
Abstract
Translocations resulting in a kinase fusion are well reported in many tumor types and indeed can be defining, particularly in the case of hematopoietic malignancies. The recent reports of multiple protein kinase fusions within melanocytic neoplasms, particularly in those with spitzoid morphology, have heralded a new era of classification of these melanocytic tumors. Seen within approximately half of all spitzoid neoplasms and present within the full spectrum of benign, atypical, and malignant lesions, kinase fusions likely represent an early oncogenic event contributing to cell proliferation and growth. Although the presence of a kinase fusion does not seem to correlate with the biologic potential of a given lesion, documentation of a kinase fusion will likely be important, particularly in the case of spitzoid melanoma, as numerous specific kinase receptor inhibitors have shown promise as therapeutic agents in a subset of cases with kinase fusions. Interrogation of non-spitzoid melanomas for similar kinase fusions as a potential driving oncogenic mechanism has revealed some similarities and some differences. This review will focus on the kinase fusions described to date in spitzoid neoplasms and how subsequent studies have informed current melanoma research.
Similar content being viewed by others


Main
Spitzoid neoplasms represent one of the more diagnostically challenging subset of melanocytic neoplasms within dermatopathology. Characterized by large, epithelioid, and spindled melanocytes, even benign Spitz nevi often possess histopathologic features associated with malignancy in other contexts. Accurate classification and assessment of risk in these neoplasms is difficult, with even experts failing to uniformly anticipate tumors that behave in an aggressive fashion.1 Recent reports document the frequent presence of kinase fusions within this class of tumors,2, 3, 4 thereby providing new avenues for discovery regarding the biology of these unique melanocytic tumors. Although kinase fusions are likely early oncogenic events in the development of spitzoid neoplasms and therefore—at this juncture—do not necessarily provide information regarding the biologic potential of these neoplasms, they may offer potential therapeutic targets for a subset of spitzoid melanocytic neoplasms that behave aggressively and metastasize. Moreover, documentation of kinase fusions in spitzoid tumors has naturally led to the investigation into similar mechanisms that could be relevant in non-spitzoid melanoma. This review will focus on the kinase fusions described to date in spitzoid neoplasms and how subsequent studies have informed current melanoma research.
Kinase fusions in spitzoid neoplasms
In 2014, Wiesner and colleagues documented the presence of a kinase fusion in approximately half of all spitzoid neoplasms investigated.2 Discovery of the kinase fusions was accomplished using targeted massively parallel DNA sequencing, with validation of the data performed by breakapart fluorescent in situ hybridization (FISH) targeting the translocated kinases. In total, 55% (41 of 74) Spitz nevi, 56% (18 of 32) atypical Spitz tumors, and 39% (13 of 33) Spitzoid melanomas were found to harbor a kinase fusion. Gene fusions were detected involving ROS1 (17.1%, n=24), ALK (10%, n=14), NTRK1 (16.4%, n=23), and RET (2.9%, n=4), all of which are membrane-bound receptor tyrosine kinases, and in the serine–threonine kinase BRAF (5%, n=7).2 Subsequent studies also identified MET kinase fusions in a small number of atypical Spitz tumors and spitzoid melanomas.5 Numerous fusion partners were identified, and the presence of a kinase fusion was found to be mutually exclusive with the presence of an HRAS or BAP-1 mutation, previously documented and well-characterized mutations that occur in a subset of Spitz nevi.2, 6 Spitzoid neoplasms with a documented translocation occurred in patients younger than tumors without a rearrangement.2 For the translocations involving receptor tyrosine kinases, moderate to strong, diffuse immunoreactivity for the kinase protein often correlated with the presence of a kinase fusion.2 Early efforts have been made to characterize specific histologic features that may correlate with the presence of a specific kinase fusion. As many of the kinases, particularly the receptor tyrosine kinases, share homology in their catalytic domain, it may be determined that specific histologic features are driven by the fusion partners rather than the kinases themselves. The N-terminal fusion partner often possesses the ability to determine subcellular localization, and this cellular distribution of the fusion product then determines the function of the gene product.7, 8 Ultimately, this function may result in cellular activity that drives certain histologic parameters. Continued investigation is warranted, particularly as certain fusion partners (such as DCTN1 and TPM3) have been detected fused to multiple of the described tyrosine kinases.
The constitutively activated kinases that result from rearrangement have been shown to activate multiple downstream signaling cascades, including the ERK/MAPK, PI3K/AKT/mTOR, and JAK-STAT pathways; activation of the PI3K/AKT/mTOR pathway has been hypothesized as contributing to the enlarged cell size of spitzoid melanocytes as this pathway is a key regulator of cell size.6 The frequency of kinase fusions in spitzoid neoplasms and their presence over the entire biologic spectrum of spitzoid neoplasms (benign, atypical, and malignant) suggests that kinase fusions represent an important and early driver in the formation of spitzoid tumors, with subsequent genetic insults, such as a mutation in the promoter region of TERT, hypothesized to provide transformative properties that result in aggressive clinical behavior.9 As an early oncogenic event, the presence of a kinase fusion therefore is unlikely to provide prognostic information regarding the biologic potential of the lesion. Indeed, a recent outcomes study of atypical spitzoid neoplasms in a pediatric cohort documented fusion kinases in 36% of cases, but the presence of a kinase fusion was not able to predict the likelihood of recurrence.10 On the other hand, even if documentation of a kinase fusion does not provide prognostic data, it certainly represents a potential therapeutic target. The use and continued emergence of numerous specific kinase inhibitors represent opportunities for targeted therapy when a kinase fusion is present in aggressive-behaving spitzoid melanomas, although admittedly, additional genomic alterations found in malignant spitzoid neoplasms may render the tumor resistant to a kinase inhibitor alone as a treatment. Kinase inhibitors could also be investigated for a role in treating residual disease in Spitz nevi and atypical Spitz tumors, thereby potentially decreasing the need for surgical re-excision.
The subsequent sections will discuss specific kinase fusions and their known function. A summary of the kinase fusions and other related genetic aberrations are found in Table 1.
Alk fusions
Anaplastic lymphoma kinase (ALK) is a key player in numerous cancer types, with fusions of this kinase being documented in anaplastic large cell lymphoma, lung adenocarcinoma, inflammatory myofbroblastic tumor, and other tumor types, including recently in a majority of epithelioid fibrous histiocytomas.11 Detected in 10–17% of spitzoid tumors,2, 10 ALK fusions are found in benign Spitz nevi, atypical Spitz tumors, and slightly less commonly in spitzoid melanoma.2
Several different fusion partners have been described in spitzoid neoplasms. The most common reported partners are tropomyosin 3 (TPM3) and dynactin1 (DCTN1); GTF3C2, NPM1, TPR, and CLIP1 translocations also have been documented.2, 3, 4, 12 In all cases, rearrangement links the kinase domain of ALK to an N-terminal partner gene. Interestingly, the DCNT1-ALK fusions that have been described to date were all classified as atypical Spitz tumors.4 At least a subset of Spitz tumors with ALK fusions have been tested with the melanoma FISH assay and none were found to meet the criteria for melanoma.4 Array chromosomal genomic hybridization performed on tumors with ALK fusions generally lacks widespread chromosomal gains or losses, although partial loss of chromosome 2 (encompassing the ALK and DCTN loci) or chromosome 1p are sometimes detected.3, 6 Expression of a fusion construct between ALK and DCTN1 in cell culture was shown to increase phosphorylation of the kinase fusion itself compared to controls, suggesting constitutive activation of the chimeric protein. It also results in the activation of MAPK and PI3K/AKT/mTOR signaling cascades, as measured by phosphorylation of proteins in the pathway and their effector proteins, and this activation can be suppressed by treatment with the ALK inhibitor crizotinib.2
Spitz tumors harboring an ALK rearrangement are some of the best characterized spitzoid neoplasms with kinase fusions to date. These tumors have been reported to occur in patients ranging from 5 months to 64 years of age, with median ages of 12 and 17 in two studies respectively.3, 4 One study showed a female predominance in ALK-rearranged tumors;3 another found no differences in sex predilection.4 Clinically, the lesions are solitary with a propensity to occur on the extremities. Many tumors lack clinical (and histologic) pigmentation, leading to a clinical differential diagnosis that includes vascular lesions, xanthogranuloma, keloid, or verruca in addition to melanocytic nevus and their variants.3, 4
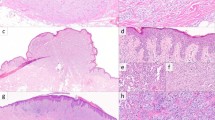
Histopathologically, spitzoid neoplasms with an ALK fusion are usually polypoid and exophytic, compound melanocytic lesions. Architecturally, the majority of lesions have been shown to demonstrate a plexiform growth pattern, with fascicles of melanocytes arranged radially with convergence toward the base in the dermis. A bulbous to nodular ‘dumbbell’ growth pattern has been documented in some lesions, but most commonly, these tumors show a wedge-shaped base with an infiltrative border at the periphery.3, 4 Cytologically, the melanocytes are often plump and spindled (fusiform) rather than epithelioid, and some authors have described a fibrillary quality to the cytoplasm as well as clefting between individual melanocytes3 (Figure 1). Nuclei generally demonstrate smooth, rounded contours, with distinct nucleoli. ALK-DCTN1 fusion lesions in particular have been noted to more often display mitotic activity, epithelioid morphology, and nuclear pleomorphism, and are less likely to display a fascicular and plexiform architecture.4 About half of lesions show an associated lymphocytic infiltrate and a similar proportion display dermal mitotic figures. When pigment was noted in the tumors, it has been described as coarse and granular rather than fine or ‘dusty’.3

A large polypoid, markedly atypical spitzoid neoplasm (a; 20x H&E). Higher magnification reveals plump spindled and epithelioid melanocytes arranged in short fascicles and nests with evidence of clefting between melanocytes (b; 200x H&E). The tumor demonstrates diffuse strong immunoreactivity for ALK and highlights the plexiform to packeted architecture (c; 100x).
The presence of an ALK fusion can be readily detected through the use of immunohistochemistry, as fusion positive cases nearly always demonstrate strong and diffuse cytoplasmic immunoreactivity for ALK, in contrast to fusion-negative cases which are negative. Immunohistochemistry often highlights the infiltrative nature of the lesions.3 Detection of an ALK rearrangement can also be performed by FISH, PCR, or next-generation sequencing, but the ease and reliability of the immunohistochemical staining make this the optimal method of detection.6
Although data are limited, to date no deaths or recurrences have been reported in patients with an ALK kinase fusion. Sentinel lymph node positivity has been seen in several patients harboring the fusion, but all patients with follow up thus reported in the literature have no evidence of residual disease.4
Ros1 fusions
Located on chromosome 6q22, ROS1 encodes a receptor tyrosine kinase in the insulin receptor family. Fusions involving ROS1 have been documented in lung adenocarcinomas, glioblastomas, inflammatory myofibroblastic tumors, and cholangiocarcinoma.6, 8 ROS1 fusions were detected in 17% of spitzoid neoplasms, being more common in benign Spitz nevi (25%) than in atypical Spitz tumors or spitzoid melanomas.2 Nine different partner genes were discovered, all of which fused to the preserved tyrosine kinase coding domain of ROS1. Expression of a fusion construct in cell lines showed increased phosphorylation of the fusion protein, and increased activation of downstream MAPK and PI3K signaling cascades compared to controls. Crizotinib, a tyrosine kinase inhibitor with activity against ROS1 and ALK, was able to inhibit phosphorylation of the fusion kinase as well as several downstream signaling molecules.2
In contrast to spitzoid tumors with an ALK kinase fusion, spitzoid neoplasms with a ROS1 fusion seem to lack specific architectural or cytologic features to indicate the presence of a fusion. Like Spitz nevi in general, these neoplasms are characterized by a well-circumscribed proliferation of epidermal and dermal nests of epithelioid to spindled melanocytes, often with epidermal hyperplasia and Kamino bodies.6 While detection of ROS1 using immunohistochemistry is quite specific for the presence of a kinase fusion protein, staining is often weak.6 Moreover, the currently available antibodies lack sensitivity, and other molecular methods such as FISH, PCR, or next-generation sequencing may be necessary to accurately capture the presence of a ROS1 rearrangement.6
Ret fusions
Fusions involving the RET kinase gene are rare in melanocytic neoplasms, comprising ~5% of spitzoid neoplasms, including a spindle cell nevus of Reed. Fusion partners identified include KIF5B (located on chromosome 10p11) and GOLGA5 (located on chromosome 14q32);2 RET fusions with both of these fusion partners have also been documented as oncogenic in other cancer types including lung cancer and thyroid cancer.6 Transfection of a GOLGA5-RET fusion construct into cultured cells led to increased phosphorylation of the fusion kinase as well as members and targets of the ERK/MAPK and PI3K/AKT/mTOR signaling cascades compared to controls, and this effect was suppressed by the RET inhibitors cabaozantinib and vandetanib.2
The rarity of RET fusions limits any generalizations about predictable clinical or histologic features in these tumor types. Immunohistochemistry for RET was specific for the presence of a kinase fusion.2
Ntrk1 fusions
NTRK1 encodes the receptor tyrosine kinase TRKA, whose activation is triggered by a number of secreted growth factors. In addition to spitzoid neoplasms, NTRK1 fusions have been described in adenocarcinoma of the lung, glioblastoma, pontine glioma, papillary thyroid carcinoma, and soft tissue neoplasms and sarcomas. Most kinase fusions involve a preserved NTRK1 kinase domain and loss of at least a portion of the transmembrane domain fused to a partner gene in the 5′ region.13 In spitzoid neoplasms, NTRK1 fusions were detected in ~16% of cases, being equally common in benign Spitz nevi, atypical Spitz tumors, and spitzoid melanomas. The most commonly identified fusion partner is LMNA,6 with TP53 representing another fusion partner.2 Translocations involving TPM3 have also been documented.12 Transfection of cell lines with a NRTK1-LMNA construct resulted in phosphorylation of the kinase itself, phospholipase gamma-1 (PLCγ1), and signaling molecules in and downstream of the ERK/MAPK and PI3K/AKT/mTOR pathways. Activation of these pathways was abolished with the NTRK1 inhibitor AZ-23.2
Tumors with NTRK1 kinase fusions demonstrate classic spitzoid features. No unique histopathologic features predict the presence of the fusion,6 although the two cases harboring an NTRK1 fusion in one study were both characterized by deep extension of melanocytes and ‘confluent cellular nodules with rounded pushing margins’ at the base of the lesion.12 The plexiform or fascicular growth pattern seen in neoplasms with ALK rearrangements is statistically less common in NTRK1 fusion- positive neoplasms, and a larger proportion (nearly half) of NTRK1-rearranged lesions display epithelioid predominant cell populations compared to the rarity of this finding in ALK-rearranged tumors.14 Immunohistochemistry appears to be a reliable mechanism to identify the presence of a fusion kinase, as strong cytoplasmic immunoreactivity for NTRK1 was seen in all cases with the rearrangement.2 Weak background expression due to low endogenous levels of the protein may be seen in neoplasms lacking the fusion kinase and should not be overly interpreted as positive staining.6 One patient with an NTRK1 translocation has had documented spitzoid melanoma present beyond the sentinel lymph node (tumor present on completion lymphadenectomy), however this tumor also demonstrated homozygous deletion of chromosomal 9p21 by FISH, which has been shown to correlate with aggressive disease.10
Braf fusions
BRAF fusions are found in a small percentage of spitzoid neoplasms, estimated between 5 and 10%,2, 10 and encompassing the spectrum of benign to malignant. In contrast to the majority of kinases discussed thus far, BRAF encodes a serine/threonine kinase rather than a receptor tyrosine kinase, placing its normal protein localization within the cytoplasm rather than associated with the plasma membrane. B-Raf signaling may be activated by growth factor binding to receptor tyrosine kinases upstream as well as other intracellular signals. Point mutations in BRAF (the most common being a valine to glutamate substitution at codon 600) are common in benign nevi and in a subset of melanomas and lead to constitutive activation of the kinase. Similarly, BRAF kinase fusions in spitzoid neoplasms are also predicted to lead to constitutive kinase activation, as the autoinhibitory, N-terminal RAS binding domain is lost in the rearrangement.2 Several different binding partners have been identified. Wild-type BRAF amplifications were also identified by FISH in a minority of spitzoid neoplasms.2
Spitzoid neoplasms with a BRAF fusion lack unique defining histopathologic characteristics;6 melanomas harboring a BRAF fusion tend to have epithelioid (spitzoid) morphology and are discussed in greater depth below. Immunohistochemistry is not useful in detecting the presence of a fusion owing to the detectable expression of wild-type BRAF in most melanocytic neoplasms,6 therefore FISH or next-generation sequencing are the recommended modalities for documenting a fusion.
Met fusions
MET kinase rearrangements were not detected in the original description of kinase fusions in spitzoid neoplasms, so it stands to reason their presence is extremely rare, estimated at 0.5% of cases. They have been detected in six spitzoid melanocytic neoplasms to date.5 The tumors occurred in female patients ranging in age from 9 to 29 and were diagnosed as atypical Spitz tumor, favor benign (n=2), atypical Spitz tumor (n=2), and spitzoid melanoma (n=2).5 The histologic features of these lesions shared the usual morphology of spitzoid tumors including epidermal hyperplasia, large junctional nests, and spindle and epithelioid melanocytes;5 too few MET fusion-positive spitzoid lesions have been identified to determine if there are unique histologically defining features to suggest the presence of this kinase fusion. As with other the kinase fusions, the fusion involved the intact kinase domain of MET fused to a variety of partner genes. Although unique fusion partners were described in each of the six cases, some of these fusion partners (such as DCTN1) have been documented previously as fusion partners with other kinases. The fusion product results in the loss of the transmembrane domain of MET, thus rendering the fusion protein non-responsive to growth factor stimulation when expressed in cell lines (due to its new cytoplasmic localization). However, expression of MET fusion constructs in cell lines was shown to result in increased phosphorylation of the kinase itself and activation of downstream signaling cascades, as well as abrogation of these changes upon application of an inhibitor of MET signaling.5 In contrast to benign, non-spitzoid melanocytic nevi which showed weak to absent expression of MET by immunohistochemistry, the three tumors for which tissue was available showed strong and diffuse immunoreactivity to MET, as well as increased phosphorylated MET protein.5
Kinase fusions in non-spitzoid melanocytic neoplasms
The detection of kinase fusions in such a large proportion of spitzoid neoplasms has naturally led to an interest in investigating whether similar oncogenic mechanisms are involved in other types of melanocytic lesions, particularly in non-spitzoid melanoma. Detection of kinase fusions in malignant melanocytic lesions could lead to new therapeutic options, as many specific kinase inhibitors are already in widespread clinical use for other tumor types. In addition, as sequencing capabilities become more sophisticated and widespread, discovery based sequencing projects have discovered (and likely will continue to discover) kinase fusions in other melanocytic tumor types. The following section describes research to date regarding kinase fusion detection in non-spitzoid melanocytic neoplasms and insight regarding additional mechanisms of melanoma tumorigenesis that has derived from these studies.
Prkca fusions
To date, only a single melanocytic neoplasm has been reported in the literature which harbored a fusion of the alpha subunit of protein kinase C (PRKCA).15 An infant girl had a large scalp mass removed at age 5 months which had been present at birth but had grown rapidly. Histopathologic analysis revealed an epithelioid melanocytic neoplasm with marked melanin pigmentation, mitotic figures, and mild pleomorphism. The final diagnosis rendered was ‘severely atypical melanocytic proliferation of indeterminate malignant potential’.15 RNA sequencing of the tumor identified the kinase fusion transcript between PRKCA and the ATP-ase ATP2B4; other kinase fusions or hot spot mutations commonly found in melanocytic neoplasms were not detected. Subsequent testing by breakapart FISH for PRKCA rearrangement confirmed the rearrangement in the tumor, but not in eight other kinase fusion-negative spitzoid lesions tested.15
The protein product of PRKCA is a serine–threonine kinase that can activate Raf signaling upstream of the ERK/MAPK cascasde. The fusion transcript—as with other kinase fusions described in melanocytic neoplasms—preserved the kinase domain of PRKCA but resulted in a loss the autoinhibitory domain, speculated to result in constitutive activity of the chimeric fusion protein.16 The tumor bore considerable histologic similarity to the pigment-producing neoplasms termed pigmented epithelioid melanocytoma and the epithelioid blue nevi seen in Carney complex, a genodermatosis and cancer predisposition syndrome most often caused by a germline loss of function mutation in the tumor suppressor protein kinase A receptor subunit PRKAR1A. The authors suggested that Carney complex families lacking a PRKAR1A mutation may benefit from screening for PRKCA alterations.15
Alk and melanoma
Naturally, following the discovery of kinase fusions in spitzoid neoplasms, investigations into whether or not similar pathogenic mechanisms could be at work in non-spitzoid melanoma have been pursued. Recently, a newly described isoform of ALK-ALKATI- has been characterized that also results in immunoreactivity for ALK by immunohistochemistry. This ALK isoform results from an alternative transcription initiation site of the ALK gene and was discovered first through RNA sequencing of metastatic melanoma and thyroid carcinoma.17 ALKATI initiates transcription within intron 19, thereby encompassing a portion of intron 19 as well as the kinase domain within exons 20–29 in its transcript, but lacking exons 1–19 as seen in the wild-type transcript (interestingly, exons 20–29 are also encompassed in tumors with ALK translocations).17 This newly discovered isoform maintains the kinase domain but lacks the extracellular and transmembrane domain of wild-type ALK. Functional activity of the isoform was confirmed as ALKATI was found to promote growth factor independent proliferation in cell culture systems and to promote tumorigenesis in a mouse model system, and ALK inhibitors (crizotinib, ceritinib, and TAE-684) were able to reverse or inhibit these effects. Moreover, a patient with ALKATI expressing metastatic melanoma who had progressed on prior conventional therapies was treated with crizotinib and showed reduction in tumor burden and symptomatic improvement within 6 weeks, adding clinical evidence that tumors harboring this alternative isoform may benefit from ALK inhibitors as treatment.17
Using ALK immunoreactivity as a screen for ALK translocation or other ALK expression aberration, Busam and colleagues stained tissue microarrays and found ALK expression in 7 of 303 primary melanomas and in 9 metastatic melanomas.18 ALK-expressing tumor samples from the microarray were subsequently examined with immunohistochemistry performed on full sections of the tumor. In contrast to the homogenous staining reported in ALK-translocated Spitzoid tumors, ALK expression in non-Spitzoid melanomas was frequently heterogenous, both within the tumor and between tumors. ALK expression was seen predominantly within the cytoplasm, with rare nuclear labeling. The melanomas expressing ALK were of all subtypes but tended toward the nodular subtype. Many of the tumors were amelanotic and composed of epithelioid melanocytes. Upon further investigation, none of the ALK-expressing melanomas harbored an ALK translocation. Rather, all tumors with sufficient RNA for analysis were found to express the truncated ALK isoform, ALKATI.18 Metastatic tumors expressing ALKATIwhose primary melanoma was available for evaluation showed ALKATI expression in the primary tumor as well, suggesting that expression of the alternative isoform is an early oncogenic event, and expression of ALKATI also appeared to be independent of other known mutational events in melanoma, including BRAF and NRAS mutations.18 Given that the variable intensity and distribution of ALK expression might have led to an underestimation of the frequency of ALKATI expression in non-spitzoid melanomas using tissue microarrays, the authors estimated that 2–10% of melanomas might be expected to express ALKATI.18 In 2013, a Chinese group analyzed 58 cases of acral and mucosal melanomas and detected ALK immunoreactivity and ALK breakpoints by FISH in 4 acral melanomas; the authors suggested that the results indicated potential kinase fusion.19 Interestingly, similar to cases with ALKATI expression, ALK immunoreactivity was variable within tumor cells in the reported cases, and despite the positive FISH results, no fusion partners could be identified by PCR.19
Braf fusions in melanoma
Although ALK kinase fusions do not seem to be a common oncogenic driver in non-spitzoid melanomas, a fusion kinase involving BRAF, leading to constitutive kinase activation, has been documented in a subset of melanomas by several groups, with ~3% of melanomas estimated to harbor such fusions.2, 20, 21, 22, 23 As described previously, such fusions are also present in a similar percentage (~5%) of spitzoid neoplasms, both benign and atypical.
Using array chromosomal genomic hybridization, Botton and colleagues identified amplifications of the BRAF kinase domain in 10 of 848 melanocytic neoplasms (5 of which were diagnosed as melanoma±spitzoid morphology), which corresponded to a rearrangement of the gene. In each of the six cases sequenced, the BRAF kinase domain was fused to different, N-terminal gene partners. Ras-independent, constitutive activation of the kinase fusion was predicted because the translocation involved preservation of the kinase domain and loss of the autoinhibitory domain of the protein. A BRAF kinase fusion was also detected in a melanoma cell line, which indeed was confirmed to have constitutive BRAF kinase activation.20 Hutchinson et al.21 also described detection of BRAF kinase fusion in 2 of 52 melanomas interrogated with targeted next-generation sequencing, as well identifying 2 other potential BRAF fusions from analysis of whole-genome and whole-exome sequencing data of tumors available on the Cancer Genome Atlas melanoma data set. The translocation partners were different in each case and unique from the partners discovered by Botton et al. In both studies, melanomas with BRAF kinase fusions were found to lack other known melanoma driver mutations.20, 21
In a recent, broader screen of many tumor types which included 531 melanomas, BRAF fusions were detected in 14 melanomas, with nine of those cases demonstrating an epithelioid and spindled (Spitzoid) morphology.23 This study also confirmed that BRAF kinase fusions are almost always mutually exclusive of other mutations in the MAP kinase pathway, although one melanoma harbored both a BRAF kinase fusion and a V600E mutation.23 In vitro experiments have found that BRAF fusion-expressing cell lines are not sensitive to vemurafinib, the mutation specific V600E BRAF inhibitor,20, 21 but have suggested that melanomas with BRAF kinase fusions are sensitive to inhibition by MEK inhibitors.21 Indeed, early reports have documented clinical response to trametinib in two patients with metastatic melanoma harboring unique BRAF kinase fusions.22 Sorafenib, a multikinase and wild-type RAF inhibitor, has also been reported to have clinical utility.20
Conclusions
The detection of kinase fusions in approximately half of spitzoid melanocytic neoplasms has provided a framework for better understanding of spitzoid tumor development. As these fusions may be detected across the entire spectrum from benign to malignant, kinase fusions are thought to represent an early oncogenic event. Though many kinases and a variety of different binding partners have been identified, these gene fusions result in a common end point of enhanced and constitutive kinase activation leading to increased cellular growth, proliferation, and survival of tumor cells. As specific kinase fusions become more readily identified and better characterized, it will be possible to determine if specific histologic features align with the presence of particular kinase fusions or with certain fusion partners. As the catalytic domains of several of these kinases are genetically similar (for example ALK and ROS), we may find that the fusion partner drives some of the histologic or biologic differences between tumors. The biologic potential of spitzoid neoplasms is notoriously difficult to predict and, with further study of larger cohorts, the presence or absence of a kinase fusion may provide additional insight into the predicted outcome of a particular lesion. Already, kinase inhibitors are used with good effect in the treatment of other cancer types and are showing preliminary success in a subset of patients with aggressive melanocytic lesions that have a documented kinase fusion. Undoubtedly, further work in the field will continue to reveal mechanisms important for the development and growth of these tumors and will assist in the accurate diagnosis of these challenging melanocytic neoplasms.
References
Gerami P, Busam K, Cochran A et al, Histomorphologic assessment and interobserver diagnostic reproducibility of atypical spitzoid melanocytic neoplasms with long-term follow-up. Am J Surg Pathol 2014; 38: 934–940.
Wiesner T, He J, Yelensky R et al, Kinase fusions are frequent in Spitz tumours and spitzoid melanomas. Nat Commun 2014; 5: 3116.
Yeh I, de la Fouchardiere A, Pissaloux D et al, Clinical, histopathologic, and genomic features of Spitz tumors with ALK fusions. Am J Surg Pathol 2015; 39: 581–591.
Busam KJ, Kutzner H, Cerroni L et al, Clinical and pathologic findings of Spitz nevi and atypical Spitz tumors with ALK fusions. Am J Surg Pathol 2014; 38: 925–933.
Yeh I, Botton T, Talevich E et al, Activating MET kinase rearrangements in melanoma and Spitz tumours. Nat Commun 2015; 6: 7174.
Wiesner T, Kutzner H, Cerroni L et al, Genomic aberrations in spitzoid melanocytic tumours and their implications for diagnosis, prognosis and therapy. Pathology 2016; 48: 113–131.
Hallberg B, Palmer RH . The role of the ALK receptor in cancer biology. Ann Oncol 2016; 27: iii4–iii15.
Uguen A, De Braekeleer M . ROS1 fusions in cancer: a review. Future Oncol 2016; 12: 1911–1928.
Lee S, Barnhill RL, Dummer R et al, TERT promoter mutations are predictive of aggressive clinical behavior in patients with spitzoid melanocytic neoplasms. Sci Rep 2015; 5: 11200.
Lee CY, Sholl LM, Zhang B et al, Atypical spitzoid neoplasms in childhood: a molecular and outcome study. Am J Dermatopathol 2016 (e-pub ahead of print 15 November 2016).
Doyle LA, Marino-Enriquez A, Fletcher CD et al, ALK rearrangement and overexpression in epithelioid fibrous histiocytoma. Mod Pathol 2015; 28: 904–912.
Wu G, Barnhill RL, Lee S et al, The landscape of fusion transcripts in spitzoid melanoma and biologically indeterminate spitzoid tumors by RNA sequencing. Mod Pathol 2016; 29: 359–369.
Vaishnavi A, Le AT, Doebele RC . TRKing down an old oncogene in a new era of targeted therapy. Cancer Discov 2015; 5: 25–34.
Kiuru M, Jungbluth A, Kutzner H et al, Spitz tumors: comparison of histological features in relationship to immunohistochemical staining for ALK and NTRK1. Int J Surg Pathol 2016; 24: 200–206.
Bahrami A, Lee S, Wu G et al, Pigment-synthesizing melanocytic neoplasm with protein kinase C alpha (PRKCA) fusion. JAMA Dermatol 2016; 152: 318–322.
Wiesner T . Genomic rearrangements in unusual and atypical melanocytic neoplasms. JAMA Dermatol 2016; 152: 260–262.
Wiesner T, Lee W, Obenauf AC et al, Alternative transcription initiation leads to expression of a novel ALK isoform in cancer. Nature 2015; 526: 453–457.
Busam KJ, Vilain RE, Lum T et al, Primary and metastatic cutaneous melanomas express ALK through alternative transcriptional initiation. Am J Surg Pathol 2016; 40: 786–795.
Niu HT, Zhou QM, Wang F et al, Identification of anaplastic lymphoma kinase break points and oncogenic mutation profiles in acral/mucosal melanomas. Pigment Cell Melanoma Res 2013; 26: 646–653.
Botton T, Yeh I, Nelson T et al, Recurrent BRAF kinase fusions in melanocytic tumors offer an opportunity for targeted therapy. Pigment Cell Melanoma Res 2013; 26: 845–851.
Hutchinson KE, Lipson D, Stephens PJ et al, BRAF fusions define a distinct molecular subset of melanomas with potential sensitivity to MEK inhibition. Clin Cancer Res 2013; 19: 6696–6702.
Menzies AM, Yeh I, Botton T et al, Clinical activity of the MEK inhibitor trametinib in metastatic melanoma containing BRAF kinase fusion. Pigment Cell Melanoma Res 2015; 28: 607–610.
Ross JS, Wang K, Chmielecki J et al, The distribution of BRAF gene fusions in solid tumors and response to targeted therapy. Int J Cancer 2016; 138: 881–890.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The author declares no conflict of interest.
Additional information
Translocations resulting in kinase fusions have been recently described in melanocytic neoplasms, particularly those with spitzoid morphology. Seen within approximately half of all spitzoid neoplasms and present within benign, atypical, and malignant lesions, kinase fusions likely represent an early oncogenic event. This review article focuses on kinase fusions described to date in spitzoid neoplasms and how subsequent studies have informed current melanoma research.
Rights and permissions
About this article

Cite this article
Shalin, S. A review of kinase fusions in melanocytic tumors. Lab Invest 97, 158–165 (2017). https://doi.org/10.1038/labinvest.2016.122
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/labinvest.2016.122
This article is cited by
-
Novel gene fusion discovery in Spitz tumours and its relevance in diagnostics
Virchows Archiv (2023)
-
Expanding spectrum of “spitzoid” lesions: a small series of 4 cases with MAP2K1 mutations
Virchows Archiv (2021)
