
Abstract
Recent advances in DNA sequencing technologies and subsequent progress in genome-wide association study (GWAS) are rapidly changing the landscape of human diseases. Our knowledge on disease–gene linkage has been exponentially growing, and soon we will obtain complete maps of SNPs and mutations linked to nearly all major disease conditions. These studies will undoubtedly lead us to a more comprehensive understanding of how multiple genetic modifications link to human pathobiology. But what comes next after we discover these genetic linkages? To truly understand the mechanisms of how polygenic modifications identified through GWAS lead to disease conditions, we need an experimental interface to study their pathobiological effects. In this study, induced pluripotent stem cells (iPSCs), retaining all the genetic information from patients, will likely serve as a powerful resource. Indeed, pioneering studies have demonstrated that disease-specific iPSCs are useful for understanding disease mechanisms. Moreover, iPSC-derived cells, when recapitulating some disease phenotypes in vitro, can be a fast track screening tool for drug discovery. Further, with GWAS information, iPSCs will become a valuable tool to predict drug efficacy and toxicity for individuals, thus promoting personalized medicine. In this review, we will discuss how patient-specific iPSCs will become a powerful biomedical interface in clinical translational research.
Similar content being viewed by others


Main
Induced pluripotent stem cells (iPSCs) are pluripotent stem cells artificially generated by transiently expressing a set of exogenous transcription factors in somatic cells (Table 1). As Takahashi and Yamanaka1 originally reported the method for iPSC induction in 2006, the field has been rapidly expanding with great expectation and with some concern for their appropriate use. Essentially, the clinical implications of iPSCs are twofold; first, as a cellular resource for transplantation therapy, and second, as a system to model human diseases. Although the former direction is years away and unlikely to be an immediate concern for most experimental pathologists, the latter potential may be more relevant. We feel now is a good time to overview the present status of iPSC research for used in disease mechanism studies in this pathology-oriented journal, to discuss the potential value of iPSCs in future disease biology studies, and also to address the limitations and obstacles that need to be overcome. As many excellent reviews have been published to date (eg, see Kiskinis and Eggan,2 Stadtfeld and Hochedlinger,3 Saha and Jaenisch,4 Marchetto et al,5 and Yoshida and Yamanaka6), here we will try to avoid redundancy as much as possible and bring a newer perspective to iPSC use in modeling clinical diseases.
iPSCs TO MODEL CLINICAL DISEASES?
When the technology to generate human iPSCs first became available,7, 8 immediate attention was placed on their potential for use in cell-based transplantation. Using in vitro differentiation, iPSCs, like embryonic stem cells (ESCs), can provide an unlimited source of useful cell types for transplantation. The use of iPSCs in research has been largely welcomed by society because they lack the substantial ethical concern of cellular origin, which plagues ESCs. The fact that the cells are autologous for patients could be another advantage in transplantation. A major drawback of iPSCs for transplantation use is their carcinogenic potential, although recent progress in reprogramming technologies is overcoming the problem (see Table 1).
Soon after human iPSC technology was introduced, however, researchers also began to realize an additional and possibly greater value for the cells as a system to model human diseases. iPSCs can be generated from skin biopsies or blood samples of patients, and can be differentiated in vitro into cell types that are not easily accessible in patients, such as neurons and cardiomyocytes. As iPSCs retain all the genomic information from the original patients, iPSCs could be used to study how genetic aberrancies in the patient manifest in target cells in vitro.
One reason for advancement of hematopoietic disease understanding from molecular studies is ease of accessibility of blood or bone marrow samples for in vitro studies. Successful development of molecular-targeted drugs, such as imatinib, for the treatment of chronic myeloid leukemia represent a triumphal example of a successful outcome of long-term molecular study. In contrast to the blood, other patient tissues such as brain and heart are not easily accessible, which has been a substantial disadvantage for pathobiology studies in neural and cardiac disorders. Such drawbacks could be partly overcome by iPSC technology.
Skeptics can argue, of course, against this rather simple and bold scheme.4, 5 First, the cells obtained from in vitro differentiation of iPSC may be very different from equivalent cell types seen in real organs and tissues. Second, the cells will not likely fully or even closely recapitulate in vivo disease conditions, which are a consequence of complex systems with multiple cell types, and are due to the long-term effects by gene mutations. This is particularly a concern for late onset diseases. However, even when we see a part of the disease phenotypes or molecular changes in iPSC-derived cells, the system will be beneficial for defining and understanding disease mechanisms.
PIONEERING STUDIES OF DISEASE-SPECIFIC iPSCs
Despite some existing concerns, many pioneering studies have been conducted, some of which indeed demonstrate advantages to using patient iPSCs to understanding disease mechanisms and/or to identify novel therapeutic approaches. Table 2 summarizes the literature in which disease-specific iPSCs were generated. It should be noted that some papers listed in the table were not designed for disease biology study but rather intended for use in cell-based transplantation therapies in the future.
The first phase of research focused on demonstrating that iPSCs can be successfully generated from patients, and initial studies of this type were published as early as 2008. In an inaugural paper, Park et al9 showed that they were able to generate human iPSCs from patients with a variety of genetic disorders, and these cells showed a similar pluripotent differentiation capacity equivalent to control iPSCs derived from normal individuals. Meanwhile, Dimos et al10 generated iPSCs from ALS patients and differentiated them into motor neurons, in vitro, to demonstrate the potential of iPSC technology to produce a large amount of a disease-relevant cell type for research.
The second phase was to prove the concept that disease-iPSC-derived cells can indeed recapitulate some disease-specific effects in vitro. The first paper of this kind was published in January 2009 by Ebert et al.11 Here the authors generated iPSCs from patients with spinal muscular atrophy (SMA) who have mutations in the survival motor neuron 1 (SMN1) gene. Interestingly, deletion of SMN1 is partially compensated by a redundant SMN2 gene in human patients, which can also generate a full-length SMN protein but only at a lower level. Notably, other model animals such as worms, flies and mice lack the SMN2 gene, indicating that a model system in humans is essential for fully understanding disease mechanisms. Further, as targeted SMN2 gene activation is a potential mechanism for curing the disease, only a human cell system would be useful for the research. The study demonstrated that motor neurons derived from SMA–iPSCs harbor deficits in morphology, survival and synapsin staining, which represents SMA clinical pathology in part. In addition, the authors demonstrated that drugs, which were previously known to induce SMN2-derived SMN protein, indeed increased the level of SMN protein in SMA–iPSCs, implying the iPSC system would be useful for future drug discovery. Similar to the SMA study, recapitulation of neural disease phenotypes in vitro using the iPSC system has been nicely demonstrated with familial dysautonomia12 and Rett syndrome.13 Further, iPSCs have been generated from long QT syndrome patients,14, 15 where studies demonstrated prolonged action potentials in patient-iPSC-derived cardiomyocytes and their arrhythmogenicity, recapitulating the disease phenotype in vitro. Notably, all of the studies described above also demonstrated a reversal of the observed phenotypes by previously known drugs, indicating the system is compatible with drug discovery.
The third phase would be to prove that iPSC studies will indeed lead to novel insights for disease biology, and/or identification of novel therapeutic approaches. The study by Agarwal et al16 regarding dyskeratosis congenita (DC) may be the first in this category. DC is a disorder of telomere maintenance in which DKC1 mutation leads to destabilization of telomerase RNA component (TERC). Of interest, reprogramming into a pluripotency status increased the level of TERC despite the presence of DKC1 mutation and restored the telomere length. The discovery further led the authors to study and identify previously unidentified mechanisms of TERC upregulation, which could lead to a new therapeutic approach in the future. In Freidreich's ataxia,17 an extension of GAA/TTC nucleotide repeat was seen during iPSC generation and prolonged culture, which was partially prevented by knocking down of the MSH2 gene. These data also provided some newer insights regarding the disease progression.
The fourth phase, which has not yet been achieved, will be to demonstrate that iPSC research indeed leads to disease prevention or cure by discovery of effective therapeutic approaches or drugs. This would be the time when the technology truly reaches Nobel Prize status.
WHERE WILL WE SEE THE TRUE VALUE OF iPSCs?
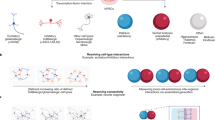
As discussed above, disease biology studies using iPSCs are on the way, and are progressing steadily with encouraging speed. Where will iPSCs likely show the most value in the near future? First, diseases that do not have high quality or appropriate animal models would benefit from iPSC study. In such cases, iPSC research has a great chance to facilitate disease understanding and/or drug discoveries. Indeed, we could say that iPSCs, which can be generated directly from patients relatively easily, are a fast track research tool when compared with other model systems in which we need to induce gene modifications (Figure 1). Moreover, iPSCs are a fast track research tool in clinical translational research for an additional reason. As a cellular system, disease iPSCs or iPSC-derived cells are directly applicable to drug screening. Importantly, we can achieve these schemes not only within relatively short time but also with relatively low cost when compared with the classical approaches in which we first identified the causes and then generated models to recapitulate them. These features may be particularly good news for research of rare diseases, which lack a large budget. In addition to the studies published (Table 2), a considerable number of research labs around the world are currently generating iPSCs from a variety of rare diseases. It may not be long until we hear promising discoveries of novel approaches or drugs to cure some of these diseases.

Fast track iPSC studies may facilitate clinical translational research.
Since the first transgenic and knockout mouse studies were published in 1980 and 1989, respectively, mice have been extensively used to model human diseases. There is no doubt their contributions to medicine are and will be countless, and indeed the development of knockout mouse technology was awarded the 2007 Nobel Prize. However, we also know that many human diseases are complex polygenic diseases, which are not easily recapitulated by gene modifications in mice. In the era of genome-wide association study (GWAS), when we accumulate our knowledge of polygenetic linkage to diseases, an alternative model to recapitulate the polygenic modifications is highly desired. In other words, in order to truly understand mechanisms of how polygenic modifications identified through GWAS lead to disease conditions, we need an experimental interface to study their pathobiological effects. In this context, iPSCs, retaining all the genetic information from patients, should have another indispensable value (Figure 1).
Personalized medicine is another field in which iPSCs are expected to make a contribution. GWAS information and clinical databases should be sufficient to predict drug efficacy and toxicity for individuals with drugs already widely used in clinics. However, for new drugs that have not been tested in clinics yet, or those tested in a small number of patients, the information would not be sufficient. Here a large iPSC library accompanying GWAS information will be very powerful. Toxicity or efficacy of new drugs on liver, eg, can be tested using hepatocytes derived from an iPSC library of normal and diseased individuals (eg, α1-antitrypsin deficiency).
Several issues should be overcome if we truly want to advance the field quickly. First, it will be critical to network iPSC labs around the world to create an iPSC library of both normal and diseased cells using a common quality standard. Second, a systematic approach to develop an iPSC library in conjunction with a clinical database, tissue bank and GWAS would be most useful. Third, further development of efficient in vitro iPSC differentiation protocols into many more cell types is essential for progress in the field. Forth, continuous effort to recapitulate phenotypes of late-onset diseases in vitro, at least partly, would be critical to extend their applications. Lastly, reducing complexity of culture methods will be important to make the system more easily applicable to high throughput screening.
CONCLUDING REMARKS
Here we discussed the potential and progress of iPSC research for disease biology studies and drug discovery. Admittedly, the major strength of iPSCs is likely the fact that these cells are derived directly from human patients. In this sense, we may be able to say that iPSCs are an alternative or additional resource for clinical tissue banks that are becoming increasingly valuable in clinical translational research particularly in this GWAS era. Although iPSCs are not comparable at all with tissues for their ability to give us histological information, iPSC provide an unlimited source of live cells from patients, even cell types that cannot be easily or frequently obtained alive. In addition, although tissue banks provide more static disease information, an iPSC system can allow for the dynamic study of gene aberrations during the process of development or cell differentiation. Lastly, as a live cell system, it is feasible to apply iPSCs to drug discovery, efficacy and toxicity testing.
Disease-specific iPSCs are a new system to model human diseases, which can become very powerful in multiple directions as discussed above. We have no intention here, however, to conclude that iPSCs are a superior model for human diseases compared with others. Animal models such as mice, rats, fruit flies, yeast etc have contributed enormously to, and will remain crucial for understanding disease biology and/or drug discovery without a doubt. Each model has its strengths and weaknesses, or advantages and drawbacks. As has always been true, the combination of multiple model systems would be the most powerful way to understand human disease biology. However, the value of iPSCs as a first universal system to use human cells for modeling a variety of human diseases should not be overlooked. Further progress in patient iPSC research may lead us to remember an old yet fundamental truth in medicine; ‘we can learn best from patients’.
References
Takahashi K, Yamanaka S . Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006;126:663–676.
Kiskinis E, Eggan K . Progress toward the clinical application of patient-specific pluripotent stem cells. J Clin Invest 2010;120:51–59.
Stadtfeld M, Hochedlinger K . Induced pluripotency: history, mechanisms, and applications. Genes Dev 2010;24:2239–2263.
Saha K, Jaenisch R . Technical challenges in using human induced pluripotent stem cells to model disease. Cell Stem Cell 2009;5:584–595.
Marchetto MC, Winner B, Gage FH . Pluripotent stem cells in neurodegenerative and neurodevelopmental diseases. Hum Mol Genet 2010;19:R71–R76.
Yoshida Y, Yamanaka S . Recent stem cell advances: induced pluripotent stem cells for disease modeling and stem cell-based regeneration. Circulation 2010;122:80–87.
Takahashi K, Tanabe K, Ohnuki M, et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007;131:861–872.
Yu J, Vodyanik MA, Smuga-Otto K, et al. Induced pluripotent stem cell lines derived from human somatic cells. Science 2007;318:1917–1920.
Park IH, Arora N, Huo H, et al. Disease-specific induced pluripotent stem cells. Cell 2008;134:877–886.
Dimos JT, Rodolfa KT, Niakan KK, et al. Induced pluripotent stem cells generated from patients with ALS can be differentiated into motor neurons. Science 2008;321:1218–1221.
Ebert AD, Yu J, Rose Jr FF, et al. Induced pluripotent stem cells from a spinal muscular atrophy patient. Nature 2009;457:277–280.
Lee G, Papapetrou EP, Kim H, et al. Modelling pathogenesis and treatment of familial dysautonomia using patient-specific iPSCs. Nature 2009;461:402–406.
Marchetto MC, Carromeu C, Acab A, et al. A model for neural development and treatment of Rett syndrome using human induced pluripotent stem cells. Cell 2010;143:527–539.
Itzhaki I, Maizels L, Huber I, et al. Modelling the long QT syndrome with induced pluripotent stem cells. Nature 2011;471:225–229.
Moretti A, Bellin M, Welling A, et al. Patient-specific induced pluripotent stem-cell models for long-QT syndrome. N Engl J Med 2010;363:1397–1409.
Agarwal S, Loh YH, McLoughlin EM, et al. Telomere elongation in induced pluripotent stem cells from dyskeratosis congenita patients. Nature 2010;464:292–296.
Ku S, Soragni E, Campau E, et al. Friedreich's ataxia induced pluripotent stem cells model intergenerational GAATTC triplet repeat instability. Cell Stem Cell 2010;7:631–637.
Okita K, Nakagawa M, Hyenjong H, et al. Generation of mouse induced pluripotent stem cells without viral vectors. Science 2008;322:949–953.
Yu J, Hu K, Smuga-Otto K, et al. Human induced pluripotent stem cells free of vector and transgene sequences. Science 2009;324:797–801.
Warren L, Manos PD, Ahfeldt T, et al. Highly efficient reprogramming to pluripotency and directed differentiation of human cells with synthetic modified mRNA. Cell Stem Cell 2010;7:618–630.
Zhou H, Wu S, Joo JY, et al. Generation of induced pluripotent stem cells using recombinant proteins. Cell Stem Cell 2009;4:381–384.
Lin T, Ambasudhan R, Yuan X, et al. A chemical platform for improved induction of human iPSCs. Nat Methods 2009;6:805–808.
Silva J, Barrandon O, Nichols J, et al. Promotion of reprogramming to ground state pluripotency by signal inhibition. PLoS Biol 2008;6:e253.
Thomson JA, Itskovitz-Eldor J, Shapiro SS, et al. Embryonic stem cell lines derived from human blastocysts. Science 1998;282:1145–1147.
Yu J, Thomson JA . Pluripotent stem cell lines. Genes Dev 2008;22:1987–1997.
Kim K, Doi A, Wen B, et al. Epigenetic memory in induced pluripotent stem cells. Nature 2010;467:285–290.
Stadtfeld M, Apostolou E, Akutsu H, et al. Aberrant silencing of imprinted genes on chromosome 12qF1 in mouse induced pluripotent stem cells. Nature 2010;465:175–181.
Polo JM, Liu S, Figueroa ME, et al. Cell type of origin influences the molecular and functional properties of mouse induced pluripotent stem cells. Nat Biotechnol 2010;28:848–855.
Murry CE, Keller G . Differentiation of embryonic stem cells to clinically relevant populations: lessons from embryonic development. Cell 2008;132:661–680.
Kobayashi T, Yamaguchi T, Hamanaka S, et al. Generation of rat pancreas in mouse by interspecific blastocyst injection of pluripotent stem cells. Cell 2010;142:787–799.
Nakagawa M, Koyanagi M, Tanabe K, et al. Generation of induced pluripotent stem cells without Myc from mouse and human fibroblasts. Nat Biotechnol 2008;26:101–106.
Soldner F, Hockemeyer D, Beard C, et al. Parkinson's disease patient-derived induced pluripotent stem cells free of viral reprogramming factors. Cell 2009;136:964–977.
Hotta A, Cheung AY, Farra N, et al. Isolation of human iPS cells using EOS lentiviral vectors to select for pluripotency. Nat Methods 2009;6:370–376.
Ye L, Chang JC, Lin C, et al. Induced pluripotent stem cells offer new approach to therapy in thalassemia and sickle cell anemia and option in prenatal diagnosis in genetic diseases. Proc Natl Acad Sci USA 2009;106:9826–9830.
Raya A, Rodriguez-Piza I, Guenechea G, et al. Disease-corrected haematopoietic progenitors from Fanconi anaemia induced pluripotent stem cells. Nature 2009;460:53–59.
Maehr R, Chen S, Snitow M, et al. Generation of pluripotent stem cells from patients with type 1 diabetes. Proc Natl Acad Sci USA 2009;106:15768–15773.
Wang Y, Jiang Y, Liu S, et al. Generation of induced pluripotent stem cells from human beta-thalassemia fibroblast cells. Cell Res 2009;19:1120–1123.
Ye Z, Zhan H, Mali P, et al. Human-induced pluripotent stem cells from blood cells of healthy donors and patients with acquired blood disorders. Blood 2009;114:5473–5480.
Kazuki Y, Hiratsuka M, Takiguchi M, et al. Complete genetic correction of ips cells from Duchenne muscular dystrophy. Mol Ther 2010;18:386–393.
Urbach A, Bar-Nur O, Daley GQ, et al. Differential modeling of fragile X syndrome by human embryonic stem cells and induced pluripotent stem cells. Cell Stem Cell 2010;6:407–411.
Carette JE, Pruszak J, Varadarajan M, et al. Generation of iPSCs from cultured human malignant cells. Blood 2010;115:4039–4042.
Carvajal-Vergara X, Sevilla A, D’Souza SL, et al. Patient-specific induced pluripotent stem-cell-derived models of LEOPARD syndrome. Nature 2010;465:808–812.
Rashid ST, Corbineau S, Hannan N, et al. Modeling inherited metabolic disorders of the liver using human induced pluripotent stem cells. J Clin Invest 2010;120:3127–3136.
Hargus G, Cooper O, Deleidi M, et al. Differentiated Parkinson patient-derived induced pluripotent stem cells grow in the adult rodent brain and reduce motor asymmetry in Parkinsonian rats. Proc Natl Acad Sci USA 2010;107:15921–15926.
Somers A, Jean JC, Sommer CA, et al. Generation of transgene-free lung disease-specific human induced pluripotent stem cells using a single excisable lentiviral stem cell cassette. Stem Cells 2010;28:1728–1740.
Chamberlain SJ, Chen PF, Ng KY, et al. Induced pluripotent stem cell models of the genomic imprinting disorders Angelman and Prader-Willi syndromes. Proc Natl Acad Sci USA 2010;107:17668–17673.
Zhang N, An MC, Montoro D, et al. Characterization of human Huntington's disease cell model from induced pluripotent stem cells. PLoS Curr 2010;2:RRN1193.
Cooper O, Hargus G, Deleidi M, et al. Differentiation of human ES and Parkinson's disease iPS cells into ventral midbrain dopaminergic neurons requires a high activity form of SHH, FGF8a and specific regionalization by retinoic acid. Mol Cell Neurosci 2010;45:258–266.
Yang J, Cai J, Zhang Y, et al. Induced pluripotent stem cells can be used to model the genomic imprinting disorder Prader-Willi syndrome. J Biol Chem 2010;285:40303–40311.
Zhang J, Lian Q, Zhu G, et al. A human iPSC model of Hutchinson Gilford Progeria reveals vascular smooth muscle and mesenchymal stem cell defects. Cell Stem Cell 2011;8:31–45.
Tolar J, Park IH, Xia L, et al. Hematopoietic differentiation of induced pluripotent stem cells from patients with mucopolysaccharidosis type I (Hurler syndrome). Blood 2011;117:839–847.
Tolar J, Xia L, Riddle MJ, et al. Induced pluripotent stem cells from individuals with recessive dystrophic epidermolysis bullosa. J Invest Dermatol 2011;131:848–856.
Pessach IM, Ordovas-Montanes J, Zhang SY, et al. Induced pluripotent stem cells: a novel frontier in the study of human primary immunodeficiencies. J Allergy Clin Immunol 2011; doi:10.1016/j.jaci.2010.11.008 (in press).
Acknowledgements
We thank Dr Tetsuo Ashizawa and Dr Anthony Yachnis (University of Florida) for helpful discussion and critical readings of the manuscript. This work was supported in part by NIH grant RC1 GM091238 and University of Florida Faculty Enhancement Opportunity Award to NT. KEH is a recipient of predoctoral fellowship of T32 CA009126.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
This review outlines the current status of pluripotent stem cell research in pathobiology and discusses how patient-specific iPSCs will become a powerful biomedical interface in clinical translational research, particularly in the era of genome-wide association studies.
Rights and permissions
About this article
Cite this article
Hankowski, K., Hamazaki, T., Umezawa, A. et al. Induced pluripotent stem cells as a next-generation biomedical interface. Lab Invest 91, 972–977 (2011). https://doi.org/10.1038/labinvest.2011.85
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/labinvest.2011.85
Keywords
This article is cited by
-
Puromycin-based purification of cells with high expression of the cytochrome P450 CYP3A4 gene from a patient with drug-induced liver injury (DILI)
Stem Cell Research & Therapy (2022)
-
Ammonia-based enrichment and long-term propagation of zone I hepatocyte-like cells
Scientific Reports (2021)
-
Distinctive features of single nucleotide alterations in induced pluripotent stem cells with different types of DNA repair deficiency disorders
Scientific Reports (2016)
-
A practical guide to induced pluripotent stem cell research using patient samples
Laboratory Investigation (2015)
-
Generation of pluripotent stem cells without the use of genetic material
Laboratory Investigation (2015)
