times.
Statistical Analysis
PFS and overall survival (OS) were analyzed by Kaplan–Meier method. The resulting curves were compared using the log-rank test. Student’s t-test was applied to compare continuous variables between two groups. Correlation between molecular and clinicopathological parameters was evaluated by Student’s t-test or Freeman–Halton extension of Fisher’s exact test. All P-values obtained were two-sided, and P<0.05 was considered statistically significant.
RESULTS
Transcript Level of KIF14 in MB as Determined by Quantitative RT-PCR
Taqman-based quantitative RT-PCR was applied to study the abundance of KIF14 in 32 primary tumors and seven MB cell lines. The clinicopathological features of the tumors are shown in Supplementary Table 1. A total of six commercially purchased normal cerebella were served as controls. As shown in Figure 1, KIF14 mRNA level was noticeably increased by 2.0–355.8-fold in primary tumors (P<0.01) and by 4.6–88.4-fold in cell lines (P<0.01) as compared to the average of normal cerebella. KIF14 transcript abundance was not associated with clinicopathological parameters (Supplementary Table 2). Survival data were available in 19 samples, but we did not observe a link between KIF14 transcript level and clinical outcome, possible due to the small sample size. Thus, to examine the clinical implication of KIF14 upregulation in a large cohort of MB tumors, we retrieved expression profiling data set from the study by Cho et al18 (http://www.broadinstitute.org/pubs/medulloblastoma/cho), in which 129 MB had overall survival data. Using median expression as a cutoff, we found high KIF14 mRNA level was significantly associated with poor overall survival (Figure 2; P=0.049).

Expression of KIF14 in 32 primary MBs (gray bar), seven cell lines (white bar) and six normal cerebella (black bar). The transcript level of KIF14 was detected by RT-PCR. Expression was normalized to GAPDH. Upregulation of KIF14 was observed in primary tumors and cell lines. From left to right, the cell lines were D283, D458, D341, D425, ONS-76, D384, and DAOY.

Overall survival curve of patients according to KIF14 mRNA level in Cho et al study. A total of 129 MB with clinical outcome was included in the analysis. Patients with high KIF14 transcript level displayed significantly shorter overall survival (P=0.049).
KIF14 is Highly Expressed in Primary MB Samples and Associated with Clinical Features
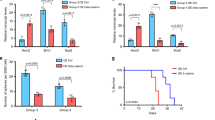
Immunohistochemical detection was performed to examine KIF14 expression in an independent cohort of 93 MB. Expression was graded as low and high according to the percentage of cells and intensity of the expression as described in ‘Materials and Methods’. Positive staining for KIF14 was observed both in the nucleus and cytoplasm. Representative figures are shown in Figure 3. Patients were separated into low KIF14 expression and high KIF14 expression based on the IHC staining score. High expression was detected in 46/93 (49.4%) samples. Molecular subgroup affiliation in these MB samples was conducted as described previously.15, 16 Furthermore, tumors were evaluated for CTNNB1 mutational analysis to ensure all cases with nuclear β-catenin staining indeed represented WNT tumors (Supplementary Figure 1). We detected a strong correlation between KIF14 expression and molecular subgroups (Table 1; P=0.024). Low KIF14 expression was frequently detected in WNT subgroup that is known to exhibit favorable clinical outcomes.3 In contrast, high KIF14 expression were frequently detected in tumors of SHH and non-WNT/SHH subgroups (Supplementary Figure 2). No association was observed between KIF14 expression and age, sex, histological variants, and clinical presentations (Table 1). Survival analysis revealed that KIF14 upregulation was significantly associated with shorter PFS (Figure 4a; P=0.0063) and overall survival (Figure 4b; P=0.0083). Among molecular subgroups, high KIF14 expression was strongly associated with shorter overall survival in WNT tumors and shorter PFS in SHH tumors (Figure 5), but the number of cases in the WNT subgroup is rather small and further study of a large cohort is needed to validate the finding.

Representative images of (left) hematoxylin and eosin (H&E) staining and (right) immunohistochemical staining of KIF14 in MB primary tumors. The right shows representative images of weak (top) and strong (bottom) KIF14 staining.

Prognostic significance of KIF14 expression in 93 MB patients. Immunohistochemistry (IHC) was employed to evaluate KIF14 expression. Tumors were divided into low and high KIF14 expressions according to the final IHC score, which was calculated by multiplying the intensity and extent of positivity stained cells (as mentioned in ‘Materials and Methods’). Tumors with IHC score between 0 and 4.5 were considered as low expression, and those with score >4.5 were classified as high expression. Survival analysis by Kaplan–Meier estimate and log-rank test revealed a significant association of high KIF14 expression with shorter (a) progression-free survival (P=0.0063) and (b) overall survival (P=0.0083).

Progression-free survival (left) and (right) overall survival curves of MB patients according to KIF14 expression and molecular subgroups. Molecular subgroup affiliation was determined as mentioned in 93 MB samples15 with the additional of CTNNB1 mutational analysis. High KIF14 expression was significantly associated with shorter progression-free survival in SHH subgroup (P=0.029) and shorter overall survival in WNT subgroup (P=0.041).
Gain of Chromosome 1q is a Frequent Event in MB
To determine if chromosome 1q gain contributes to KIF14 overexpression in MB, we conducted dual-colored FISH with a locus-specific DNA probe (RP11-92G12 mapped to 1q32.1 covering KIF14) and a control probe (centromeres of chromosome 1) on our cohort of 93 MB. We applied the criteria set by Adamson et al17 for analysis. Seventeen out of ninety-three (18.3%) MB samples showed evidence of increased copy number for chromosome 1q. Examples of chromosome 1q gain and chromosome 1q-non-gain tumors are shown in Figure 6. Important, unbalanced copy-number gain was significantly associated with KIF14 overexpression (Table 2; P=0.03). Our finding was in concordance with previous multi-platform genome-wide and expression arrays analysis conducted by Robinson et al19 demonstrating frequent chromosome 1q gain in 17/64 (26.6%) informative MB, and a correlation between chromosome 1q gain and increased KIF14 level. We also found that chromosome 1q gain was enriched in SHH and non-WNT/SHH tumors, whereas none of the WNT tumors displayed chromosome 1q abnormality (Table 2). The finding is in agreement with Northcott et al20 showing that chromosome 1q is a recurrent somatic copy-number aberration in SHH, Group 3, and Group 4 tumors. The association between gain of chromosome 1q and clinical features or presentations is provided in Table 2.

Representative fluorescent in situ hybridization (FISH) findings for (left) chromosome 1q gain and (right) chromosome 1q-non-gain MB samples. Locus-specific probes BAC clone RP11-92G12 on 1q32.1 was labeled in red and the centromere 1 probe was labeled in green. The criteria previously published were employed to assess chromosome 1q gain.17
Regarding the clinical significance of gain of chromosome 1q, we did not detect a significant association between survival (PFS and OS) and chromosome 1q abnormality (Supplementary Figure 3). Similar finding showing the lack of chromosome 1q abnormality and adverse clinical outcome has been reported.21 The results suggest that chromosome 1q aberration contributes to KIF14 upregulation and it is a common event in MB.
Transient Silencing KIF14 Inhibited Cell Proliferation and Induced Apoptosis In Vitro
To evaluate the biological significance of KIF14 in MB tumorigenesis, we transiently transfected two gene-specific siRNAs to knockdown KIF14 in two MB cell lines. The siKIF14#1 and siKIF14#2 targeted exon 25 and exon 12, respectively. Gene silencing efficiency was confirmed by RT-PCR at 24, 48, 72, and 96 h post transfection (Supplementary Figure 4; P<0.01), and by western blot analysis at 48 h after transfection (Supplementary Figure 5; P<0.01). Significant gene repression was consistently achieved during the course of study period. The effects of silencing KIF14 on cell proliferation was examined by MTT-based assay in a consecutive 4 days post transfection. As depicted in Figure 7, KIF14 repression prominently inhibited cell proliferation of MB cells. The effect was detected as early as 24 h post transfection when KIF14 siRNA treatment reduced cell proliferation by ~35% in DAOY and ~30% in ONS-76 (P<0.05). Continuously, decline in cell proliferation was observed in the following days. By 96 h post transfection, cell proliferation in KIF14 siRNA-treated cells was repressed by 82.3–90.3% in DAOY and 74.0–82.0% in ONS-76 compared to their respective control cells (P<0.01). The result indicated biological roles of KIF14 in regulating MB cell growth.

Effects of transient transfection of siRNAs against KIF14 on cell proliferation of (a) DAOY and (b) ONS-76 cells. MB cells were transfected with two gene-specific siRNAs against KIF14 (siKIF14#1 and siKIF14#2), and a negative control (siNegative control). Cell proliferation was measured by MTT-based assay in a 4 consecutive days post transfection. Compared with the negative control, depletion of KIF14 markedly inhibited cell proliferation of MB cells. Data represent mean±s.d. of three independent experiments (*P<0.05; **P<0.01).
To investigate the contribution of apoptosis in the decrease of cell proliferation mediated by KIF14 gene silencing, MB cells were examined for apoptosis by Annexin V-FITC/PI dual-staining technique, which detects early apoptotic (annexin V+ and PI−), late apoptotic (annexin V+ and PI+), and necrotic cells (annexin V− and PI+).22 In two MB cell lines, a clear enhancement in early and late apoptotic cells was noted (Figure 8). In DAOY, transfection of two gene-specific siRNAs increased the percentage of early apoptotic cells by 2.1–2.3-fold and late apoptotic cells by 1.5–2.2-fold at 72 h post transfection (Figure 8; P<0.01). Similarly, compared to the control cells, treatment with siRNAs against KIF14 in ONS-76 markedly increased the percentage of early and late apoptotic cells by 1.8–2.8-fold and 1.9–2.0-fold, respectively (Figure 8; P<0.05). We did not detect a significant change in the abundance of necrotic cells that have lost the integrity of plasma membrane and were positive for PI. The results indicated that downregulation of KIF14 promoted cell apoptosis.

Flow cytometry analysis of DAOY and ONS-76 cells transfected with siRNAs against KIF14 for 72 h. (a) Representative figures showing population of viable (annexin V−, PI−), early apoptotic (annexin V+, PI−), late apoptotic (annexin V+, PI+), and necrotic (annexin V−, PI+) cells. (b) Bar chart illustrating increased percentage of early and late apoptotic cells in KIF14-depleted cells. The bars indicated mean±s.d. of three independent experiments (*P<0.05; **P<0.01).
Stable Knockdown of KIF14 Reduced Cell Proliferation and Viability In Vitro
To investigate the effects of KIF14 gene silencing in detail, we established stable cell lines by transfecting two short hairpin RNA (shRNA) constructs (named shKIF14-816 and shKIF14-817) targeting different regions of KIF14 into two MB cell lines. The plasmids carried the puromycin-resistance gene and shRNA was expressed from a human U6 promoter. Two independent stable clones from each vector-mediated shRNA were selected in DAOY and ONS-76 cells. Quantitative RT-PCR and western blot analysis confirmed that KIF14 expression in these stable clones were substantially downregulated (Supplementary Figures 6 and 7). At transcript level, KIF14 expression was decreased by 76.4–85.8% in DAOY and 78.8–87.3% in ONS-76 (P<0.01). At protein level, KIF14 abundance was repressed by at least 70% in selected KIF14 knockdown clones (P<0.05).
To assess the inhibitory effect of silencing KIF14 on cell proliferation, we conducted MTT-based assay. The time-course study revealed that cell proliferation of stably KIF14 knockdown clones of DAOY was significantly impaired at 24 h and onward (Figure 9a; P<0.05). Similar change on cell proliferation was detected in stably KIF14 knockdown ONS-76 clones at 72 and 96 h (Figure 9b; P<0.01). Compared to the controls, the most noticeable effect was detected at 96 h when cell proliferation was reduced by 42.3–57.4% in DAOY clones and by 34.8–42.8% in ONS-76 clones.

Effect of stably KIF14 knockdown on cell proliferation of MB cells. (a) DAOY and (b) ONS-76 cells stably expressing shRNAs targeting KIF14 or control shRNA (empty vector) were examined for cell proliferation by MTT-based assay at 24, 48, 72, and 96 h of incubation. Stably KIF14 knockdown cells showed remarkably reduction in cell growth. Data represent mean±s.d. of three independent experiments (*P<0.05; **P<0.01).
We then ought to confirm the effect on cell proliferation by Trypan blue exclusion assay. Stable clones were stained with Trypan blue and viable cells were counted for a consecutive 4 days. Stable KIF14 knockdown cells demonstrated a marked decreased in cell viability (Figure 10). At 48 h and onward, four KIF14 knockdown DAOY clones showed significant reduction in cell number compared to the two control clones. At 96 h, a maximum of 64.4% decreased in cell number was detected (Figure 10a; P<0.05). Likewise, compared to control clones, cell viability of KIF14 knockdown ONS-76 clones showed substantial reduction at 48, 72, and 96 h after incubation (Figure 10b; P<0.05). Taken together, the results highlight the biological importance of KIF14 in controlling MB cell growth.

Stable knockdown of KIF14 impaired cell viability of MB cells. Cell viability was determined by cell counting in (a) DAOY and (b) ONS-76. Numbers are average of three independent experiments (*P<0.05; **P<0.01).
KIF14 Depletion Impaired Clonogenic Capacity
Given that there is correlation between soft agar clonogenicity and metastatic propensity,23 we then examined the effect of KIF14 depletion to form colony by anchorage-independent growth assay. The single cell of stable clones of DAOY cells was allowed to grow in soft agar for 3–4 weeks, and the number of colony with >100 μm in diameter was stained and counted. As shown in Figure 11, four stable clones with KIF14 knockdown yielded significant fewer colonies than the control clones. Compared to the control clone 3, the number of colonies in KIF14 knockdown clones was decreased by 57.9–94.0% (P<0.05). Similarly, compared to the control clone 5, a maximum of 94.3% reduction in colony formation was achieved (P<0.05). Anchorage-independent cell growth assay was not conducted in ONS-76 stable clones due to the inability to form colony in soft agar. The results pointed out that downregulation of KIF14 suppressed pro-metastatic activities in MB cells.

Stable knockdown of KIF14 retarded anchorage-independent growth of DAOY cells. Stable KIF14 knockdown cells were grown in soft agar for 3–4 weeks. Colonies were formed from single cells and the colonies with diameter >100 μm were counted under a microscope. The bars represent the average number of colonies formed in three independent experiments. Representative photographs of crystal violet stained soft agar plates are shown on the top of bar chart (*P<0.05).
Stable shRNA-Mediated Knockdown of KIF14 Reduced Cell Migration and Invasion Potential
To delineate the impact of silencing KIF14 on cell migration, stably KIF14 knockdown MB clones and their respective control clones were analyzed by Boyden chamber assay. Cells with motile capacity could migrate through the pores of the transwell filters because of the attraction to 10% FBS in the lower chamber. As shown in Figure 12, the number of cells migrated through the microporous membrane due to the attraction of FBS in the lower chamber was markedly decreased in KIF14 silenced clones as compared to the control clones (P<0.05). ShRNA-mediated KIF14 knockdown reduced migration capacity by 51.5–86.0% in DAOY (Figure 12a) and by 48.7–80.7% in ONS-76 (Figure 12b).

Effects of stably KIF14 knockdown on migration. Stable cells of (a) DAOY and (b) ONS-76 expressing shRNAs against KIF14 were assessed for migration capability by transwell assay. Migration was assessed by counting the number of cells migrated across uncoated transwell membranes. Stable KIF14 knockdown cells displayed marked reduction in migration potential compared with the control cells. The bars represent the average number of cells in three independent experiments (*P<0.05; **P<0.01).
We also exploited whether suppression of KIF14 could hamper cell invasion. Using a pre-coated Matrigel chamber, we found that shRNA-mediated KIF14 knockdown also prevented invasion of stable KIF14 knockdown cells. The invasiveness was decreased by 42.0–84.1% in KIF14 knockdown DAOY clones (Figure 13a; P<0.05) and 47.8–86.1% in KIF14 knockdown ONS-76 clones (Figure 13b; P<0.05). These data pointed out the involvement of KIF14 in the course of migration and invasion of MB cells.

Effects of stably KIF14 knockdown on invasion. (a) DAOY and (b) ONS-76 cells were evaluated for invasion potential by Matrigel invasion assay. Invasion was determined by counting the number of invaded cells in five random fields per chamber. Invasion ability was decreased remarkably in stable KIF14 knockdown cells. Data indicate mean±s.d. of three independent experiments (*P<0.05; **P<0.01).
Knockdown KIF14 Diminished Three Dimensional Culture Growth In Vitro
Emerging evidence indicates that a subset of cancer cells, called cancer stem cells, possess stemness properties that appears to be a driving force for the initiation and maintenance of tumorigenicity.24 The tumor sphere assay in which cells are propagated in suspension under non-adherent, serum-free culture conditions has been widely employed to examine the stem cell potential, and the abundance and growth of tumor sphere are indicative of cancer stem cell capacity.25, 26 Under standard conditioned tumor sphere medium, DAOY cells were capable of forming typically spherical tumor sphere.27
We therefore examined the effects of silencing KIF14 on tumor sphere formation. Stable KIF14 knockdown cells and the control cells were plated in serum-free neurobasal medium containing B27, EGF, and FGF, and l-glutamine for 7 days. The number of tumor sphere was then quantified. We observed the number of tumor sphere formed by stable KIF14 knockdown clones was profoundly decreased compared to those of control clones (Figure 14; P<0.05). The inhibition efficiencies of tumor sphere formation were between 35.7 and 79.7% compared to controls. The results indicated a functional role of KIF14 in controlling the population of stem cell-like tumor-initiating cells.

Effects of stable knockdown of KIF14 on tumor sphere-forming potential. DAOY cells expressing shRNAs against KIF14 were cultured in serum-free medium on non-adhesive plates for 7 days. The number of tumor spheres was then counted. Stable knockdown of KIF14 led to a decrease in tumor sphere formation. Bar graph indicates the average number of tumor spheres±s.d. in three independent experiments (*P<0.05; **P<0.01). Shown above the bar graph are the representative pictures of tumor spheres in stable KIF14 knockdown cells.
DISCUSSION
MB is the most common malignant solid tumor in childhood, accounting for one quarter of all CNS tumors in children below the age of 21.28 The WHO classification of MB in year 2016 has a parallel molecular classification in addition to a histopathological classification.29 The later recognizes four histopathological variants, classical, desmoplastic/nodular, large cell/anaplastic, and MB with extensive nodularity. The former recognizes four molecular groups, namely WNT-activated, SHH-activated, Group 3, and Group 4. Group 3 and Group 4 are also known as the non-WNT/non-SHH, showing overlapping and similar features in transcriptome, methylation, and microRNA profiling analyses.29
Although improvements in treatment design to include combination of maximal surgical resection, craniospinal irradiation, and chemotherapy have greatly improved prognosis in average-risk patients, significant morbidity and poor clinical outcomes remain the major problems in high-risk patients. The 5-year survival rate for average-risk patients is approaching 80%;30 whereas the 5-year survival rate for high-risk patients remains ~50%.31, 32 Still, metastatic and recurrent tumors represent a tremendous challenge and they show dismal clinical outcomes with the 5-year survival to be around 30–50%.33, 34
Kinesin superfamily proteins (or KIFs) were first discovered in 1985 as a class of microtubules-associated motor proteins with conserved ATP-binding domain (known as motor domain) for ATPase activity.35, 36 Subsequent studies revealed that KIFs are highly conserved among mammals.37 In human, 45 KIFs have been identified and they are classified into 15 subfamilies (kinesin 1–kinesin 14b) according to the phylogenetic study.38 KIFs regulate various cellular processes including intracellular transport, cargo trafficking, signal transduction, and cell division.39, 40, 41, 42
KIF14 belongs to the kinesin subfamily 3.38, 43 It encodes a conserved motor domain located in the N terminal and a forkhead-associated domain (FHA) at the C terminal.43, 44 It participates in various biological processes, such as cell cycle progression, cytokinesis, chromosome segregation, congression, and alignment of chromosomes.14, 45 During cytokinesis, KIF14 interacts with the central spindle microtubule-bundling protein PRC1 (protein-regulating cytokinesis 1), and depletion of KIF14 by siRNA results in failure of cytokinesis.46 Furthermore, KIF14 has been implicated in brain development. Previous study demonstrated mice with KIF14 mutation displayed brain malformation with hypomyelination and cytoarchitecture disruption in cerebellum, cerebral cortex, and the hippocampus.47
KIF14 is a candidate oncogene on long arm of chromosome 1 for many cancer types.48 Increased KIF14 expression was detected in glioma, hepatocellular carcinoma, cervical, and breast cancers, and its expression was positively associated with staging.8, 9, 10, 13, 49 High level of KIF14 was also significantly correlated with chemoresistance in cervical and breast cancers.7, 9 Importantly, overexpression of KIF14 was associated with adverse clinical outcome, tumor metastasis, and tumor recurrence.9, 49, 50, 51
Functional study indicated that siRNA-mediated knockdown of KIF14 attenuated cell proliferation, inhibited cell cycle progression, and induced apoptosis in glioma.13 Similar finding was observed when cell proliferation and migration of liver cancer cells were suppressed upon KIF14 silencing by RNA interference technique.14 In contrast, in vitro studies revealed overexpression of KIF14 led to increased tumor proliferation and anchorage-independent growth.7, 14 In vivo model also demonstrated upregulation of KIF14 resulted in increased tumor growth rate and accelerated tumor initiation.12
This study demonstrated the increased mRNA abundance of KIF14 in MB. This finding was also in line with previous study showing a profound upregulation of KIF14 in MB compared with fetal brain.48 Although we did not detect a link between KIF14 transcript level and clinical outcome in our series possibly owing to the small sample size, subsequent analysis of a published, large transcriptome data set revealed a tight association between increased KIF14 mRNA abundance and inferior overall survival. Nevertheless, further examination is needed to delineate the clinical significance of KIF14 transcript level in MB.
Notably, our data demonstrated high KIF14 protein level in MB tumors. We found high KIF14 expression is predominantly detected in SHH and non-WNT/non-SHH subgroups. Importantly, we observed a positive correlation between KIF14 expression and poor prognosis, and provide the first evidence that expression of a mitotic kinesin is a prognostic marker for clinical outcome in MB.
Given that cumulative studies employed different cytogenetic and molecular technologies including chromosomal karyotype, array CGH and SNP array have demonstrated the presence of gain and amplification of chromosome 1q in MB.20, 52, 53 We therefore postulated genomic aberration played a role in KIF14 dysregulation. We found 18.3% of MB samples displayed gain of chromosome 1q and increased KIF14 expression was tightly associated with genomic copy-number changes. Our data suggest that genomic abnormality is an important mechanism driving KIF14 aberration. Discrepancy in the prognostic value of gain of chromosome 1q in MB has been indicated in two studies. On one hand, chromosome 1q gain was identified as a negative prognostic marker.54 In another study comprising 73 cases, evidence for relationship between chromosome 1q gain and poor survival was not detected.21 In our study, we found no difference in survival in patients with and without chromosome 1q aberration.
Although functional studies have demonstrated the oncogenic potential of KIF14 in various cancers, its role in MB tumorigenesis has never been examined. In this study, we studied the roles of KIF14 in MB biology by silencing KIF14 through siRNA- and shRNA-mediated approaches. The former method allowed us to detect the effects in transiently transfected cells, whereas the latter method was designed to monitor the impact in stable transfectants. Our quantitative RT-PCR and western blot analyses confirmed successful and effective knockdown of KIF14. We then observed that downregulation of KIF14 in both transient and stable transfection experiments led to decrease in cell viability, suggesting the involvement of KIF14 in controlling cell proliferation in MB. Flow cytometry analysis further indicated an increase in apoptotic cells in KIF14-depleted cells. Therefore, we speculated that the proliferation inhibitory effect in KIF14-depleted cells was a result of apoptosis induction. Our findings were in agreement with the study by Huang et al13 showing the impairment of cell proliferation and increase in apoptosis in glioma cells treated with KIF14-specific siRNAs.
We also showed KIF14 knockdown was able to impair colony formation ability and migration and invasion, and tumor sphere formation in stable KIF14 knockdown cells. These results were in concordance with other studies revealed silencing KIF14 attenuated anchorage-independent growth in glioma, and suppressed migration and invasion in breast cancer.13, 55 In addition, the inhibitory effect on tumor sphere generation in KIF14-depleted MB cells suggests KIF14 is important in regulating the growth of a subpopulation of MB cells that displayed tumor-initiating and stem cell-like properties. Yet, the mechanisms for repressing tumor sphere formation remain elusive.
In conclusion, we show here overexpression of KIF14, and the tight correlation between KIF14 upregulation and inferior clinical outcome in MB, suggesting KIF14 is a potential negative prognostic marker in MB. We also provide evidence that KIF14 participates in the control of cell proliferation, apoptosis, anchorage-independent growth, migration and invasion, and tumor sphere formation of MB cells. At present, most MB patients do not have well-defined drug targets therapy. Our results provide preliminary support for further investigation of KIF14 as a potential target for MB treatment.
References
Parsons DW, Li M, Zhang X et al, The genetic landscape of the childhood cancer medulloblastoma. Science 2011;331:435–439.
MacDonald TJ, Rood BR, Santi MR et al, Advances in the diagnosis, molecular genetics, and treatment of pediatric embryonal CNS tumors. Oncologist 2003;8:174–186.
Kool M, Korshunov A, Remke M et al, Molecular subgroups of medulloblastoma: an international meta-analysis of transcriptome, genetic aberrations, and clinical data of WNT, SHH, Group 3, and Group 4 medulloblastomas. Acta Neuropathol 2012;123:473–484.
Taylor MD, Northcott PA, Korshunov A et al, Molecular subgroups of medulloblastoma: the current consensus. Acta Neuropathol 2012;123:465–472.
Pizer BL, Clifford SC . The potential impact of tumour biology on improved clinical practice for medulloblastoma: progress towards biologically driven clinical trials. Br J Neurosurg 2009;23:364–375.
Rath O, Kozielski F . Kinesins and cancer. Nat Rev Cancer 2012;12:527–539.
Singel SM, Cornelius C, Zaganjor E et al. KIF14 promotes AKT phosphorylation and contributes to chemoresistance in triple-negative breast cancer. Neoplasia 2014;16:247–256.
Thériault BL, Cybulska P, Shaw PA et al, The role of KIF14 in patient-derived primary cultures of high-grade serous ovarian cancer cells. J Ovarian Res 2014;7:123.
Wang W, Shi Y, Li J et al. Up-regulation of KIF14 is a predictor of poor survival and a novel prognostic biomarker of chemoresistance to paclitaxel treatment in cervical cancer. Biosci Rep 2016;36:e00315.
Corson TW, Gallie BL . Kif14 mRNA expression is a predictor of grade and outcome in breast cancer. Int J Cancer 2006;119:1088–1094.
Corson TW, Zhu CQ, Lau SK et al, KIF14 messenger RNA expression is independently prognostic for outcome in lung cancer. Clin Cancer Res 2007;13:3229–3234.
O'Hare M, Shadmand M, Sulaiman RS et al, Kif14 overexpression accelerates murine retinoblastoma development. Int J Cancer 2016;139:1752–1758.
Huang W, Wang J, Zhang D et al, Inhibition of KIF14 suppresses tumor cell growth and promotes apoptosis in human glioblastoma. Cell Physiol Biochem 2015;37:1659–1670.
Xu H, Choe C, Shin SH et al, Silencing of KIF14 interferes with cell cycle progression and cytokinesis by blocking the p27(Kip1) ubiquitination pathway in hepatocellular carcinoma. Exp Mol Med 2014;46:e97.
Zhang ZY, Xu J, Ren Y et al, Medulloblastoma in China: clinicopathologic analyses of SHH, WNT, and non-SHH/WNT molecular subgroups reveal different therapeutic responses to adjuvant chemotherapy. PLoS ONE 2014;9:e99490.
Ellison DW, Dalton J, Kocak M et al, Medulloblastoma: clinicopathological correlates of SHH, WNT, and non-SHH/WNT molecular subgroups. Acta Neuropathol 2011;121:381–396.
Adamson DC, Shi Q, Wortham M et al, OTX2 is critical for the maintenance and progression of Shh-independent medulloblastomas. Cancer Res 2010;70:181–191.
Cho YJ, Tsherniak A, Tamayo P et al, Integrative genomic analysis of medulloblastoma identifies a molecular subgroup that drives poor clinical outcome. J Clin Oncol 2011;29:1424–1430.
Robinson G, Parker M, Kranenburg TA et al, Novel mutations target distinct subgroups of medulloblastoma. Nature 2012;488:43–48.
Northcott PA, Shih DJ, Peacock J et al, Subgroup-specific structural variation across 1,000 medulloblastoma genomes. Nature 2012;488:49–56.
Clifford SC, O'Toole K, Ellison DW . Chromosome 1q gain is not associated with a poor outcome in childhood medulloblastoma: requirements for the validation of potential prognostic biomarkers. Cell Cycle 2009;8:787.
Wlodkowic D, Telford W, Skommer J et al, Apoptosis and beyond: cytometry in studies of programmed cell death. Methods Cell Biol 2011;103:55–98.
Li L, Price JE, Fan D et al, Correlation of growth capacity of human tumor cells in hard agarose with their in vivo proliferative capacity at specific metastatic sites. J Natl Cancer Inst 1989;81:1406–1412.
Beck B, Blanpain C . Unravelling cancer stem cell potential. Nat Rev Cancer 2013;13:727–738.
Ghods AJ, Irvin D, Liu G et al, Spheres isolated from 9L gliosarcoma rat cell line possess chemoresistant and aggressive cancer stem-like cells. Stem Cells 2007;25:1645–1653.
Lee SH, Lee CR, Rigas NK et al, Human papillomavirus 16 (HPV16) enhances tumor growth and cancer stemness of HPV-negative oral/oropharyngeal squamous cell carcinoma cells via miR-181 regulation. Papillomavirus Res 2015;1:116–125.
Harris PS, Venkataraman S, Alimova I et al, Polo-like kinase 1 (PLK1) inhibition suppresses cell growth and enhances radiation sensitivity in medulloblastoma cells. BMC Cancer 2012;12:80.
Gajjar A, Packer RJ, Foreman NK et al, Children’s oncology group’s 2013 blueprint for research: central nervous system tumors. Pediatr Blood Cancer 2013;60:1022–1026.
Ellison DW, Eberhart CG, Pietsch T, et al. Medulloblastoma. In:Louis DN, Ohgaki H, Wiestler OD, et al (eds). WHO Classification of Tumours of the Central Nervous System. IARC Press: Lyon, France, 2016, pp 184–200..
Gatta G, Zigon G, Capocaccia R et al, Survival of European children and young adults with cancer diagnosed 1995-2002. Eur J Cancer 2009;45:992–1005.
Moots PL, O'Neill A, Londer H et al, Preradiation chemotherapy for adult high-risk medulloblastoma: a trial of the ECOG-ACRIN Cancer Research Group (E4397). Am J Clin Oncol 2017; doi:10.1097/COC.0000000000000326.
Nazemi KJ, Shen V, Finlay JL et al, High incidence of veno-occlusive disease with myeloablative chemotherapy following craniospinal irradiation in children with newly diagnosed high-risk CNS embryonal tumors: a report from the Children's Oncology Group (CCG-99702). Pediatr Blood Cancer 2016;63:1563–1570.
Müller K, Mynarek M, Zwiener I et al, Postponed is not canceled: role of craniospinal radiation therapy in the management of recurrent infant medulloblastoma—an experience from the HIT-REZ 1997 & 2005 studies. Int J Radiat Oncol Biol Phys 2014;88:1019–1024.
Vivekanandan S, Breene R, Ramanujachar R et al, The UK experience of a treatment strategy for pediatric metastatic medulloblastoma comprising intensive induction chemotherapy, hyperfractionated accelerated radiotherapy and response directed high dose myeloablative chemotherapy or maintenance chemotherapy (Milan strategy). Pediatr Blood Cancer 2015;62:2132–2139.
Marx A, Hoenger A, Mandelkow E . Structures of kinesin motor proteins. Cell Motil Cytoskeleton 2009;66:958–966.
Vale RD, Reese TS, Sheetz MP . Identification of a novel force-generating protein, kinesin, involved in microtubule-based motility. Cell 1985;42:39–50.
Miki H, Setou M, Kaneshiro K et al, All kinesin superfamily protein, KIF, genes in mouse and human. Proc Natl Acad Sci USA 2001;98:7004–7011.
Hirokawa N, Tanaka Y . Kinesin superfamily proteins (KIFs): Various functions and their relevance for important phenomena in life and diseases. Exp Cell Res 2015;334:16–25.
Hirokawa N . Kinesin and dynein superfamily proteins and the mechanism of organelle transport. Science 1998;279:519–526.
Niwa S . Kinesin superfamily proteins and the regulation of microtubule dynamics in morphogenesis. Anat Sci Int 2015;90:1–6.
Kevenaar JT, Bianchi S, van Spronsen M et al, Kinesin-binding protein controls microtubule dynamics and cargo trafficking by regulating kinesin motor activity. Curr Biol 2016;26:849–861.
Lum L, Zhang C, Oh S et al, Hedgehog signal transduction via smoothened association with a cytoplasmic complex scaffolded by the atypical kinesin, costal-2. Mol Cell 2003;12:1261–1274.
Miki H, Okada Y, Hirokawa N . Analysis of the kinesin superfamily: insights into structure and function. Trends Cell Biol 2005;15:467–476.
Arora K, Talje L, Asenjo AB et al, KIF14 binds tightly to microtubules and adopts a rigor-like conformation. J Mol Biol 2014;426:2997–3015.
Zhu C, Zhao J, Bibikova M et al, Functional analysis of human microtubule-based motor proteins, the kinesins and dyneins, in mitosis/cytokinesis using RNA interference. Mol Biol Cell 2005;16:3187–3199.
Gruneberg U, Neef R, Li X et al, KIF14 and citron kinase act together to promote efficient cytokinesis. J Cell Biol 2006;172:363–372.
Fujikura K, Setsu T, Tanigaki K et al, Kif14 mutation causes severe brain malformation and hypomyelination. PLoS ONE 2013;8:e53490.
Corson TW, Huang A, Tsao MS et al, KIF14 is a candidate oncogene in the 1q minimal region of genomic gain in multiple cancers. Oncogene 2005;24:4741–4753.
Yang T, Zhang XB, Zheng ZM . Suppression of KIF14 expression inhibits hepatocellular carcinoma progression and predicts favorable outcome. Cancer Sci 2013;104:552–557.
Thériault BL, Pajovic S, Bernardini MQ et al, Kinesin family member 14: an independent prognostic marker and potential therapeutic target for ovarian cancer. Int J Cancer 2012;130:1844–1854.
Wang Q, Wang L, Li D, Deng J et al, Kinesin family member 14 is a candidate prognostic marker for outcome of glioma patients. Cancer Epidemiol 2013;37:79–84.
Cohen N, Betts DR, Tavori U et al, Karyotypic evolution pathways in medulloblastoma/primitive neuroectodermal tumor determined with a combination of spectral karyotyping, G-banding, and fluorescence in situ hybridization. Cancer Genet Cytogenet 2004;149:44–52.
Rossi MR, Conroy J, McQuaid D et al, Array CGH analysis of pediatric medulloblastomas. Genes Chromosomes Cancer 2006;45:290–303.
Lo KC, Ma C, Bundy BN et al, Gain of 1q is a potential univariate negative prognostic marker for survival in medulloblastoma. Clin Cancer Res 2007;13:7022–7028.
Ahmed SM, Thériault BL, Uppalapati M et al, KIF14 negatively regulates Rap1a-Radil signaling during breast cancer progression. J Cell Biol 2012;199:951–967.
Acknowledgements
This study was supported by the National Natural Science Foundation of China (reference number 81472373).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on the Laboratory Investigation website
Kinesin family member 14 (KIF14) is a member of kinesin family proteins located on chromosome 1q. This study describes the tight association of upregulation of KIF14 and genomic gain of chromosome 1q in medulloblastoma. The authors show that KIF14 affects medulloblastoma tumorigenesis by enhancing cell viability, colony formation, migration and invasion. These results indicate that KIF14 is a potential therapeutic target for MB.
Supplementary information
Rights and permissions
About this article

Cite this article
Li, KW., Qi, Y., Xia, T. et al. The kinesin KIF14 is overexpressed in medulloblastoma and downregulation of KIF14 suppressed tumor proliferation and induced apoptosis. Lab Invest 97, 946–961 (2017). https://doi.org/10.1038/labinvest.2017.48
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/labinvest.2017.48
This article is cited by
-
RNA-seq analysis reveals changes in mRNA expression during development in Daphnia mitsukuri
BMC Genomics (2024)
-
MSN, MWCNT and ZnO nanoparticle-induced CHO-K1 cell polarisation is linked to cytoskeleton ablation
Journal of Nanobiotechnology (2021)
-
Inhibiting microcephaly genes as alternative to microtubule targeting agents to treat brain tumors
Cell Death & Disease (2021)
-
The Drosophila Forkhead/Fox transcription factor Jumeau mediates specific cardiac progenitor cell divisions by regulating expression of the kinesin Nebbish
Scientific Reports (2021)
-
Observations on spontaneous tumor formation in mice overexpressing mitotic kinesin Kif14
Scientific Reports (2018)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}