
Abstract
A pro-fibrotic role of matrix metalloproteinase-9 (MMP-9) in tubular cell epithelial–mesenchymal transition (EMT) is well established in renal fibrosis; however studies from our group and others have demonstrated some previously unrecognized complexity of MMP-9 that has been overlooked in renal fibrosis. Therefore, the aim of this study was to determine the expression pattern, origin and the exact mechanism underlying the contribution of MMP-9 to unilateral ureteral obstruction (UUO), a well-established model of renal fibrosis via MMP-9 inhibition. Renal MMP-9 expression in BALB/c mice with UUO was examined on day 1, 3, 5, 7, 9, 11 and 14. To inhibit MMP-9 activity, MMP-2/9 inhibitor or MMP-9-neutralizing antibody was administered daily for 4 consecutive days from day 0–3, 6–9 or 10–13 and tissues harvested at day 14. In UUO, there was a bi-phasic early- and late-stage upregulation of MMP-9 activity. Interestingly, tubular epithelial cells (TECs) were the predominant source of MMP-9 during early stage, whereas TECs, macrophages and myofibroblasts produced MMP-9 during late-stage UUO. Early- and late-stage inhibition of MMP-9 in UUO mice significantly reduced tubular cell EMT and renal fibrosis. Moreover, MMP-9 inhibition caused a significant reduction in MMP-9-cleaved osteopontin and macrophage infiltration in UUO kidney. Our in vitro study showed MMP-9-cleaved osteopontin enhanced macrophage transwell migration and MMP-9 of both primary TEC and macrophage induced tubular cell EMT. In summary, our result suggests that MMP-9 of both TEC and macrophage origin may directly or indirectly contribute to the pathogenesis of renal fibrosis via osteopontin cleavage, which, in turn further recruit macrophage and induce tubular cell EMT. Our study also highlights the time dependency of its expression and the potential of stage-specific inhibition strategy against renal fibrosis.
Similar content being viewed by others


Main
Matrix metalloproteinase-9 (MMP-9), also known as gelatinase B, belongs to a large family of zinc-dependent matrix-degrading enzymes called matrix metalloproteinases (MMPs) that have an important role in both physiological and pathological conditions in vivo.1 MMPs are well known for their anti-fibrotic effect due to their ability to degrade and remodel extracellular matrix (ECM) proteins, however it has been recognized that MMPs can also exert pro-fibrotic effect under various pathological conditions.
Myofibroblasts are the effector cells responsible for the production and secretion of ECM in renal fibrosis. It has been reported that myofibroblasts can derive from the activation of renal interstitial fibroblasts, perivascular fibroblasts and pericytes, from bone marrow-derived stem cells and from mesenchymal transition of endothelial cells and tubular epithelial cells (TECs).2, 3, 4, 5 Tubular cell epithelial–mesenchymal transition (EMT) is the transition of TEC into cells of mesenchymal phenotype, where TEC lose their epithelial characteristics and acquire a mesenchymal phenotype with the ability to secrete ECM. While EMT of primary TEC or cell lines in response to transforming growth factor-β1 (TGF-β1) has been well established,6, 7, 8, 9 the exact contribution of tubular cell EMT to kidney fibrosis is still under debate.10, 11, 12, 13 Iwano et al3 demonstrated that EMT of TEC contributed to over a third of myofibroblasts in renal fibrosis by using proximal TEC genetically tagged mice in a UUO model. However, by using the same Cre/lox technology in the same UUO model, Humphry et al11 found no evidence of TEC migrating to interstitium and expressing myofibroblast markers Nevertheless, a role for TEC in kidney fibrosis has been proven by their overexpression of MMP-214 or TGF-β1.13 Hertig et al15 showed that TEC do not need to go through a complete transition to myofibroblasts to contribute to renal fibrosis. It is possible that TEC undergoing EMT-like changes may become local producers of ECM.16 Migration across basement membrane as in EMT of cancer (type III) or embryonic cells (type I) may not be a feature of the EMT of epithelial cells (type II) in fibrotic diseases.17
MMP-9 is well established to have a role in tubular cell EMT,9, 18 in fact induction of tubular cell EMT in vitro and in vivo is associated with increased expression of MMP-9. Although MMP-9 knockout (KO) has been shown to reduce tubular cell EMT and renal fibrosis in murine unilateral ureteral obstruction (UUO) model,19 interpretation of results obtained from the KO mice may have oversimplified the biological complexity of MMP-9. Moreover, the use of MMP-KO model has yielded conflicting results in various disease models.20
Recent studies from our group have demonstrated that MMP-9 itself is capable of inducing the entire course of tubular cell EMT via the disruption of E-cadherin/β-catenin-mediated cell-to-cell adhesion complex, independent of epithelial cell–basement membrane disruption.6, 21 Moreover, results from our in vitro study has highlighted a prominent role and potentially an important source of MMP-9 from macrophages and TEC in tubular cell EMT induction, which has not previously been addressed in renal fibrosis.6 Beside tubular cell EMT induction, MMP-9 is known to be capable of cleaving osteopontin (OPN),22 a macrophage chemoattractant and macrophages are well known to have a role in renal fibrosis.23, 24, 25 However, whether a protection in renal fibrosis was due to a reduction of macrophage infiltration in MMP-9 KO model is unclear. Furthermore, MMPs including MMP-9 has been shown to exhibit an opposing role at different stages of disease progression including renal disease.26, 27, 28
Overall, the above observations highlight the complex biological function of MMP-9 that has previously been overlooked in renal fibrosis. Therefore, the aim of this study was to determine the expression pattern, origin and the exact mechanism underlying the contribution of MMP-9 in UUO model via MMP-9 inhibition.
MATERIALS AND METHODS
Animal and UUO
Six-week-old BALB/c mice were purchased from Australian Research Council and experiments were performed in accordance with protocols approved by Animals Ethics Committee of Sydney West Area Health Service. UUO was performed by left proximal ureteral ligation under anesthesia as described elsewhere.29 A total of five mice were used in each control and experimental group. Both obstructed and contralateral unobstructed kidneys were harvested at 2 weeks after UUO. For the UUO time-course study, tissues were harvested on day 1, 3, 5, 7, 9 and 14.
To inhibit MMP-9 activity, an MMP-2/9 inhibitor (2 mg/kg) (Chemicon, Billerica, MA, USA) or MMP-9-neutralizing antibody (6-6B) (2 mg/kg) (Calbiochem) was administered intraperitoneally for 4 consecutive days 0–3, 6–9 or 10–13 after UUO and tissues were harvested at 2 weeks after UUO. DMSO (Sigma) and mouse IgG1 isotype control antibody (Calbiochem) were used as vehicle for MMP-2/9 inhibitor and MMP-9-neutralizing antibody, respectively.
Cell Culture
Primary proximal TEC were obtained from the cortex of MMP-9 wild type (WT) or MMP-9 KO BALB/c mice using an established protocol adapted from Doctor et al.30 Briefly, kidney cortex was minced and digested in collagenase IV and proximal TEC were separated by centrifugation using Percoll solution and cultured in K1 medium supplemented with 5% fetal calf serum (FCS). After 24 h, proximal TEC were washed and cultured in serum-free K1 medium for 4–5 days before treatment with TGF-β (5 ng/ml) or conditioned medium derived from non-stimulated or lipopolysaccharide (LPS)-stimulated MMP-9 WT or KO macrophages for 24 h.
Splenocytes from MMP-9 WT or MMP-9 KO BALB/c mice were harvested and washed in cold RPMI-1640. Tissue was triturated and the resulting cell suspension was filtered through a 40-μm nylon mesh and incubated on a petri dish at 37 °C for 30 min. Adherent cells were harvested and purified by magnetic-activated cell-sorting CD11b+ Microbeads. Purified macrophages were cultured in RPMI-1640 supplemented with 10% FCS at 37 °C under humidified conditions with 5% CO2. After 24 h, macrophages were washed and cultured in complete medium with or without LPS (100 ng/ml) for 24 h, washed three times and cultured in serum-free RPMI-1640 for 48 h. After 48 h, medium was collected and filtered through a 0.22-μm filter.
Murine tubular epithelial cell line (C1.1) was cultured in K1 medium (Dulbecco’s Modified Eagle Medium: HAM’s F12; 1:1 v/v) (Invitrogen, Carlsbad, CA, USA) containing 25 μg/ml of epidermal growth factor (Sigma, St Louis, MO, USA), 25 μmol/L of HEPES (Invitrogen), hormone mixture and 5% FCS at 37 °C with 5% CO2. For treatment, C1.1 cells were cultured under low confluency for 24 h in complete medium, rinsed in phosphate-buffered saline (PBS) and then in serum-free medium with TGF-β (3 ng/ml) alone or with MMP-2/9 inhibitor (0.5, 1, 2.5 or 5 μM).
Indirect Double Immunofluorescence
For indirect double immunofluorescence, cells cultured on glass coverslips were washed with PBS, fixed with cold methanol at −20 °C for 10 min, and blocked with 2% bovine serum albumin (BSA) in PBS for 20 min at room temperature. Cells were incubated with primary rabbit polyclonal anti-E-cadherin (1:100, Abcam) and monoclonal anti-mouse α-SMA (1:200, Sigma) in 2% BSA in PBS for 1 h at room temperature. After washing with PBS, cells were incubated with secondary anti-rabbit Alexa Fluor-488 (1:600, Invitrogen) and anti-mouse Alexa Fluor-546 (1:600, Invitrogen) for 40 min at room temperature. Cells were then washed with PBS and counterstained with 4′, 6-diamidino-2 phenylindole (DAPI) (Invitrogen) for 5 min before mounting with fluorescent-mounting medium. The number of E-cadherin-negative or α-SMA-positive stained cells was quantitated by fluorescence microscopy.
Histology and Immunohistochemistry
Kidney tissues were fixed and embedded in paraffin. Sections were cut at 5 μm, deparaffinized, rehydrated, blocked with 3% H2O2 in methanol (v/v) for 10 min and followed by 2% BSA (Sigma) in PBS for 1 h at room temperature. For detection of β-catenin, antigen retrieval was performed using antigen decloaker (Biocare Medical) for 10 min at 95 °C. Primary antibodies for OPN (1:100, Abcam), snail (0.2 μg/ml, Abcam), α-SMA (1:200, Sigma), F4/80 (1:200, Abcam) and β-catenin (1:100, BD bioscience) were applied for 1 h at room temperature. Secondary antibody was applied for 40 min at room temperature. Antibody binding was visualized with 3′3-diaminobenzidine (DAB) (Dako) and sections were counterstained with hematoxylin (Sigma). Isotype control antibody was used as primary antibody for negative control, and no staining was observed. The presence of interstitial fibrosis was determined by Gomori trichrome staining.
Histological Analysis
For histological quantitation, a minimum of 15 non-overlapping images of cortex were taken under × 200 magnification for each kidney unless otherwise specified, and quantitated in a blinded manner. Quantitation for interstitial α-SMA and Gomori trichrome staining was performed by a modified grid-counting method adapted from Vielhauer et al.31
To quantitate the number of infiltrating macrophages and Snail- or OPN-positive cells, the number of F4/80-positive cells in the interstitium and Snail-positive TEC or OPN-positive cells (including TEC and interstitial cells) were counted, and results expressed as the average number of the respective positive cells per high power field (HPF). To quantitate β-catenin translocation in TEC, the number of TEC with cytoplasmic or nuclear staining was counted. The result was divided by the total number of cells positively stained for β-catenin (including membrane, cytoplasmism and nucleus) to obtain the percentage of β-catenin translocation.
To quantitate macrophage migration in vitro, the number of migrated J774 macrophages was counted by immunofluorescence microscopy. A minimum of 20 consecutive non-overlapping fields of view was taken under × 200 magnification. Results are expressed as the average number of cells per HPF from an average of a duplicate per experiment and a minimum of four independent experiments.
To quantitate the percentage of E-cadherin or α-SMA-positive cells in primary TEC culture, a minimum of 10 consecutive non-overlapping fields of view was taken under green fluorescence filter (for E-cadherin), red fluorescence filter (for α-SMA) and blue fluorescence filter (for DAPI) at × 200 magnification. Quantitation of the number of E-cadherin-negative or α-SMA-positive cells was performed using merged images of E-cadherin and α-SMA staining, and the total cell number was determined by DAPI staining. Results are expressed as percentage of E-cadherin-negative or α-SMA-positive cells per HPF from an average of duplicate experiments and a minimum of four independent experiments.
Western Blot Analysis
Kidney cortex was homogenized in RIPA buffer and protein concentration was determined using Bio-Rad protein assay. Lysates from equal numbers of cultured cells were obtained by Tris-Glycine SDS sample buffer (Gradipore, Frenchs Forest, Australia).
Equal quantities of protein or lysates from equal numbers of cells were loaded per lane on 12-well NUPAGE 4 to 12% Bris-Tris gel (Invitrogen) for electrophoresis under reduced conditions. After electrophoresis, proteins were electron transferred onto PVDF membrane for 2 h using Mini Trans-Blot Electrophoretic Transfer Cell (Bio-Rad, Hercules, CA). For immunodetection, membranes were blocked in 5% (wt/vol) skimmed milk in PBS overnight at 4 °C and were incubated with primary antibodies against MMP-9 (1:1000), OPN (1 μg/ml), β-catenin (1:2000), α-SMA (1 μg/ml) and β-actin (1:4000, Sigma) for 1 h at room temperature. Membranes were washed and incubated with horseradish peroxidase (HRP)-conjugated secondary antibody (1:2000, Santa Cruz Biotechnology) for 40 min at room temperature. Protein bands were visualized with an enhanced chemiluminescence detection kit. Band intensity was quantified by densitometry using Image J software.
Gelatin Zymography
Equal quantities of protein from kidney cortex homogenates were loaded and electrophoresed through 10% Novex zymogram gelatin gels with Tris-Glycine SDS Running Buffer (Invitrogen) under constant voltage of 125 V for 90 min. After electrophoresis, the gel was incubated in Zymogram renaturing and developing buffer, stained with Coomassie Blue R-250 (Bio-Rad) and destained as described previously.6 Gelatinolytic activity of MMP-9 was visualized as a clear band on a blue background. Band intensity was quantified by densitometry using Adobe Photoshop 8 software, as described previously.6
Macrophage Migration Assay
Migration of murine macrophage cell line (J774) was assayed by a modification of Boyden chamber method using 24-well Transwell inserts with a 5-μm diameter porous membrane (Costar, Cambridge, MA, USA). Briefly, Transwell inserts were coated with BSA or OPN (50 ng/ml) with or without recombinant MMP-9 (200 ng/ml) (BIOMOL International, Plymouth Meeting, PA, USA) in RPMI medium, incubated at 37 °C overnight and blocked with 10 mg/ml BSA in RPMI for 1 h at 37 °C, and 600 μl of RPMI was added into the lower chamber with recombinant CCL-2 (50 ng/ml) or vehicle. An aliquot of 0.1 × 106 J774 macrophages in 200 μl of RPMI medium was loaded onto the Transwell insert and incubated for 5 h. Non-migrated J774 macrophages were gently removed on the upper side of the membrane with cotton swabs and the undersurface of the membrane was fixed with cold methanol at −20 °C for 10 min, stained with DAPI (Invitrogen) and the number of migrated J774 macrophages was counted by immunofluorescence microscopy.
In situ Hybridization
To probe MMP-9 mRNA, all buffers were treated with 0.1% diethylpyrocarbonate or made in 0.1% diethylpyrocarbonate-treated distilled H2O. For detection of MMP-9 mRNA in kidney tissues, formalin-fixed paraffin-embedded kidney tissues were cut at 2 μM, deparafinized, rehydrated and blocked in 3% H2O2 in methanol for 15 min at room temperature. The sections were incubated in 0.3% triton-X in PBS for 15 min at room temperature, washed in PBS, digested with proteinase-K for 20 min at 37 °C and washed in PBS. The sections were then immersed in cold 20% acetic acid (v/v) for 20 s, dehydrated and air dried. Double digoxigenin (DIG)-labeled custom locked nucleic acid detection MMP-9 mRNA probe or sense control probe (Table 1) (LNA mRNA, Exiqon, Woburn, MA, USA) (40 μM) was diluted in hybridization buffer (4 × SCC, 10 mM DTT, 10% dextran sulfate, 2 × Denhart solution, 50% formamide) and denatured at 95 °C for 2 min. The sections were then hybridized with double-DIG-labeled MMP-9 mRNA probe or sense control probe at 51 °C for 22 h in a humidified chamber. After hybridization, sections were rinsed once in 2 × SCC and washed 3 times in 0.2 × SCC for 5 min each at 45 °C. For immunodetection of DIG-labeled probes, sections were blocked in 2% BSA in PBS for 1 h at room temperature and incubated with HRP-conjugated anti-DIG antibody (1:100, Abcam) in 2% BSA in PBS at 37 °C for 2 h in a humidified chamber. After washing with PBS, probe binding was visualized with DAB.
Real-time PCR
Kidney cortex was homogenized, RNA was extracted using RNeasy Mini Kit (Qiagen, Hilden, Germany) and reverse-transcribed into cDNA using Superscript First-strand synthesis system (Invitrogen). Real-time PCR amplification with primers (Table 2) was carried out in Corbett Rotorgene 3000 Real-time Thermo cycler using a PCR mixture containing primers, cDNA and SYBR green mastermix (Invitrogen). Housekeeping gene β-actin was used as endogenous control.
Statistical Analysis
Results are expressed as mean±s.e.m. Statistical significance was evaluated using two-tail t-test for comparison between two groups and a value of P<0.05 was regarded as statistically significant.
RESULTS
Renal MMP-9 Expression after UUO
MMP-9 expression was detectable by western blot analysis on days 1 and 3, rose further on day 5, was low on days 7 and 9 and peaked on days 11 and 14 (Figure 1a). There was a similar time course for gelatinolytic activity of MMP-9 on days 1, 3, 5, 11 and 14 in UUO kidneys (Figures 1b and c). MMP-9 expression and gelatinolytic activity were undetectable in sham control kidneys throughout the time course (Figures 1a and b, respectively).

Bi-phasic, early- and late-stage expression of matrix metalloproteinase-9 (MMP-9) in kidney after unilateral ureteral obstruction (UUO). Representative western blot staining of MMP-9 (a) and gelatin zymography of MMP-9 gelatinolytic activity (b) in sham-operated (sham control) or UUO kidney at day 1, 3, 5, 7, 9, 11 and 14 after surgery. Each lane contains an equal amount of protein from kidney cortex. (c) Gelatinolytic activity of MMP-2 and MMP-9 were quantified by densitometry of gelatin zymographs. Data are expressed as mean±s.e.m. n=5 for each experimental group. *P<0.05, **P<0.01 and ***P<0.001 vs sham control.
Sources of Renal MMP-9 after UUO
To determine the source of MMP-9, immunofluorescence staining and in situ hybridization were performed. Immunofluorescence staining showed double staining of MMP-9 with F4/80 and MMP-9 with α-SMA in UUO kidney (Figures 2a and b, respectively). Co-localization of MMP-9 mRNA-positive cells with immunohistochemical staining for F4/80 or α-SMA on consecutive kidney tissue sections showed three main sources of MMP-9 after UUO, namely TEC, macrophages and myofibroblasts (Figure 2c). Quantitative analysis based on MMP-9 mRNA-positive cells showed an early stage increase, mid stage reduction and late-stage increase in the number of MMP-9 mRNA-positive cells after UUO (Figure 2d). TEC were the predominant source of MMP-9 at early stage and a major source of MMP-9 at mid and late stage after UUO (Figures 2e and f, respectively). Interstitial cells were the major source of MMP-9 during late stage after UUO (Figure 2e), with approximately one-third of all MMP-9 mRNA-positive cells originated from macrophages or myofibroblasts (Figure 2f).

Tubular epithelial cell (TEC), macrophages (F4/80) and myofibroblasts (α-SMA) are major sources of matrix metalloproteinase-9 (MMP-9) in kidney after unilateral ureteral obstruction (UUO). Immunofluorescence staining of (a) MMP-9 (red), F4/80 (green) and merged (arrows), and (b) MMP-9 (red), α-SMA (green) and merged (asterisks) in UUO kidney at day 14 after UUO. (c) Co-localization of MMP-9 mRNA-positive cells with myofibroblasts (+) and macrophages (*) in UUO kidney at day 3, 7 and 14 after surgery. Quantitative analysis of the number of (d) total MMP-9 mRNA-positive cells, (e) interstitial vs tubular origin of MMP-9 mRNA-positive cells and (f) myofibroblasts, macrophages and TEC (based on morphology) which co-localized with MMP-9 mRNA-positive cells at day 3, 7 and 14 after surgery. Data are expressed as mean±s.e.m. n=5 for each experimental group. †P<0.001 vs D7 (mid); ‡P<0.001 vs D14 (late); #P<0.05 and ##P<0.01 vs tubular; *P<0.05, **P<0.01 and ***P<0.001 vs other. HPF, high power field.
Effect of MMP-9 Inhibition on OPN Expression and MMP-9-cleaved OPN in UUO Kidney
The effect of MMP-9 inhibition on OPN expression in UUO kidneys was assessed by immunohistochemistry. Compared with sham control kidneys, OPN expression was upregulated in UUO kidneys, but there were no significant differences in the expression of OPN in UUO kidneys of mice treated with MMP-9 Ab or MMP-2/9 inhibitor or their controls at early, mid or late stage of the disease (data not shown).
Because MMP-9 is capable of cleaving OPN,32 we then sought to determine the effect of MMP-9 inhibition on MMP-9-cleaved OPN (34, 32 and 5 kDa) by western blot analysis. Kidneys of mice treated with MMP-9 Ab or MMP-2/9 inhibitor during early and late stage, but not mid stage after UUO showed a significant reduction in MMP-9-cleaved OPN (32 kDa) compared with isotype Ab or vehicle-treated UUO kidneys (Figures 3a and b). MMP-9-cleaved OPN corresponding to protein size 34 and 5 kDa was undetectable (data not shown).

Early- and late-stage inhibition of matrix metalloproteinase-9 (MMP-9) reduces MMP-9-cleaved osteopontin (OPN) in unilateral ureteral obstruction (UUO) kidney. (a) Representative western blot staining of OPN in UUO kidney of mice treated with isotype or MMP-9 Ab, vehicle or MMP-2/9 inhibitor during early, mid or late stage after UUO. (b) OPN expression was quantified by densitometry and results are presented as the relative ratio of OPN to β-actin. Data are expressed as mean±s.e.m. n=5 for each experimental group. *P<0.05 and **P<0.01 vs respective control.
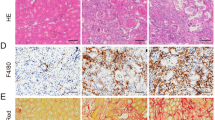
Effect of MMP-9 Inhibition on Macrophage Infiltration in UUO Kidney
The effect of reduced MMP-9-cleaved OPN on macrophage infiltration was assessed by immunohistochemistry. Macrophage infiltration was increased in UUO kidneys compared with sham control kidneys, but was significantly reduced in UUO kidneys of mice treated with MMP-9 Ab or MMP-2/9 inhibitor at early and late stage, but not mid stage of the disease compared with UUO kidneys of mice treated with isotype Ab or vehicle (Figures 4a and b). Expression of chemokine (C-C motif) ligand 2 (CCL-2) mRNA was upregulated in UUO kidney but unaffected by early-, mid- or late-stage inhibition of MMP-9 activity with MMP-2/9 inhibitor or MMP-9 Ab (Figure 4c).

Early- and late-stage inhibition of matrix metalloproteinase-9 (MMP-9) reduces macrophage infiltration in unilateral ureteral obstruction (UUO) kidney, independent of CCL-2 expression. (a) Representative immunohistochemical staining for F4/80 in kidney sections of sham-operated and UUO kidney of mice treated with isotype or MMP-9 Ab, vehicle or MMP-2/9 inhibitor at early, mid or late stage after UUO. (b) The number of infiltrating F4/80 cells in sham-operated and UUO kidney of mice treated with isotype or MMP-9 Ab, vehicle or MMP-2/9 inhibitor during early, mid or late stage after UUO. (c) The mRNA expression level of CCL-2 from cortex of sham-operated and UUO kidney of mice treated with isotype or MMP-9 Ab, vehicle or MMP-2/9 inhibitor during early, mid or late stage after UUO was measured by real-time RT-PCR. Results are presented as the relative ratio of CCL-2 to β-actin. Data are expressed as mean±s.e.m. n=5 for each experimental group. *P<0.05, **P<0.01, ***P<0.001 vs respective control.
Effect of MMP-9-cleaved OPN on Macrophage Migration In vitro
To determine whether MMP-9-cleaved OPN can enhance macrophage migration, a migration assay was performed using recombinant full-length OPN or recombinant full-length OPN pre-treated or not with recombinant MMP-9. As a negative control for OPN, BSA was used. Migration of J774 macrophages towards CCL-2 was significantly enhanced when cell culture inserts were coated with OPN compared with BSA. The number of migrated J774 macrophages was increased further when cell culture inserts were coated with OPN pre-treated with recombinant MMP-9 (Figures 5a and b).

Matrix metalloproteinase-9 (MMP-9)-cleaved osteopontin enhances macrophage migration in vitro. J774 macrophages were subjected to chemotaxis assays in modified Boyden chambers. Transwells were pre-coated with bovine serum albumin (BSA) treated or not with MMP-9 (as control) or with recombinant osteopontin (OPN) treated or not with MMP-9. Following J774 macrophage attachment, CCL-2 or vehicle was used as the chemotactic signal for migration. (a) Nuclei of migrated J774 macrophages were stained with 4′, 6-diamidino-2 phenylindole, (b) quantitated and expressed as number of macrophages per high power field (HPF). Experiments were repeated four times in duplicate. Data are expressed as mean±s.e.m. *P<0.05, **P<0.01.
Effect of MMP-9 Inhibition on Fibrosis in UUO Kidney
We next explored the effect of MMP-9 inhibition on renal fibrosis in UUO kidneys by immunohistochemistry for α-SMA and Gomori trichrome. Increased interstitial α-SMA staining was observed in UUO kidneys compared with sham control kidneys (Figure 6a). Interstitial α-SMA was increased in UUO kidneys on day 9 compared with day 7 and increased further on day 11 compared with day 9, consistent with progression of fibrosis throughout the course of UUO (Figure 6a). In UUO, kidneys of mice treated at early and late stage, but not mid stage with MMP-9 Ab or MMP-2/9 inhibitor there was a significant reduction in interstitial α-SMA and Gomori trichrome staining compared with isotype Ab or vehicle-treated UUO kidneys (Figures 6b and c and 7a and b).

Early- and late-stage inhibition of matrix metalloproteinase-9 (MMP-9) reduces interstitial myofibroblasts in unilateral ureteral obstruction (UUO) kidney. (a) Representative immunohistochemical staining and quantitative analysis for α-SMA in sham-operated kidney and UUO kidney sections on day 7, 9, 11 and 14 after surgery. (b) Representative immunohistochemical staining and (c) quantitative analysis for α-SMA of kidney sections from sham-operated kidney and UUO kidney of mice treated with isotype or MMP-9 Ab, vehicle or MMP-2/9 inhibitor during early, mid or late stage after UUO. Data are expressed as mean±s.e.m. n=5 for each experimental group. ††P<0.01 vs day 7 UUO; †††P<0.001 vs day 9 UUO; *P<0.05, ***P<0.001 vs respective control.

Early- and late-stage inhibition of matrix metalloproteinase-9 (MMP-9) reduces interstitial fibrosis in unilateral ureteral obstruction (UUO) kidney. (a) Representative Gomori trichrome staining (blue) and (b) quantitative analysis of interstitial Gomori trichrome staining of kidney sections from sham-operated kidney or UUO kidney of mice treated with isotype or MMP-9 Ab, vehicle or MMP-2/9 inhibitor during early, mid or late stage after UUO. Data are expressed as mean±s.e.m. n=5 for each experimental group. *P<0.05, **P<0.01, ***P<0.001 vs respective control.
Effect of MMP-9 Inhibition on β -catenin Translocation and Expression in UUO Kidney
To determine whether the reduction in myofibroblast (α-SMA) in UUO kidney after MMP-9 inhibition during early and late stage could be due to a reduction in tubular cell EMT, β-catenin expression and localization were examined. Recent in vitro studies from our group showed that MMP-9 is capable of translocating β-catenin from cell membrane to cytoplasm or nucleus during the course of tubular cell EMT.6 The overall expression of β-catenin was unaffected in UUO kidneys of mice treated with MMP-9 Ab or MMP-2/9 inhibitor at all stages of the disease progression as compared with UUO kidneys of isotype Ab or vehicle-treated mice (data not shown). However, an increased in cytoplasmic or nuclear staining of β-catenin observed in UUO kidneys was significantly reduced in UUO kidneys of mice treated with MMP-9 Ab or MMP-2/9 inhibitor during early and late stage, but not mid stage as compared with isotype Ab or vehicle-treated mice (Figures 8a and b).

Early- and late-stage inhibition of matrix metalloproteinase-9 (MMP-9) reduces the translocation of β-catenin in tubular epithelial cell (TEC) in unilateral ureteral obstruction (UUO) kidney. (a) Representative immunohistochemical staining for β-catenin and (b) quantitative analysis of β-catenin translocation in sham-operated kidney and UUO kidney of mice treated with isotype or MMP-9 Ab, vehicle or MMP-2/9 inhibitor during early, mid or late stage after UUO. β-catenin localized at the cell membrane under normal conditions and translocated into cytoplasm or nucleus in UUO as indicated by strong cytoplasmic or nuclear staining (arrows). Data are expressed as mean±s.e.m. n=5 for each experimental group. *P<0.05 vs respective control.
Effect of MMP-9 Inhibition on Snail Expression in UUO Kidney
To confirm the role of MMP-9 in inducing tubular cell EMT induction in UUO kidneys, we examined the effect of MMP-9 inhibition on the expression of the EMT-associated transcription factor Snail. Consistent with our other findings, an increase in immunohistochemical staining for Snail in TEC, and an upregulated expression of Snail mRNA in UUO kidney was significantly reduced in UUO kidney of mice treated with MMP-2/9 inhibitor or MMP-9 Ab at early or late stage, but not mid stage of the disease as compared with UUO kidneys of isotype or vehicle-treated mice (Figures 9a and b).

Early- and late-stage inhibition of matrix metalloproteinase-9 (MMP-9) reduces epithelial–mesenchymal transition transcription factor Snail expression in unilateral ureteral obstruction (UUO) kidney. (a) Representative immunohistochemistry and quantitative analysis of Snail staining in tubular cells and (b) mRNA expression of Snail of sham-operated kidney and UUO kidney of mice treated with isotype or MMP-9 Ab, vehicle or MMP-2/9 inhibitor during early, mid or late stage after UUO. Real-time RT-PCR results are presented as the relative ratio of Snail to β-actin. Data are expressed as mean±s.e.m. n=5 for each experimental group. *P<0.05, **P<0.001, ***P<0.001 vs respective control.
A Role for TEC or Macrophage-derived MMP-9 in Tubular Cell EMT Induction
To directly examine the role of endogenous MMP-9 from TEC or macrophages in the induction of tubular cell EMT, primary TEC or macrophages derived from MMP-9 WT or KO mice were used. TEC derived from MMP-9 WT or KO mice were treated with TGF-β. Immunofluorescence staining showed that TGF-β-induced increases in α-SMA-positive and E-cadherin-negative cells were significantly reduced in MMP-9 KO TEC compared with MMP-9 WT TEC (Figure 10a). This result is supported by a separate experiment where an increase in α-SMA induced by TGF-β in C1.1 TECs was reduced by a potent MMP-9 inhibitor in a dose-dependent manner (Figure 10b).

Loss of matrix metalloproteinase-9 (MMP-9) reduces transforming growth factor-β (TGF-β) or macrophage-induced tubular cell epithelial–mesenchymal transition in MMP-9 knockout (KO) tubular epithelial cell (TEC) or MMP-9 KO macrophages, respectively. (a) Representative double immunofluorescence staining of E-cadherin (green) and α-SMA (red) of TGF-β-treated TEC derived from MMP-9 wild type (WT) or KO mice. (b) Western blot analysis of α-SMA in C1.1 cells treated with TGF-β in absence or presence of MMP-2/9 inhibitor. (c) Representative double immunofluorescence staining of E-cadherin (green) and α-SMA (red) of MMP-9 WT TEC treated with resting (M0) or activated (M1) macrophage (MΦ) conditioned medium derived from MMP-9 WT or MMP-9 KO mice. Loss of E-cadherin and acquisition of α-SMA expression in TEC were quantified and expressed as a percentage of cells. (d) Real-time RT-PCR analysis of MMP-2, 3, 7 and 9 expression in TGF-β-treated TEC and activated macrophage (M1) from MMP-9 WT or KO mice. Data are expressed as mean±s.e.m. n=4 for each experimental group. *P<0.05, **P<0.01, ***P<0.001 vs respective control.
To determine the role of macrophage-derived MMP-9 in the induction of tubular cell EMT, conditioned medium derived from LPS-stimulated MMP-9 WT or MMP-9 KO macrophages was cultured with MMP-9 WT TEC. Conditioned medium derived from LPS-stimulated MMP-9 KO macrophages increased the percentage of α-SMA-positive and E-cadherin-negative cells, whereas the percentage of these cells was significantly lower than with conditioned medium derived from LPS-stimulated MMP-9 WT macrophages (Figure 10c). Previous studies have reported compensatory upregulation of MMPs in MMP-KO animals. To demonstrate whether other MMPs were upregulated in MMP-9 KO mice, we examined the expression of MMP-2, MMP-3, MMP-7 and MMP-9 mRNA in TGF-β treated MMP-9 WT or KO TEC, and LPS-stimulated MMP-9 WT or KO macrophages. MMP-2 and MMP-3 mRNA were significantly upregulated in KO TEC as compared with WT TEC treated with TGF-β (Figure 10d). Only MMP-2 mRNA was significantly upregulated in LPS-stimulated KO macrophages as compared with WT macrophages (Figure 10d).
DISCUSSION
Although a role of MMP-9 has previously been established in renal fibrosis, its pathological complexity has largely been overlooked and the exact mechanism underlying its contribution to renal fibrosis remains unclear. In the present study, we revealed several previously unrecognized role of MMP-9 in the pathogenesis of renal fibrosis. Our results showed a bi-phasic early- and late-stage upregulation of MMP-9 in UUO model; MMP-9 recruit macrophages via OPN cleavage; early- and late-stage inhibition of MMP-9 reduced tubular cell EMT and renal fibrosis.
MMP production is tightly regulated and its expression may be cell specific and depend on signals that are temporarily expressed.33 In this study, we showed a bi-phasic early- and late-stage upregulation of renal MMP-9 during the course of UUO. This observation recalls two previous reports of bi-phasic upregulation of MMP-9 during the course of experimental herpes simplex virus (HSV) type-1 keratitis,34 and in a rat model of chronic allograft nephropathy.35 Interestingly, we showed that early increase in MMP-9 expression was predominantly from TEC and further increase in MMP-9 expression at later stage arises from inflammatory cells, specifically macrophages and myofibroblasts in UUO kidney. This observation not only suggests TEC and macrophages as an important source of MMP-9 in renal fibrosis but also implicates its role in tubular cell EMT induction as result from our previous and present in vitro studies demonstrated a critical role of both TEC and macrophage MMP-9 in tubular cell EMT induction.6 However, the study of Koesters et al13 demonstrates that TEC may indirectly contribute to renal fibrosis via secretion of TGF-β, a well-recognized pro-fibrogenic growth factor and also an inducer of MMP-9.36 In support of their study, our current result showed that MMP-9 recruits macrophages via OPN cleavage and that macrophages may contribute to renal fibrosis by at least partial tubular cell EMT induction.
Whereas upregulation of MMPs is often associated with disease severity,37 the pathogenic relevance of this association is uncertain because, for example in cancer progression,38 studies have shown an opposing role for MMPs at different stages of disease. In the current study, we showed that inhibition of MMP-9 activity by an MMP-2/9 inhibitor or MMP-9 Ab, specifically at early or late stages of UUO, was protective against the development of renal fibrosis. This result is consistent with a role of MMP-9 in renal fibrosis, which was previously reported,19, 39 however the mechanism underlying its contribution was not fully explained. To the best of our knowledge, our study is the first to demonstrate a stage-specific expression of MMP-9 in renal fibrosis and its relevance to stage-specific therapy.
Previous studies by Yang et al19, 39 demonstrated that direct or indirect inhibition of MMP-9 activity resulted in a reduction in tubular cell EMT and renal fibrosis in obstructive nephropathy. Findings from our current study further extended those observations where early- and late-stage inhibition of MMP-9 not only reduced tubular cell EMT and renal fibrosis but also reduced MMP-9-cleaved OPN and macrophage infiltration.
Although OPN is well known to have a role in macrophage infiltration,40 and in vitro studies have demonstrated an enhanced chemotactic function of MMP-cleaved OPN,32, 41 no previous in vivo study has shown whether MMP-cleaved OPN is also chemotactic. Earlier studies demonstrated that the expression of OPN correlates well with macrophage infiltration in various experimental models of renal injury.42, 43 Persy et al44 showed that the loss of OPN expression in OPN-deficient mice resulted in a significant reduction in macrophage infiltration and renal fibrosis in a murine model of ischemia reperfusion injury. In our current study, reduction of macrophage infiltration in UUO following MMP-9 inhibition was associated with a reduction in expression of MMP-cleaved OPN corresponding to the protein size of 32 kDa and not the expression of chemokine CCL-2, which was previously found to be a key macrophage chemoattractant in UUO model.31, 45 This 32-kDa-cleaved OPN fragment has been demonstrated to be a byproduct of MMP-9 cleavage.32 Our result suggests that it is MMP-9-cleaved OPN, rather than OPN expression, that determines its role in macrophage infiltration.
Numerous studies have highlighted an important role for macrophages in causing renal injury and fibrosis.46, 47, 48, 49 For example, specific ablation of macrophages in UUO kidney of diphtheria toxin (DT)-treated CD11b-DT receptor transgenic mice,47, 49 even at early stage resulted in a reduction in renal fibrosis.49 Moreover Duffield et al46 demonstrated an important role for macrophages in maintaining the myofibroblast population in renal fibrosis. Consistent with both observations, our current study showed that reduction in macrophage infiltration at early and late stage of UUO was associated with a reduction in myofibroblast numbers and renal fibrosis. Moreover, results from our in vitro and in situ hybridization suggest macrophage may have a direct role in renal fibrosis via tubular cell EMT induction. We showed that tubular cell EMT induced by conditioned medium derived from MMP-9 KO macrophages was reduced compared with medium derived from wild-type macrophages. Furthermore, in situ hybridization showed macrophage as a major source of MMP-9 late after UUO.
Overall, our current study demonstrates an important role of both TEC and macrophage MMP-9 in renal fibrosis. Our result suggests that MMP-9 of both TEC and macrophage origin may directly or indirectly contribute to the pathogenesis of renal fibrosis via induction of tubular cell EMT and OPN cleavage, which in turn further recruit macrophage. This study also highlights the time dependency of its expression and the potential of stage-specific inhibition strategy against renal fibrosis.
References
Chakraborti S, Mandal M, Das S et al. Regulation of matrix metalloproteinases: an overview. Mol Cell Biochem 2003;253:269–285.
Zeisberg EM, Potenta SE, Sugimoto H et al. Fibroblasts in kidney fibrosis emerge via endothelial-to-mesenchymal transition. J Am Soc Nephrol 2008;19:2282–2287.
Iwano M, Plieth D, Danoff TM et al. Evidence that fibroblasts derive from epithelium during tissue fibrosis. J Clin Invest 2002;110:341–350.
Lin SL, Kisseleva T, Brenner DA et al. Pericytes and perivascular fibroblasts are the primary source of collagen-producing cells in obstructive fibrosis of the kidney. Am J Pathol 2008;173:1617–1627.
Li Y, Kang YS, Dai C et al. Epithelial-to-mesenchymal transition is a potential pathway leading to podocyte dysfunction and proteinuria. Am J Pathol 2008;172:299–308.
Tan TK, Zheng G, Hsu TT et al. Macrophage matrix metalloproteinase-9 mediates epithelial-mesenchymal transition in vitro in murine renal tubular cells. Am J Pathol 2010;176:1256–1270.
Zheng G, Lyons JG, Tan TK et al. Disruption of E-cadherin by matrix metalloproteinase directly mediates epithelial-mesenchymal transition downstream of transforming growth factor-beta1 in renal tubular epithelial cells. Am J Pathol 2009;175:580–591.
Zeisberg M, Hanai J, Sugimoto H et al. BMP-7 counteracts TGF-beta1-induced epithelial-to-mesenchymal transition and reverses chronic renal injury. Nat Med 2003;9:964–968.
Liu Y . Epithelial to mesenchymal transition in renal fibrogenesis: pathologic significance, molecular mechanism, and therapeutic intervention. J Am Soc Nephrol 2004;15:1–12.
Liu Y . New insights into epithelial-mesenchymal transition in kidney fibrosis. J Am Soc Nephrol 2010;21:212–222.
Humphreys BD, Lin SL, Kobayashi A et al. Fate tracing reveals the pericyte and not epithelial origin of myofibroblasts in kidney fibrosis. Am J Pathol 2010;176:85–97.
Hinz B, Phan SH, Thannickal VJ et al. Recent developments in myofibroblast biology: paradigms for connective tissue remodeling. Am J Pathol 2012;180:1340–1355.
Koesters R, Kaissling B, Lehir M et al. Tubular overexpression of transforming growth factor-beta1 induces autophagy and fibrosis but not mesenchymal transition of renal epithelial cells. Am J Pathol 2010;177:632–643.
Cheng S, Pollock AS, Mahimkar R et al. Matrix metalloproteinase 2 and basement membrane integrity: a unifying mechanism for progressive renal injury. FASEB J 2006;20:1898–1900.
Hertig A, Anglicheau D, Verine J et al. Early epithelial phenotypic changes predict graft fibrosis. J Am Soc Nephrol 2008;19:1584–1591.
Fragiadaki M, Witherden AS, Kaneko T et al. Interstitial fibrosis is associated with increased COL1A2 transcription in AA-injured renal tubular epithelial cells in vivo. Matrix Biol 2011;30:396–403.
Fragiadaki M, Mason RM . Epithelial-mesenchymal transition in renal fibrosis - evidence for and against. Int J Exp Pathol 2011;92:143–150.
Liu Y . Renal fibrosis: new insights into the pathogenesis and therapeutics. Kidney Int 2006;69:213–217.
Wang X, Zhou Y, Tan R et al. Mice lacking matrix metalloproteinase-9 gene reduces renal interstitial fibrosis in obstructive nephropathy. Am J Physiol Renal Physiol 2010;299:F973–82.
Kruger A . Functional genetic mouse models: promising tools for investigation of the proteolytic internet. Biol Chem 2009;390:91–97.
Zheng G, Lyons JG, Tan TK et al. Disruption of E-cadherin by matrix metalloproteinase directly mediates epithelial-mesenchymal transition downstream of transforming growth factor-{beta}1 in renal tubular epithelial cells. Am J Pathol 2009;175:580–591.
Gürsoy G, Acar Y, Alagöz S . Osteopontin:A multifunctional molecule. J Med Med Sci 2010;1:55–60.
Nomiyama T, Perez-Tilve D, Ogawa D et al. Osteopontin mediates obesity-induced adipose tissue macrophage infiltration and insulin resistance in mice. J Clin Invest 2007;117:2877–2888.
Ophascharoensuk V, Giachelli CM, Gordon K et al. Obstructive uropathy in the mouse: role of osteopontin in interstitial fibrosis and apoptosis. Kidney Int 1999;56:571–580.
O'Regan AW, Hayden JM, Body S et al. Abnormal pulmonary granuloma formation in osteopontin-deficient mice. Am J Respir Crit Care Med 2001;164:2243–2247.
Fanjul-Fernandez M, Folgueras AR, Cabrera S et al. Matrix metalloproteinases: evolution, gene regulation and functional analysis in mouse models. Biochim Biophys Acta 2010;1803:3–19.
Lutz J, Yao Y, Song E et al. Inhibition of matrix metalloproteinases during chronic allograft nephropathy in rats. Transplantation 2005;79:655–661.
Zeisberg M, Khurana M, Rao VH et al. Stage-specific action of matrix metalloproteinases influences progressive hereditary kidney disease. PLoS Med 2006;3:e100.
Moriyama T, Kawada N, Ando A et al. Up-regulation of HSP47 in the mouse kidneys with unilateral ureteral obstruction. Kidney Int 1998;54:110–119.
Doctor RB, Chen J, Peters LL et al. Distribution of epithelial ankyrin (Ank3) spliceoforms in renal proximal and distal tubules. Am J Physiol 1998;274 (1 Pt 2):F129–138.
Vielhauer V, Anders HJ, Mack M et al. Obstructive nephropathy in the mouse: progressive fibrosis correlates with tubulointerstitial chemokine expression and accumulation of CC chemokine receptor 2- and 5-positive leukocytes. J Am Soc Nephrol 2001;12:1173–1187.
Takafuji V, Forgues M, Unsworth E et al. An osteopontin fragment is essential for tumor cell invasion in hepatocellular carcinoma. Oncogene 2007;26:6361–6371.
Parks WC, Wilson CL, Lopez-Boado YS . Matrix metalloproteinases as modulators of inflammation and innate immunity. Nat Rev Immunol 2004;4:617–629.
Yang YN, Bauer D, Wasmuth S et al. Matrix metalloproteinases (MMP-2 and 9) and tissue inhibitors of matrix metalloproteinases (TIMP-1 and 2) during the course of experimental necrotizing herpetic keratitis. Exp Eye Res 2003;77:227–237.
Inkinen KA, Soots AP, Krogerus LA et al. Fibrosis and matrix metalloproteinases in rat renal allografts. Transpl Int 2005;18:506–512.
Strutz F, Zeisberg M, Ziyadeh FN et al. Role of basic fibroblast growth factor-2 in epithelial-mesenchymal transformation. Kidney Int 2002;61:1714–1728.
Roy R, Yang J, Moses MA . Matrix metalloproteinases as novel biomakers and potential human therapeutic targets for human cancer. J Clin Oncol 2009;27:5287–5297.
Lopez-Otin C, Matrisian LM . Emerging roles of proteases in tumour suppression. Nat Rev Cancer 2007;7:800–808.
Yang J, Shultz RW, Mars WM et al. Disruption of tissue-type plasminogen activator gene in mice reduces renal interstitial fibrosis in obstructive nephropathy. J Clin Invest 2002;110:1525–1538.
Mazzali M, Kipari T, Ophascharoensuk V et al. Osteopontin--a molecule for all seasons. QJM 2002;95:3–13.
Agnihotri R, Crawford HC, Haro H et al. Osteopontin, a novel substrate for matrix metalloproteinase-3 (stromelysin-1) and matrix metalloproteinase-7 (matrilysin). J Biol Chem 2001;276:28261–28267.
Panzer U, Thaiss F, Zahner G et al. Monocyte chemoattractant protein-1 and osteopontin differentially regulate monocytes recruitment in experimental glomerulonephritis. Kidney Int 2001;59:1762–1769.
Hartner A, Porst M, Gauer S et al. Glomerular osteopontin expression and macrophage infiltration in glomerulosclerosis of DOCA-salt rats. Am J Kidney Dis 2001;38:153–164.
Persy VP, Verhulst A, Ysebaert DK et al. Reduced postischemic macrophage infiltration and interstitial fibrosis in osteopontin knockout mice. Kidney Int 2003;63:543–553.
Grande MT, Perez-Barriocanal F, Lopez-Novoa JM . Role of inflammation in tubulo-interstitial damage associated to obstructive nephropathy. J Inflamm (Lond) 2010;7:19.
Duffield JS, Tipping PG, Kipari T et al. Conditional ablation of macrophages halts progression of crescentic glomerulonephritis. Am J Pathol 2005;167:1207–1219.
Henderson NC, Mackinnon AC, Farnworth SL et al. Galectin-3 expression and secretion links macrophages to the promotion of renal fibrosis. Am J Pathol 2008;172:288–298.
Lange-Sperandio B, Trautmann A, Eickelberg O et al. Leukocytes induce epithelial to mesenchymal transition after unilateral ureteral obstruction in neonatal mice. Am J Pathol 2007;171:861–871.
Machida Y, Kitamoto K, Izumi Y et al. Renal fibrosis in murine obstructive nephropathy is attenuated by depletion of monocyte lineage, not dendritic cells. J Pharmacol Sci 2010;114:464–473.
Acknowledgements
This work was supported by the National Health and Medical Research Council (NHMRC) project Grant 632688, Australia; a Kidney Health Australia Biomedical Scholarship; a Bridging Grant and an Early Career Research and New Staff Grant from the University of Sydney.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
MMP-9 promotes epithelial to mesenchymal transition (EMT) of renal tubular epithelial cells (TECs), leading to renal fibrosis. Two waves of MMP-9 expression occur; the first from TECs and the second from TECs, macrophages, and myofibroblasts. Initially, MMP-9 cleaves osteopontin, stimulating the influx of macrophages, again amplifying MMP-9 production. Inhibition of MMP-9 at early and late stages in renal fibrosis could therefore result in improved renal function.
Rights and permissions
About this article
Cite this article
Kui Tan, T., Zheng, G., Hsu, TT. et al. Matrix metalloproteinase-9 of tubular and macrophage origin contributes to the pathogenesis of renal fibrosis via macrophage recruitment through osteopontin cleavage. Lab Invest 93, 434–449 (2013). https://doi.org/10.1038/labinvest.2013.3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/labinvest.2013.3
Keywords
This article is cited by
-
Structural insights into regulation of CCN protein activities and functions
Journal of Cell Communication and Signaling (2023)
-
A spatially anchored transcriptomic atlas of the human kidney papilla identifies significant immune injury in patients with stone disease
Nature Communications (2023)
-
Glucosidase inhibitor, Nimbidiol ameliorates renal fibrosis and dysfunction in type-1 diabetes
Scientific Reports (2022)
-
Olfactory receptors contribute to progression of kidney fibrosis
npj Systems Biology and Applications (2022)
-
Up-regulation of matrix metalloproteinases-9 in the kidneys of diabetic rats and the association with neutrophil gelatinase-associated lipocalin
BMC Nephrology (2021)
