
Abstract
Acute lung injury (ALI) and acute respiratory distress syndrome (ARDS) is characterized by increased pulmonary permeability with high mortality. Resolvin D1 (RvD1), which has potent anti-inflammatory and pro-resolving activity, can attenuate pulmonary edema in the animal model of ALI. However, the mechanism underlying the protection of RvD1 on pulmonary edema is still unknown. Here we explore the effects and mechanism of RvD1 on the disruption of tight junction protein that results in the permeability edema in a model of lipopolysaccharide (LPS)-induced ALI. The severity of pulmonary edema was assessed by wet-to-dry rate and Evans blue infiltration; expressions of tight junction (TJ) proteins occludin and zona occludin-1 (ZO-1) were examined by immunofluorescence staining and western blot; mRNA in lung tissue was studied by real time-PCR; the TUNEL kit was performed for the detection of apoptosis of pulmonary barrier. Twenty-four hours after LPS inhalation by mice, wet-to-dry rate and Evans blue infiltration indicated that pretreatment with RvD1 relieved the pulmonary edema and pulmonary capillary permeability. Moreover, RvD1 attenuated the LPS-induced deterioration of TJ protein ZO-1 and occludin significantly. And we found that RvD1 increased heme oxygenase-1 (HO-1) expression contributed to the protection on the deterioration of TJs. In addition, we found that RvD1 could reduce pulmonary cellular apoptosis in LPS-induced mice. In conclusion, RvD1 possesses the ability that relieves the pulmonary edema and restores pulmonary capillary permeability and reduces disruption of TJs in LPS-induced ALI of mice, at least in part, by upregulating HO-1 expression.
Similar content being viewed by others


Main
Acute lung injury (ALI) is still a severe clinical problem with a high mortality despite significant advances in antimicrobial therapy and supportive care made in the past few years.1 An important character of ALI is edema, which is due to the disruption of the paracellular alveolar barrier.2 Tight junction (TJ) proteins create a regulated paracellular barrier to the movement of water, solutes, and immune cells between both epithelial and endothelial cells.3 It has previously been demonstrated that alveolar epithelial cells express occludin and zona occludin-1 (ZO-1) as part of the TJ complex.3, 4 The trans-membrane protein occludin is an excellent candidate for the sealing protein and is bound on the cytoplasmic membrane surface to the proteins ZO-l and ZO-2.3 In addition, both apoptosis of endothelial and epithelial cells and dysfunction of TJ proteins can introduce pulmonary barriers disruption and cause edema during the ALI.5
Most important is that no standard treatment exists for permeability edema, making the research for restoring endothelial and epithelial hyper-permeability important. Resolvin D1 (RvD1) is a novel lipid mediator identified in resolving inflammation that was enzymatically derived from docosahexaenoic acid (DHA). DHA-derived resolvins act as a cytoprotective mechanism against oxidant injury6, 7 and the pro-resolving mediators, eg, lipoxin A4 and resolvins, can return the permeability/edema to normal.8 Previous studies have showed that RvD1 displays potent anti-oxidative stress,9 anti-inflammation, and pro-resolving actions that have been demonstrated in mouse models of ALI.10, 11, 12 Though they have been observed that RvD1 can reduce pulmonary edema induced by lipopolysaccharide (LPS), the effects of RvD1 on TJ protein remain elusive.
LPS, an endotoxin that resides on the outer membrane of Gram-negative bacteria, is known to enhance the formation of reactive oxygen species (ROS), inflammatory mediators, and promotes oxidative stress.13 ROS and inflammatory mediators triggers significant disruption of TJ proteins resulting in increased pulmonary permeability.5, 14
In this study, we test the hypothesis that RvD1 relieves permeability edema in LPS-induced mice by reduces disruption of TJ proteins. As a result, the deterioration of ZO-1 and occludin in mice exposed to LPS was attenuated by pretreatment with RvD1, which was reversed by a heme oxygenase-1 (HO-1) inhibitor.
MATERIALS AND METHODS
Reagents
LPS was purchased from Sigma (St Louis, MO, USA). RvD1 was from Cayman (Ann Arbor, MI, USA). The integrity of RvD1 was assured by monitoring the physical properties with High Performance Liquid Chromatography (HPLC) just before evaluating their biological activities. Zinc Protoporphyrin-9 (Znpp IX) was from (Enzo Life Sciences). GAPDH antibodies were purchased from Cell Signaling Technology (Boston, MA, USA). Mouse HO-1 antibody was from Abcam (Cambridge, MA, USA). Occludin and ZO-1 antibodys were purchased from Invitrogen (Frederick, MD, USA). Horseradish peroxidase-conjugated goat anti-rabbit antibody, bicinchoninic acid (BCA) protein assay kit and enhancer chemiluminescent (ECL) reagent were obtained from Pierce Biotechnology (Rockford, IL, USA). Protein extraction kit and TUNEL in situ apoptosis detection kit was from Key GEN Bio TECH.
Animals
Male Balb/c-mice (8–10 weeks old; weight range, 25–30 g) were obtained from Guangdong Medical Laboratory Animal Center (Guangdong, PR China). All mice received humane care according to the guidelines of the Local Institutes of Health guide for the care and use of laboratory animals. Mice were housed in a specific pathogen-free laboratory under optimum conditions (25 °C, 55% humidity and 12 h day/night rhythm) and fed with a standard laboratory diet and water. They were acclimatized for at least 1 week before use. All experimental procedures involving animals were approved by the Animal Care and Use Committee of Tongji Medical College in Huazhong University of Science and Technology.
Experimental Protocols
ALI was performed as described before.15 Briefly, animals were anesthetized with ketamine (30 mg/kg) intraperitoneally (i.p.). Sterile saline solution containing LPS was placed after intubation and aspirated into the lungs (3.0 mg/kg). Sixty mice were randomly assigned to six groups: (a) control (n=10), (b) LPS (n=10), (c) LPS+5 μg/kg RvD1 (n=10), (d) 5 μg/kg RvD1 (n=10), (e) LPS+5 μg/kg RvD1+50 mg/kg Znpp (n=10), and (f) LPS+50 mg/kg Znpp (n=10). Znpp was dissolved in 100% dimethyl sulfoxide (Calbiochem) and injected i.p. 1 h before administration of LPS. Then, RvD1 diluted in 0.9% sterile saline solution or vehicle (0.9% endotoxin-free saline) was injected though caudal vein 30 min before LPS administration. Mice (n=10) were humanely killed at 24 h after LPS administration; the lung tissues were used for further analysis. Another 40 mice were randomly assigned to four groups: (a) control (n=10), (b) LPS (n=10), (c) LPS+RvD1 (n=10), and (d) RvD1 (n=10). Each groups (a, b, c, and d) were injected Evans blue dye (EB; 2%; 5 ml/kg; Sigma, St Louis, MO, USA) for permeability analysis.
Hematoxylin and Eosin (HE) Staining
Lung samples were obtained at 24 h after LPS administration. Tissues were fixed in 4% paraformaldehyde and subsequently embedded in paraffin. Sections were stained with HE using a standard protocol and analyzed by light microscopy.
Lung Wet/Dry Weight Ratio
The lungs were dissected, weighed, and dried in an oven at 60 °C for 5 days. Then the W/D ratio was calculated by dividing the wet weight by the final dry weight.
EB
To further assess lung permeability, EB was dissolved in 0.9% saline at a final concentration of 5 mg/ml. Animals were anesthetized, weighed, and injected with 20 mg/kg EB in the vein. After 30 min, the animals were killed and the lungs perfused with 1 ml PBS containing 5 mM EDTA. The lungs were collected and frozen in liquid N2. The frozen lungs were homogenized in 2 ml PBS. The homogenate was diluted with two volume of formamide and incubated at 60 °C for 2 h, followed by centrifugation at 5000 g for 30 min. The supernatant was collected and absorbance was measured at 650 nm in a spectrophotometer. The EB concentration was determined from the standard absorbance curves evaluated in parallel. Correction for contaminating heme dye was calculated. The EB concentration was expressed as a percentage of the total dose of EB administered as described earlier.16
Immunofluorescence Microscopy
The microwave antigen repair technique was used to improve occludin and ZO-1-positive detection rate. Slides were placed in a plastic Coplin jar, filled with citrate buffer (pH 6.0), and covered with perforated cling film to minimize evaporation. Coplin jars were placed, evenly spaced, in a domestic microwave oven and irradiated for 5 min at medium power. After microwave irradiation, the slides were allowed to cool to room temperature and washed with PBS (pH 7.4) for 5 min 3 × . The sections were then blocked with 10% goat serum for 30 min at 37 °C and incubated with primary antibodies with antibodies directed against occludin (Rabbit anti-Occludin (C-term), 1:100; Invitrogen) and ZO-1 (rabbit IgG, 1:100; Invitrogen). Samples were incubated with primary antibodies at 4 °C overnight and then the second antibody with biotin for 30 min at room temperature. After drying, samples were conjugated with SABC complex SABC-CY3/(red fluorescence) for 30 min at room temperature. Before any step, there must be sufficient washes with PBS for 3 min 3 × , and all the incubation must be done in wet box. Images were captured by confocal laser scanning microscopy (Eclipse 80i, Nikon, Japan).
Real-Time-PCR
Differential expression of occludin and ZO-1 was validated by a separate comparison of new subjects using real-time-PCR. RNA was extracted (Invitrogen, Camarillo, Calif, USA) following the manufacturer’s instructions. The cDNA was generated from 1 μg total of each sample (SuperScript III; Invitrogen). The sequences of following primers were designed by Primer Premier 5.0 software (Premier Biosoft International, Palo Alto, California) and are shown in Table 1. Relative gene expression was calculated by the 2−ΔΔCT method.
Western Blot Assays
The proteins of samples were prepared according to the method described by the protein extract kit. Proteins concentrations were determined by the BCA protein assay kit (Piece Biotechnology, Rockford, USA). In all, 50-mg proteins extracts were fractionated on 8% polyacrylamide-sodiumdodecyl sulfate gel and then transferred to PVDF membrane. The membrane was blocked with 5% (w/v) fat-free milk in Tris-buffered saline containing 0.05% Tween 20 for 2 h, followed by incubation with rabbit primary polyclonal antibodies, including anti-occludin (1:150), ZO-1 (1:250), HO-1 (1:200), and anti-GAPDH (1:2000) at 4 °C overnight. After washing three times, membranes were incubated with the horseradish peroxidase-conjugated goat anti-rabbit secondary antibody (1:40 000) secondary antibody for 1 h at room temperature. The immune active bands were detected with ECL chemiluminescence detection kits (Thermo), followed by exposure to Kodakx-ray film (Kodak, Japan). The bands were quantified densitometrically as arbitrary volume integration units using ImageQuant software (Molecular Dynamics, Sunnyvale, CA). Results were corrected for the internal standard GAPDH.
TUNEL Assay
TUNEL assay was conducted by using a TUNEL detection kit according to the manufacturer’s instructions (Key GEN Bio TECH). Briefly, sections were incubated with 10 μg/ml proteinase K for 15 min at room temperature and then washed with PBS for 5min 3 × . Endogenous peroxidase was inactivated by 3% H2O2 for 10 min at room temperature and then washed with PBS. Sections were immersed in terminal deoxynucleotidyl transferase (TdT) buffer containing Equilibratin Buffer+Biotin-11-dUTP+TdT Enzyme in TdT buffer, incubated in a dark room at 37 °C for 60 min, and then washed with PBS. The sections were incubated at room temperature for 30 min with anti-horseradish peroxidase-conjugated antibody, and the signals were visualized with diaminobenzidine. From each biopsy, at least four fields were evaluated under a light microscope at a × 200 magnification for TUNEL-positive cells. The number of positive cells is calculated for analysis.
Statistical Analysis
Data are expressed as means±s.e.m. of the indicated number of independent experiments. All data were analyzed by one-way ANOVA. Pairwise testing between the different treatment groups was performed with Tukey’s post hoc test for multiple comparisons. Statistical analysis was performed using the GraphPad Prism 5.01 (GraphPad Software, San Diego, CA). P<0.05 was considered statistically significant.
RESULTS
Effect of RvD1 on LPS-Induced Pathological Changes of Lung Tissue
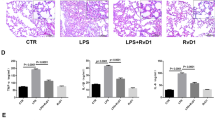
To determinate whether RvD1 exhibits protection on LPS-induced ALI, RvD1 was administrated 30 min before LPS challenge. HE staining was used to show pathological changes. There were no pathological changes in the lung tissue of control group (Figure 1a). In contrast, in the lung tissues of LPS group mice, significant infiltration of inflammatory cells, extensive thickening of the alveolar wall, demolished structure of pulmonary alveoli, and hemorrhage were observed at 24 h after LPS installation (Figure 1b). Pretreatment with RvD1 markedly alleviated LPS-induced pathological changes of lung (Figure 1c). The group pretreated with only RvD1 did not show any difference with the control group (Figure 1d).

Pathology changes in the lung. (Control group): No morphological alteration was detected; (LPS group): much inflammation cells infiltrated into intestitial and alveolar spaces and marked edema was observed; (LPS+RvD1 group): the inflammation induced by LPS was attenuated with less leukocytes and edematous fluid; (RvD1 group): no differences compared with the control group. Original magnification: × 400 (n=10).
Effect of RvD1 on LPS-Induced Pulmonary Edema and Vascular Permeability
The W/D ratio and extravasation of EB in the lung tissue were significantly elevated in the LPS group compared with the control group (Figure 2a and b), suggesting that LPS induced significant disruption of the pulmonary capillary barrier and elevation of pulmonary capillaries permeability. Notably, pretreatment with RvD1 attenuated pulmonary capillaries permeability in LPS-induced mice: the W/D ratio was reduced (Figure 2a) and the amount of EB leakage was diminished (Figure 2b).

Effect of RvD1 on LPS-induced pulmonary edema. (a) Lung tissue wet/dry weight ratio (W/D ratio). (b) The Evans blue dye in lung comparing with the total dose injected (EB lung/EB total) was used to assess lung extravascular permeability. LPS significantly increased the W/D ratio of lung weight and the Evans blue infiltration, which was attenuated by pretreatment with RvD1. The data were presented as mean±s.e.m.; (n=10). *P<0.05, **P<0.01 versus control group; ##P<0.01 versus LPS group.
Effect of RvD1 on LPS-Induced Disruption of TJ Proteins
To determine whether elevation of pulmonary capillaries permeability in LPS-induced mice is the result of disruption of TJ proteins ZO-1 and occludin, western blot analysis and immunofluorescence were used. Our results found that exposure to LPS markedly reduced the protein expression of ZO-1 and occludin in the lung tissue compared with the control group, which was blunted by pretreatment with RvD1 significantly. In addition, only administration of RvD1 without LPS did not result in any significant change of TJ proteins ZO-1 and occludin in lung tissue (Figure 3).

Effect of RvD1 on the protein expression of ZO-1 and occludin in lungs. Immunofluorescenece and western blot analysis were performed to detect the expression of tight junction proteins ZO-1 and occludin in lung tissue. (a) Twenty-four hours after exposure of LPS to mice, the expression of ZO-1 and occludin was dramatically reduced in lung tissues, which was attenuated by pretreatment with RvD1. GAPDH was used to show the equal loading volume. Protein expression/GAPDH was used to show the relative expression. Data were presented as mean±s.e.m.; (n=10). *P<0.05, **P<0.01 versus control group; #P<0.05, ##P<0.01 versus LPS group. (b) The results of immunostaining showed that ZO-1 and occludin stain diffusely in normal lung tissue, which was disrupted by exposure to LPS. And RvD1 restored the tight junction protein distribution (n=10).
Effect of RvD1 on LPS-Enhanced Expression of HO-1 in Lung Tissue
LPS binds to Toll-like receptor 4 initiating pro-inflammatory mediators and excessive ROS, which is associated with the disruption of TJ proteins. HO-1 has an important role in protecting lung tissue from injury induced by ROS. To determine whether HO-1 signal is involved in the protective effects of RvD1, a special inhibitor of heme oxygenase, Zinc Protoporphyrin-9 (Znpp IX), was exposed to mice before treatment with RvD1. We evaluated the protein and mRNA expression of HO-1 in each group. Both protein and mRNA expression of HO-1 were increased after treatment with LPS, which was enhanced by pretreatment with RvD1, and pretreatment with Znpp blocked the expression of HO-1 induced by LPS (Figure 4a, b and c). Subsequently, we evaluated the pathological change after challenge with Znpp and found that exposure to Znpp reversed the protection of RvD1 on pulmonary injury induced by LPS, which was consistent with the expression of HO-1 (Figure 4d).

Effect of RvD1 on the expression of heme oxygenase-1(HO-1). Mice were pretreated with or without Znpp (50 mg/kg) 30 min before RvD1 (5 μg/kg) or vehicle followed by LPS administration. Proteins and mRNA were extracted from lung tissues. (n=10). (a) Western blot analysis of HO-1 expression. (b) Quantification of HO-1 bands from experiments in panel a was normalized by GAPDH. (c) RT-PCR analysis of HO-1 expression. The data were presented as mean±s.e.m.; *P<0.05, **P<0.01 versus control group; #P<0.05, ##P<0.01 versus LPS group; ΔΔP<0.01 versus LPS+RvD1 group. (d) Morphological alteration was evaluated in each groups (original magnification: × 400), (n=10). Znpp blocked the protection of RvD1 on pulmonary injury induced by LPS.
HO-1-Mediated RvD1 Protection on LPS-Induced Disruption of TJ
To detect whether HO-1 devotes to protection of RvD1 on disruption of TJs induced by LPS, we used RT-PCR and western blot to analysis the expressions of ZO-1 and occludin. Results showed that Znpp can sharply attenuate the effect of RvD1 on protecting TJ proteins from disruption (Figure 5), suggesting that HO-1 mediated RvD1 protection on LPS-induced lung injury and TJ protein disruption.

Effect of HO-1 on the expression of ZO-1 and occludin in the lungs. (a) Western blot bands showed the effect of HO-1 on the protein expressions of ZO-1 and occludin. GAPDH was used to show the equal loading volume. Protein expression/GAPDH was used to show the relative expression. (b and c) Density analysis revealed that RvD1 enhanced the protein amount of ZO-1 and occludin in the LPS-treated lung tissue. But after Znpp was administered, which blocked the expression of HO-1, the protein expression of ZO-1 and occludin was reduced. Besides, the expression of ZO-1 and occludin show no difference in the LPS and LPS+Znpp groups. (d and e) RT-PCR shows the effect of HO-1 on the mRNA amount of ZO-1 and occludin. RvD1 enhanced the mRNA amount of ZO-1 and occludin in the LPS-treated lung tissue. After Znpp blocked the expression of HO-1, the mRNA expression of ZO-1 and occludin was reduced. The amount of ZO-1 and occludin show no difference in the LPS and LPS+Znpp groups. The data were presented as mean±s.e.m.; (n=10). *P<0.05, **P<0.01 versus control group; #P<0.05, ##P<0.01 versus LPS group; ΔP<0.05, ΔΔP<0.01 versus LPS+RvD1 group.
Effect of RvD1 on LPS-Induced Pulmonary Cell Apoptosis
Considering that pulmonary cell apoptosis is another important pathophysiological process of elevation of pulmonary capillary permeability in LPS-exposed mice, we evaluated the RvD1 effects on LPS-induced pulmonary cell apoptosis. Our results found that LPS resulted in a marked increase in the proportion of TUNEL-positive apoptotic cells in the lung tissue relative to the control group. The LPS-induced apoptosis was profoundly reduced in the presence of RvD1, which was abrogated by Znpp (Figure 6).

Cell apoptosis in lung tissue section by TUNEL analysis. Lung samples were obtained at 24 h after LPS administration. Tissues were fixed in 4% paraformaldehyde and subsequently embedded in paraffin. Sections were used to calculate the positive apoptosis cells in lung tissues by TUNEL. At least four fields were calculated in each section (n=10). (a) The TUNEL section showed the levels of apoptosis in lung. Original magnification: × 400. (b) Analysis of the positive cells. Data revealed that sharply increasing apoptosis of the lung induced by LPS was attenuated by RvD1. But interestingly, when exposed to Znpp, the impact of RvD1 was weakened. **P<0.01 versus control group; #P<0.05, ##P<0.01 versus LPS group; ΔP<0.05, ΔΔP<0.01 versus LPS+RvD1 group.
DISCUSSION
Although excessive edema and hyper pulmonary capillary permeability in ALI and its most severe form ARDS has been realized for many years, there are still no effective therapeutic agents.17, 18 RvD1, which possesses anti-inflammatory and pro-resolving ability in many process of inflammatory resolution, has demonstrated that it can attenuate inflammatory responses in ALI,10, 11 peritonitis,19, 20 kidney ischemia-reperfusion injury,21 and retinopathy.22 Here our study was focused on the effects of RvD1 on disruption of pulmonary barrier in ALI and was undertaken to investigate possible molecular and cellular mechanism of RvD1. Our results showed that the effect of 5 μg/kg RvD1 is potent, which is in accordance with the previous experiment.10 Our present data provide the evidence that RvD1 restores increased pulmonary permeability induced by LPS in vivo, which is via preventing deterioration of ZO-1 and occludin. It has been shown that LPS-induced inflammation is linked to increased pulmonary permeability;23 our result confirmed that administration of LPS resulted in ALI, characterized by pulmonary edema in interstitium and alveolous, and demolished structure of pulmonary alveoli and hyper-vascular permeability.
There are two pathways in lung tissue that allow solutes to traverse the barriers: trans-cellular and paracellular.24, 25 TJs serve the major functional purpose of providing a ‘barrier’ and a ‘fence’ within the membrane, by regulating paracellular permeability and maintaining cell polarity. Occludin is expressed in the TJs of endothelial cells and epithelial cells and requires the co-expression of junction protein ZO-1 for cell surface expression.4, 26 Our study, for the first time, demonstrated that RvD1 can reduce deterioration of TJ protein ZO-1 and occludin in LPS-induced mice by using western blot and immunofluorescence staining, which is at least one reason which can explain that RvD1 can relieve pulmonary edema, reduce wet-to- dry rate, and EB infiltration.
There are many factors that can influence TJ, such as infection,27, 28 cytokines,29 and ROS.30, 31 In the pathogenesis of ALI, extensive cytokines and ROS were released.32 Inflammation initiated by oxidative stress can be regulated by the RvD1.9 One of our previous studies on 15-epi-16-parafluorophenoxy lipoxin A4 (ATL), an analogue of RvD1, have found that ATL not only increased HO-1 expression but also markedly diminished all markers of lung injury.33 In addition, ATL-1 induces HO-1 in human endothelial cells, revealing an undescribed mechanism for the anti-inflammatory activity of these lipid mediators.34 Another study has discovered that protectin D1 regulates HO-1 in a renal model.35 HO-1, which is a rate-limiting enzyme that metabolizes heme accumulates in tissues because of blood red cell turnover, is correlated with the production of ROS.36 Against this background, our purpose was to find out whether RvD1 can protect TJ proteins from disruption by regulating the expression of HO-1. Our data showed that the expression of HO-1 increased in the LPS group mice. Regulation of HO-1 activity as an adaptive response to oxidative stress is to ameliorate the effects of many oxidative stressors.37 When pretreated with RvD1, mice that inhaled LPS had a high transcription and protein expression of HO-1. Znpp, which competes with the natural heme substrate, is considered to be a potent inhibitor of HO-1 activity. In the research, Znpp reduced the mRNA and protein amount of HO-1 compared with the LPS+RvD1 group and LPS group while the pathology revealed that lung injury became more serious after Znpp was injected. Besides, most important is that the expression of occludin and ZO-1 in the LPS+RvD1 group reduced sharply by pretreatment with Znpp, but the TJ proteins in the LPS+Znpp group did not show much difference compared with the LPS group. Therefore, the results indicated that Znpp itself cannot change the expression of occludin and ZO-1, and the changes attributed to the expression of HO-1. Our findings are consistent with our study that display RvD1 can protect TJ proteins from disruption by increasing the expression of HO-1.
In addition, there is more compelling evidence that increased epithelial/endothelial cell apoptosis contributes to the endothelial and epithelial injury that is characteristic of ALI/ARDS in humans. Studies have shown that ALI is associated with increased cell death in humans,38 while apoptosis inhibitors showed increased survival rodent models of ALI.5, 39 HO-1-mediated degradation of heme also releases CO. In the sub-toxic amounts produced by HO-1, CO initiates a cyto-protective signaling cascade by inducing the activity of guanylate cyclase to produce cGMP, a second messenger molecule known to protect cells from ischemic damage and enhance cell survival through its action on apoptosis.40 TNF and other inflammatory mediators have been shown to regulate a variety of additional anti-apoptotic genes, including HO-1.41 Early study clearly demonstrated that HO-1 induced by NO had a potent anti-apoptotic function in an experimental AH136B solid tumor in rats and Znpp IX induced the apoptosis of AH136B cells both in vivo and in vitro through inhibition of HO-1 activity.42 HO-1 attenuates glucose-mediated cell growth arrest and apoptosis in human micro-vessel endothelial cells.43 These may explain why mice pretreated with RvD1 showed higher expression of HO-1 and TJ proteins and decrease of apoptosis cells in TUNEL compared with the LPS group in this study. Moreover, pro-injected with Znpp mice showed more positive cells in TUNEL section compared with LPS+RvD1 group, but the apoptosis cells in the LPS group proved no marked difference with the LPS+Znpp group. According to these, RvD1 attenuated apoptosis of epithelial/endothelial cells to protect TJ may be partially due to the pathway of HO-1. In our study, we evaluated the loss of TJ proteins and apoptosis as two parallel results, and the sequence of their happening needs further study. There are many studies on the relationship between them. Su et al44 demonstrated that apoptosis results in TJ- and MLCK-independent barrier loss; however, Bruewer et al45 demonstrated that pro-inflammatory cytokines disrupt epithelial barrier function by apoptosis-independent mechanisms.
Conclusion
In summary, this study demonstrated that pretreatment with RvD1 attenuates ALI-induced pulmonary edema and disruption of TJ induced by LPS in mice. In addition, we also demonstrated that RvD1 induce HO-1 and the counteracted effects on the protection role of RvD1 between HO-1 and its antagonist, ZnPP. Therefore, the mechanism of RvD1 attenuating pulmonary edema depends on HO-1. The increasing expression of HO-1 can improve the expression of occludin and ZO-1 and reduce the apoptosis of the epithelial/endothelial cells to avoid disruption of TJ. But how these procedures are executed needs further study.
References
Matthay MA, Zimmerman GA, Esmon C et al. Future research directions in acute lung injury: summary of a National Heart, Lung, and Blood Institute Working Group. Am J Respir Crit Care Med 2003;167:1027–1035.
Crandall ED, Matthay MA . Alveolar epithelial transport. Basic science to clinical medicine. Am J Respir Crit Care Med 2001;163:1021–1029.
Anderson JM, Van Itallie CM . Tight junctions and the molecular basis for regulation of paracellular permeability. Am J Physiol 1995;269:G467–G475.
Bazzoni G, Dejana E . Endothelial cell-to-cell junctions: molecular organization and role in vascular homeostasis. Physiol Rev 2004;84:869–901.
Lucas R, Verin AD, Black SM et al. Regulators of endothelial and epithelial barrier integrity and function in acute lung injury. Biochem Pharmacol 2009;77:1763–1772.
Serhan CN, Clish CB, Brannon J et al. Novel functional sets of lipid-derived mediators with antiinflammatory actions generated from omega-3 fatty acids via cyclooxygenase 2-nonsteroidal antiinflammatory drugs and transcellular processing. J Exp Med 2000;192:1197–1204.
Serhan CN, Hong S, Gronert K et al. Resolvins: a family of bioactive products of omega-3 fatty acid transformation circuits initiated by aspirin treatment that counter proinflammation signals. J Exp Med 2002;196:1025–1037.
Serhan CN . Novel lipid mediators and resolution mechanisms in acute inflammation: to resolve or not? Am J Pathol 2010;177:1576–1591.
Spite M, Summers L, Porter TF et al. Resolvin D1 controls inflammation initiated by glutathione-lipid conjugates formed during oxidative stress. Br J Pharmacol 2009;158:1062–1073.
Wang B, Gong X, Wan JY et al. Resolvin D1 protects mice from LPS-induced acute lung injury. Pulm Pharmacol Ther 2011;24:434–441.
Eickmeier O, Seki H, Haworth O et al. Aspirin-triggered resolvin D1 reduces mucosal inflammation and promotes resolution in a murine model of acute lung injury. Mucosal Immunol 2012;6:256–266.
Kasuga K, Yang R, Porter TF et al. Rapid appearance of resolvin precursors in inflammatory exudates: novel mechanisms in resolution. J Immunol 2008;181:8677–8687.
Matsuzawa A, Saegusa K, Noguchi T et al. ROS-dependent activation of the TRAF6-ASK1-p38 pathway is selectively required for TLR4-mediated innate immunity. Nat Immunol 2005;6:587–592.
Vandenbroucke E, Mehta D, Minshall R et al. Regulation of endothelial junctional permeability. Ann N Y Acad Sci 2008;1123:134–145.
Asti C, Ruggieri V, Porzio S . Lipopolysaccharide-induced lung injury in mice. I. Concomitant evaluation of inflammatory cells and haemorrhagic lung damage. Pulm Pharmacol Ther 2000;13:61–69.
Kaner RJ, Ladetto JV, Singh R et al. Lung overexpression of the vascular endothelial growth factor gene induces pulmonary edema. Am J Respir Cell Mol Biol 2000;22:657–664.
Matthay MA, Zemans RL . The acute respiratory distress syndrome: pathogenesis and treatment. Annu Rev Pathol 2011;6:147–163.
Maybauer MO, Maybauer DM, Herndon DN . Incidence and outcomes of acute lung injury. N Engl J Med 2006;354:416–417.
Hong S, Gronert K, Devchand PR et al. Novel docosatrienes and 17S-resolvins generated from docosahexaenoic acid in murine brain, human blood, and glial cells. Autacoids in anti-inflammation. J Biol Chem 2003;278:14677–14687.
Sun YP, Oh SF, Uddin J et al. Resolvin D1 and its aspirin-triggered 17R epimer. Stereochemical assignments, anti-inflammatory properties, and enzymatic inactivation. J Biol Chem 2007;282:9323–9334.
Duffield JS, Hong S, Vaidya VS et al. Resolvin D series and protectin D1 mitigate acute kidney injury. J Immunol 2006;177:5902–5911.
Connor KM, SanGiovanni JP, Lofqvist C et al. Increased dietary intake of omega-3-polyunsaturated fatty acids reduces pathological retinal angiogenesis. Nat Med 2007;13:868–873.
Eutamene H, Theodorou V, Schmidlin F et al. LPS-induced lung inflammation is linked to increased epithelial permeability: role of MLCK. Eur Respir J 2005;25:789–796.
Minshall RD, Sessa WC, Stan RV et al. Caveolin regulation of endothelial function. Am J Physiol Lung Cell Mol Physiol 2003;285:L1179–L1183.
Simionescu N, Simionescu M, Palade GE . Open junctions in the endothelium of the postcapillary venules of the diaphragm. J Cell Biol 1978;79:27–44.
Van Itallie CM, Anderson JM . Occludin confers adhesiveness when expressed in fibroblasts. J Cell Sci 1997;110:1113–1121.
Roxas JL, Koutsouris A, Bellmeyer A et al. Enterohemorrhagic E. coli alters murine intestinal epithelial tight junction protein expression and barrier function in a Shiga toxin independent manner. Lab Invest 2010;90:1152–1168.
Guttman JA, Finlay BB . Tight junctions as targets of infectious agents. Biochim Biophys Acta 2009;1788:832–841.
Capaldo CT, Nusrat A . Cytokine regulation of tight junctions. Biochim Biophys Acta 2009;1788:864–871.
Peerapen P, Thongboonkerd V . Effects of calcium oxalate monohydrate crystals on expression and function of tight junction of renal tubular epithelial cells. Lab Invest 2011;91:97–105.
Schreibelt G, Kooij G, Reijerkerk A et al. Reactive oxygen species alter brain endothelial tight junction dynamics via RhoA, PI3 kinase, and PKB signaling. FASEB J 2007;21:3666–3676.
Cross LJ, Matthay MA . Biomarkers in acute lung injury: insights into the pathogenesis of acute lung injury. Crit Care Clin 2011;27:355–377.
Jin SW, Zhang L, Lian QQ et al. Posttreatment with aspirin-triggered lipoxin A4 analog attenuates lipopolysaccharide-induced acute lung injury in mice: the role of heme oxygenase-1. Anesth Analg 2007;104:369–377.
Nascimento-Silva V, Arruda MA, Barja-Fidalgo C et al. Novel lipid mediator aspirin-triggered lipoxin A4 induces heme oxygenase-1 in endothelial cells. Am J Physiol Cell Physiol 2005;289:C557–C563.
Hassan IR, Gronert K . Acute changes in dietary omega-3 and omega-6 polyunsaturated fatty acids have a pronounced impact on survival following ischemic renal injury and formation of renoprotective docosahexaenoic acid-derived protectin D1. J Immunol 2009;182:3223–3232.
Matsumoto H, Ishikawa K, Itabe H et al. Carbon monoxide and bilirubin from heme oxygenase-1 suppresses reactive oxygen species generation and plasminogen activator inhibitor-1 induction. Mol Cell Biochem 2006;291:21–28.
Haines DD, Lekli I, Teissier P et al. Role of haeme oxygenase-1 in resolution of oxidative stress-related pathologies: focus on cardiovascular, lung, neurological and kidney disorders. Acta Physiol (Oxf) 2012;204:487–501.
Mizuta M, Nakajima H, Mizuta N et al. Fas ligand released by activated monocytes causes apoptosis of lung epithelial cells in human acute lung injury model in vitro. Biol Pharm Bull 2008;31:386–390.
Rudkowski JC, Barreiro E, Harfouche R et al. Roles of iNOS and nNOS in sepsis-induced pulmonary apoptosis. Am J Physiol Lung Cell Mol Physiol 2004;286:L793–L800.
Panahian N, Yoshiura M, Maines MD . Overexpression of heme oxygenase-1 is neuroprotective in a model of permanent middle cerebral artery occlusion in transgenic mice. J Neurochem 1999;72:1187–1203.
Semenza GL . Hypoxia-inducible factor 1: regulator of mitochondrial metabolism and mediator of ischemic preconditioning. Biochim Biophys Acta 2011;1813:1263–1268.
Tanaka S, Akaike T, Fang J et al. Antiapoptotic effect of haeme oxygenase-1 induced by nitric oxide in experimental solid tumour. Br J Cancer 2003;88:902–909.
Abraham NG, Kushida T, McClung J et al. Heme oxygenase-1 attenuates glucose-mediated cell growth arrest and apoptosis in human microvessel endothelial cells. Circ Res 2003;93:507–514.
Su L, Nalle SC, Shen L et al. TNFR2 activates MLCK-dependent tight junction dysregulation to cause apoptosis-mediated barrier loss and experimental colitis. Gastroenterology, advance online publication, 22 April 2013; doi:10.1053/j.gastro.2013.04.011 (e-pub ahead of print).
Bruewer M, Luegering A, Kucharzik T et al. Proinflammatory cytokines disrupt epithelial barrier function by apoptosis-independent mechanisms. J Immunol 2003;171:6164–6172.
Acknowledgements
This study was supported by Grants from the National Natural Science Foundation of China (81070060 and 30930089).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Effective therapeutics are urgently needed for treatment of pulmonary edema. This paper demonstrates that resolvin D1 relieves pulmonary edema, restores pulmonary permeability and reduces disruption of tight junction proteins in lipopolysaccharide-induced acute lung injury of mice, at least in part, by up-regulation of heme oxygenase-1 expression.
Rights and permissions
About this article
Cite this article
Xie, W., Wang, H., Wang, L. et al. Resolvin D1 reduces deterioration of tight junction proteins by upregulating HO-1 in LPS-induced mice. Lab Invest 93, 991–1000 (2013). https://doi.org/10.1038/labinvest.2013.80
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/labinvest.2013.80
Keywords
This article is cited by
-
In vitro and in vivo protective potential of quercetin-3-glucuronide against lipopolysaccharide-induced pulmonary injury through dual activation of nuclear factor-erythroid 2 related factor 2 and autophagy
Archives of Toxicology (2024)
-
Resolvin-D2 targets myogenic cells and improves muscle regeneration in Duchenne muscular dystrophy
Nature Communications (2021)
-
Lung remodeling associated with recovery from acute lung injury
Cell and Tissue Research (2017)
-
Resolvin D1 mitigates energy metabolism disorder after ischemia–reperfusion of the rat lung
Journal of Translational Medicine (2016)
-
ResolvinD1 reduces apoptosis and inflammation in primary human alveolar epithelial type 2 cells
Laboratory Investigation (2016)
