
Abstract
The molecular basis of attaining columnar phenotype in Barrett's esophagus is poorly understood. One hypothesis states that factors locally produced by cells of mesenchymal origin in chronic reflux esophagitis induce metaplastic transformation. This study was performed to elucidate the factors secreted from fibroblasts that cause columnar phenotype in adjacent squamous epithelium. Human fibroblast cells were exposed to acidified medium for 20 min, followed by medium neutralization for 2 h, and then total RNA was hybridized to Sentrix Human-6 Expression BeadChips. Furthermore, esophageal mucosal biopsy specimens from reflux esophagitis patients were examined for HB-EGF expression using immunohistochemistry. In addition, cells from the human esophageal squamous epithelial cell line HET1A were treated with recombinant HB-EGF, and changes in expressions of Cdx2 and columnar markers were analyzed. The gene expression profile revealed significant upregulation of a variety of growth factors and inflammatory chemokines in response to acid exposure. Among them, HB-EGF was upregulated more than 10-fold. Biopsy specimens from reflux esophagitis patients showed a strong expression of HB-EGF in fibroblast cells underlying the damaged epithelium. Furthermore, in vitro stimulation of HET1A cells with HB-EGF increased Cdx2 in dose-dependent manners. Functional analysis of human Cdx2 promoter also revealed its upregulation by HB-EGF stimulation, showing the role of potential responsive elements (AP-1 and NF-κB) for its transcriptional activation. Moreover, the columnar markers cytokeratin 7 and villin were also upregulated by HB-EGF stimulation. HB-EGF induces several genes characteristics of columnar phenotypes of esophageal squamous epithelium in a paracrine manner.
Similar content being viewed by others


Main
Barrett's esophagus (BE) refers to a pre-malignant condition in which squamous epithelium in the distal esophagus is replaced by metaplastic columnar epithelium, which finally leads to development of esophageal adenocarcinomas in patients with longstanding gastro esophageal reflux disease (GERD) complicated with BE symptoms.1, 2 Approximately, 10% of GERD patients develop the specialized intestinal metaplasia of BE with a 30- to 125-fold increased risk for esophageal adenocarcinoma, according to the best estimates of cancer incidence of approximately 0.5–1.0% per year worldwide.3, 4, 5 A major component of the refluxate in GERD is gastric acid and it is likely that this contributes, at least in part, to the increased risk of metaplasia.6 For diagnosis of GERD, pH 4 remains the optimum threshold, as this level has shown high sensitivity and specificity for detecting increased esophageal exposure to gastric juice.6 In contrast, some studies have also suggested that a better discrimination between healthy volunteers and patients with reflux symptoms could be achieved at a pH threshold of 5.7
Although there are several reports on development of BE-associated cellular metaplasia, the actual molecular mechanism underlying this cellular transition has not been fully described. However, it has been shown that a low pH environment induces the expression of cyclooxygenase-2 and prostaglandin E2, as well as activates the extracellular signal-regulated kinase, p38 mitogen-activated protein kinase, and nuclear factor-κB pathways, which cause increased proliferation and decreased apoptosis of esophageal cells, leading to malignant progression.8, 9 Apart from these, several lines of evidence have shown the role of Cdx2, a potent transcription factor that is a member of the homeobox family of genes is also upregulated and causes metaplastic changes in involved Barrett epithelium.10, 11
Normal epithelial cells reside in a complex microenvironment composed of extracellular matrix, diffusible growth factors and cytokines, adjacent fibroblasts, and a variety of non-epithelial immune reactive cells. Epithelial–mesenchymal interactions are known to be crucial for regular epithelial growth, differentiation, and homeostasis during embryogenesis as well as in adulthood, while in some cases stromal fibroblasts cause initiation and progression of cancer to adjacent epithelium through autocrine or paracrine mechanisms.12, 13 On stimulation by a variety of stress insults, including oxidative stress, ultraviolet irradiation, ethanol, inflammatory cytokines, and various genotoxic agents, stromal fibroblasts secrete various trophic factors and extracellular matrix remodeling enzymes, leading to perturbation of the local microenvironment by modulating the growth and differentiation of adjacent normal epithelium.14, 15 With this background in mind, it is interesting that stromal influences on epithelial neoplasia have been noted in esophageal tissues as well as other organs and tissues, namely the salivary glands, mammary glands, urinary bladder, and skin, through aberrant production of diffusible growth factors from stromal fibroblasts.16, 17, 18, 19 Although several lines of evidence support the influences of stromal fibroblasts on degenerative changes of the epithelial environment, less is known regarding the pattern of fibroblast secretory proteins or factors that modulate the growth and differentiation of adjacent progenitor or matured epithelial cells in the esophagus.
Our preliminary findings showed that stimulation of embryonic fibroblasts with low pH luminal acid-induced expression of a variety of secretory factors. Thus, we analyzed the influences of these fibroblast secretory products on the characteristics of epithelial metaplasia in BE which we speculated to occur through a paracrine mechanism. Detailed insights into the nature and mechanisms by which these acid-exposed fibroblast mediators regulate the esophageal epithelial environment may lead to development of a potential therapeutic target for controlling BE or BE-associated complications.
MATERIALS AND METHODS
Cell Culture
Normal human embryonic fibroblasts (OUMS-36T-6 cells; JCRB Cell Bank, Osaka, Japan) immortalized by hTRT were cultured in Dulbecco's modified Eagle's medium (Invitrogen, Carlsbad, CA, USA) supplemented with 10% heat inactivated fetal bovine serum (FBS), penicillin (100 IU/ml), and streptomycin (100 μg/ml) at 37 °C under a humidified condition. Human esophageal epithelial cells (HET1A) immortalized by Simian virus 40 (SV40) large T antigen (Tag) were obtained from American Type Culture Collection (ATCC, Manassas, VA, USA) and maintained in BEGM medium contained in a BulletKit (Clonetics Corporation, Walkersville, MD, USA). Human esophageal edenocarcinoma cell line OE-33 obtained from ATCC, were maintained in RPMI1640 medium, with 10% FBS and antibiotics at 37 °C under a humidified condition.
Protocol for Acid Pulsing and Cell Viability Assay
For acid pulsing, the medium was acidified with 0.1 M HCl to achieve the desired pH level of 3.5, 4, 5, 6, or 7. Subsequently, a volume of distilled water was added to the control non-acidified medium (pH 7.4) to achieve an osmolality equal to that of the acidified media. OUMS cells were seeded into 10-cm plates and exposed to acidified DMEM for 20 min after reaching 60% confluence. Next, the acidified medium was changed to normal and the cells were maintained for up to 72 h. Cell viability was assessed at 0 (t0), 6 (t6), 24 (t24), 48 (t48), and 72 (t72) hours after acid exposure using MTT (Promega, Madison, WI, USA) reagent, according to the manufacturer's protocol.
Microarray Analysis: cRNA Amplification, Labeling, Hybridization, and Data Analysis
Total RNA was isolated from OUMS cells with or without acid treatment, as described above, and used for microarray analysis. DNA contamination in the extracted RNA was removed using DNaseI. RNA quality was checked by denaturing gel electrophoresis, while the quantity was assessed using NanoDrop (ND-1000). cRNA amplification and labeling with biotin were performed using an Illumina Total Prep RNA amplification kit (Ambion, Austin, TX, USA) with 250 ng of total RNA as the input material. The resulting cRNA yields were quantified with Nanodrop and 1.5 μg of each cRNA sample was hybridized to Sentrix Human-6 Expression BeadChips (33U1430042, Illumina, San Diego, CA, USA). Each of these chips contained six identical sets of 47296 unique probes, including 24385 RefSeq annotated genes. The cRNA was hybridized to the arrays for 16 h at 58°C, before being washed and stained with streptavidin-Cy3, according to the manufacturer's protocol. The resulting signals were scanned with an Illumina Bead Array Reader confocal scanner and the data were analyzed using BeadStudio version 3 OR Genespring GX 7.3 software. The genes upregulated are shown in Table 1.
RNA Isolation and Quantitative Real-Time PCR
Total RNA was extracted from each sample using a guanidine isothiocyanate-phenol-chloroform method (Isogen; Nippon Gene, Japan). Before reverse transcription (RT), all RNA samples were treated with RNase-free DNaseI (Ambion), following the manufacturer's instructions. Equal amounts of RNA were reverse transcribed into cDNA using a random primer from an Affinity Script QPCR cDNA synthesis kit (Stratagene, La Jolla, CA, USA). Real-time fluorescence PCR assays based on SYBR Green were carried out in a 30-μl final volume containing 1 × Power SYBR Green PCR master mix (Applied Biosystems, Warrington, UK), with 200 nM of forward and 200 nM of reverse primers (Table 2), with 100 ng of cDNA used as a template. Thermal cycling was performed with the following conditions: 50 °C for 2 min and 95 °C for 10 min, then 40 cycles each at 95 °C for 15 s and 60 °C for 1 min, using an ABI PRISM 7700 sequence detection system (Applied Biosystems, Foster City, CA, USA). The levels of expression of the respective genes were normalized to that of the GAPDH gene using sequence detection software (Applied Biosystems).
Enzyme Immune Assays
Culture supernatants from OUMS cells with or without acid (pH 5) treatment were subjected to enzyme immune assays for FGF6, HB-EGF, PDGFAA, PDGFBB (R&D Systems, Minneapolis, MN, USA), and IL-8 (Biosource, Camarillo, CA, USA), following the manufacturers’ protocols. Briefly, the required amount of culture supernatant was loaded into the respective wells of coated plates supplied with the enzyme immune assay kits. After incubation for the desired time period, the wells were washed and allowed to react with HRP-labeled conjugates for at least 1–2 h. Finally, the resulting signals were measured at a wavelength of 450 nm and data from each of the standard curves were analyzed using the Curve-fit software package.
Immunohistochemistry
To perform immunohistochemistry examinations of the human esophageal specimens, a goat polyclonal antibody raised against HB-EGF of human origin was used. The protocol was approved by the ethical review committee of Shimane University School of Medicine and all the subjects had given their informed consents before studies were performed. Samples from cases with and without reflux esophagitis were obtained and 6-μm sections prepared. The sections were fixed in cold acetone for 20 min and then endogenous peroxidase activity was blocked with 0.3% hydrogen peroxidase in methanol for 30 min at room temperature, followed by incubation with normal blocking serum for 30 min. Subsequently, the sections were incubated for 2 h at room temperature with the primary antibody and then processed with the corresponding commercial immunoperoxidase staining kit (Vectastain Elite ABC Kit; Vector Laboratories, Burlingame, CA, USA), following the manufacturer's instructions. The sections were counterstained with hematoxylin. Negative controls were produced by omitting the primary antibody.
Recombinant HB-EGF Treatment of HET1A Cells
To examine the effect of HB-EGF on Cdx2, HET1A cells were used. After 12 h of starvation from hEGF and bovine pituitary extract, the cells were incubated with various concentrations of HB-EGF (R&D Systems), and then mRNA and proteins were isolated to measure Cdx2 expression. Cdx2 mRNA was measured at 12 h after incubation of Het1A cells with recombinant HB-EGF, while Cdx2 protein was measured 24 h later. In another experiment, the EGF receptor-specific phosphorylation inhibitor AG1478 (Calbiochem, La Jolla, CA, USA) was used to analyze the effects of HB-EGF on HET1A cells. For this, AG1478 was added to the cells and the effects of HB-EGF on the expressions of various genes in HET1A cells were determined.
Protein Extraction and Western Blotting Analysis
For protein extraction, cells were lysed with a syringe in 20 mM Tris, pH 7.6, containing 0.1% SDS, 1% Triton-X, 1% deoxycholate, 100 μg/ml of the protease inhibitor PMSF, and a protease inhibitor cocktail (Sigma-Aldrich, St Louis, MO, USA). The lysates were centrifuged at 14 000 × g for 20 min at 4 °C. Protein concentration was determined using Coomassie Plus Reagent (PIERCE, Rockford, IL, USA). One hundred micrograms of protein per lane was loaded and processed for SDS-PAGE fractionation, then transferred to a polyvinylidene difluoride membrane (Hybond-P; Amersham, UK). After blocking the membranes for 2 h in 5% skim milk in TBS (20 mM Tris and 150 mM NaCl, pH 7.4), each was incubated with anti-Cytokeratin 7 (CK7), villin, and β-actin antibodies at an optimized dilution for (Table 3). Finally, after 1 h of incubation with respective secondary antibodies, the resulting signals were imaged using ECL (GE Healthcare, UK). For Cdx2 immunoprecipitation, 500 μg of total protein was allowed to react with the Cdx2-specific primary antibody and immunoprecipitation was performed using protein G-agarose beads (GE Healthcare, Uppsala, Sweden), after which western blotting was performed using the anti-Cdx2 antibody, as described above.
Vector Construction for CDX2 Promoter Analysis
For construction of a human Cdx2 promoter containing a reporter vector, accession No. NC_000013 was used. Position +1 refers to the transcription start site identified in the Cdx2 gene. First, 960 bp of the Cdx2 promoter (−925 to +34) were amplified by PCR and cloned into the KpnI and XhoI sites of a pGL3-basic luciferase vector (Promega) to generate pCdx2/960-Luc. Promoters of various lengths from pCdx2/960-Luc were amplified by PCR, then subcloned into the KpnI and XhoI sites of pGL3-basic, which generated pCdx2/775-Luc (−740 to +34), pCdx2/485-Luc (−450 to +34), pCdx2/255-Luc (−220 to +34), and pCdx2/76-Luc (−41 to +34). For incorporation of mutations at the putative AP-1 and NF-κB responsive elements, site-directed mutagenesis (Stratagene, USA) was performed using the following primers: 5′-ACCATTGGTGTCTTCGGAATTACTAAT-3′ for AP-1 and 5′-CCTGTGGCGGTTCTTACCCGCCTC-3′ for NF-κB, in which the underlined nucleotides indicate the base substitutions. The cloned promoter sequences were confirmed by sequencing. As an internal control for the dual-luciferase assay, pRL-TATA-Renilla-Luc was also used, which expressed Renilla luciferase under a minimal TATA promoter.
Transfection and Luciferase Assay
The effect of recombinant HB-EGF on transcriptional activation of Cdx2 was evaluated by its promoter activity. HET1A cells were cultured in collagen type I coated 24-well culture plates (5 × 104cells per well), then transfected with 0.1 μg of the individual promoter Cdx2-Luc and 0.01 μg of pRL-TATA-Renilla-Luc in each well, using Lipofectamine 2000 reagent (Invitrogen). Twelve hours after transfection of the luciferase vectors, cells were stimulated with various concentrations of HB-EGF or the vehicle (PBS-BSA solution) alone for 36 h, then the cell lysates were used for measurement of luciferase activity with a dual-luciferase reporter assay system (Promega). Data are expressed as the n-fold increase in luciferase activity in the HB-EGF stimulated samples over that of the vehicle-treated samples. To block endogenous expression of EGFR, commercially available human EGFR-specific siRNA was used (Santa Cruz Biotechnology, Santa Cruz, CA, USA). For siRNA transfection, HET1A cells were platted into the collagen type I coated 24-well culture plates (5 × 104cells per well), and then after 12–18 h, the cells were transfected with 5–20 pmols of duplex siRNAs, using the siRNA-trasfection reagent (Santa Cruz Biotechnology). Inhibition of target gene, EGFR expression can be observed at around 18–24 h after transfection as revealed by real time PCR or western blotting.
Immunofluorescence Protocol
HET1A cells were grown on glass chamber slides (BD Biosciences, Japan), then fixed in 4% paraformaldehyde and permeabilized with 0.2% Triton X-100. Heat-induced (microwave treatment at 500 W for 10 min) epitope retrieval in DakoCytomation target retrieval solution at pH 9 was performed before staining of the HET1A cells with a monoclonal mouse anti-human CK7 antibody. CK7 expression in HET1A cells was examined using an OLYMPUS, BX51 fluorescent microscope (Olympus, Japan) at a magnification of × 20.
Statistical Analysis
All data are expressed as the mean ± standard error of the mean (s.e.m.). Values were analyzed using a paired-samples t-test and multiple comparisons were carried out with ANOVA, followed by a Bonferroni test. All analyses were conducted using SPSS 10.0 for Windows. P-values of <0.05 were considered to be statistically significant.
RESULTS
Effects of Physiologic Acid Pulse on Fibroblast Cell Morphology and Viability
A low pH environment is of paramount importance for GERD and its associated symptoms. In this study, we used an in vitro system and grew fibroblast cells in acidified media for 20 min and then in normal media for 24 h. The cells were viewed under a microscope to determine whether their morphology was affected. At pH 7.4 and 5, the cells showed a similar spindle-shaped morphology, whereas at pH 4 it was completely changed and many cells were detached from the plates (Figure 1a). Next, we attempted to assess cell viability at different pH levels (7.4–3.5) and time points. As shown in Figure 1b, in normal (pH 7.4) medium, the fibroblast cells proliferated in a time-dependent manner, whereas cell viability was affected in altered pH environments. At pH 6 and 5, the cells grew well in a manner comparable to normal pH (7.4), while growth was affected at pH below 5. On the basis of the growth curves observed at different pH levels, we selected pH 5 as the optimal acidic pH condition for survival of the fibroblast cells and performed subsequent experiments.

Morphology and viability of acid-treated fibroblast cells. (a) Propidium iodide (PI) staining of OUMS-36T-6 cells at different pH levels. OUMS-36T-6 cells were grown in acidified medium for 20 min, then in normal medium for 24 h. The cells were stained with PI (2.5 μg/ml), and then viewed under a phase-contrast microscope (left panel, magnification × 20), fluorescent microscope (midle panel, magnification × 20) overlap image (right panel, magnification × 20). (b) OUMS-36T-6 cells (1 × 105/ml) were grown in 10-cm plates. When 60% confluence was reached, the medium was changed to acidified medium (pH 7.4–3.5) for 20 min. Next, the acidified medium was replaced with normal and cell proliferation assays were performed at different time points. Growth curves shown were obtained from calculating cell number vs time. Error bar indicates the standard error of mean values (s.e.m.) obtained from three independent experiments. *P<0.05 vs pH 7.4 at 0 h.
Differential Expressions of Growth Factors and Cytokines in Acid-Treated Fibroblast Cells
To determine the status of gene expression after a single pulse of acid stimulation to fibroblast cells, microarray analysis was carried out using mRNA from samples of 2 h after 20 min of acid treatment. From the large number of upregulated genes, we chose potent growth factors and cytokines that are usually involved in oncogenicity, inflammation, differentiation, and transformation, which are presented in Table 1.
Several families of fibroblast-derived growth factors, implicated as autocrine and paracrine mediators of stromal–epithelial interactions, are involved in neoplastic progression.20 As shown in Table 1, the growth factors known to initiate the oncogenic process, for example, FGF6, HB-EGF, PDGFA, PDGFB, NELL1, and TGFB3, were significantly elevated. Moreover, in the context of inflammation, IL8, CSF2, IL1β, CXCL5, stromal cell-derived factor 1α (CXCL12), and TNFSF12 were also significantly upregulated. To correlate the microarray results, we performed quantitative real-time PCR assays of selected genes of 2 h after 20 min of acid exposure (Figures 2a and b). Similarly, we also confirmed the protein level expression of some other factors (FGF6, HB-EGF, PDGFAA, PDGFBB) and cytokines (IL8, IL1β) obtained from culture supernatants of acid-treated fibroblast cells of 4 h after a 20 min of acid exposure (Figures 2c and d). Our results showed that acid treatment significantly upregulated some potent growth factors and cytokines in fibroblast cells, which may have an important role in stromal microenvironment dysregulation.

Changes in gene expression profile in OUMS-36T-6 cells after acid treatment. OUMS cells were grown in 10-cm cell culture plates. After 20 min of incubation in acidified medium, the cells were grown in normal medium for 2 h, then the expressions of selected growth factors (a) and chemokines (b) were analyzed using real-time PCR. Error bars show s.e.m. values obtained from three independent experiments. The protein contents of selected growth factors (c) and cytokines (d) from the culture supernatants of acid-treated fibroblasts were measured using an enzyme immune assay. *P<0.05 vs cells at pH 7.4.
Predominance of HB-EGF in Subepithelial Fibroblasts of Human Reflux Esophagitis Patients
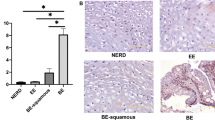
Increased production of a large number of cytokines and chemokines has been reported in mucosal biopsy specimens from GERD patients.21, 22 Furthermore, an abnormal cytokine profile was observed in colonic primary fibroblast cells.23 To support our in vitro findings of whether acid induces HB-EGF expression in fibroblast cells, we examined HB-EGF expression in biopsy specimens from the patients with reflux esophagitis as to show the in vivo evidence. For that purpose, we performed immunohistochemistry examinations of biopsy specimens obtained from 12 reflux esophagitis patients. Fundic glands of gastric epithelial specimen showed strong expression of HB-EGF and served as positive control for HB-EGF immunostaining. Two of the reflux esophagitis biopsy specimens with mucosal breaks, but without the acid suppressive medications were found to be positive for HB-EGF staining in the loosely adherent fibroblast cells, as compared with the connective tissue zone completely covered by normal esophageal squamous epithelium of healthy volunteers (Figures 3a, b and c).

HB-EGF expression in esophageal biopsy specimens from human subjects. The specimens were subjected to immunohistochemistry using the anti-human HB-EGF antibody. (a) Fundic glands of gastric epithelial specimen served as positive control for HB-EGF staining. (b) No expression of HB-EGF was found in the connective tissue cells under the squamous epithelium of normal healthy subject. (c) Esophageal specimens with chronic reflux esophagitis showed strong expression of HB-EGF in the cytoplasmic part of mesothelial cells devoid of the coverage of squamous epithelium.
HB-EGF Induces Cdx2 Expression in Human Esophageal Squamous Epithelial Cells
Recent studies of cellular transdifferentiation events in the initiation of BE have shown Cdx2 to be an important transcription factor. To delineate whether HB-EGF stimulation in epithelial cells has effects on Cdx2 expression, we treated HET1A cells with different doses of HB-EGF and measured Cdx2 expression at mRNA and protein levels. HET1A cell is a cultured cell line derived from human epithelial cells and immortalized by SV40 large Tag. Recombinant HB-EGF promoted Cdx2 expression in HET1A cells in a dose-dependent manner, with optimum induction of Cdx2 mRNA obtained at 1 ng/ml of HB-EGF. In addition, Cdx2 protein expression was optimum at the same dose of HB-EGF (Figures 4a and b). To evaluate these findings, apart from evaluating the effect of recombinant HB-EGF on Cdx2-induction in HETA1-cell line, we also aimed to check its effect on Cdx2-expression in another esophageal epithelial cell line OE-33. Similar to the results in HET1A cells, we also observed an induction of Cdx2 expression in OE-33-cells both at the mRNA and protein levels treated with different doses of recombinant HB-EGF (Figures 4c and d). Although HET1A cells are transformed by SV40 large Tag, however, in many studies it is used as a primary esophageal epithelial cell to conduct research on esophagus. Considering this facts, we carried out our further experiments using this cell line.

Recombinant HB-EGF-induced Cdx2 expression in esophageal squamous epithelial cells. HET1A or OE-33-cells were grown in six-well plates at 1 × 105 cells/ml. (a and c) After 12 h of incubation with different doses of recombinant HB-EGF, RNA was extracted and Cdx2 expression was analyzed using real-time PCR. Data are expressed as fold induction and compared with vehicle-treated cells. Error bars show s.e.m. values obtained from three independent experiments. *P<0.05 vs HB-EGF (−). (b and d) After 24 h of incubation with HB-EGF, protein was extracted and Cdx2 expression was analyzed using immunoprecipitation for Cdx2 followed by western blotting by using Cdx2 antibody. During Cdx2 immunoprecipitation, Cdx2 protein coupled with anti-Cdx2 remained bound to the beads, while the remaining supernatant was collected and used for a western blotting assay for β-actin, which served as an internal control.
As gene expression is controlled by regulatory units, we constructed a human Cdx2 promoter (Figure 5a) and analyzed the effects of HB-EGF on Cdx2 expression by analyzing its promoter function. As shown in Figure 5b, as compared with untreated cells, HB-EGF caused considerable upregulation (∼3.5-fold) of the Cdx2 promoter in HET1A-transfected cells, while its effects gradually declined with decreases in size of the parent promoter (960 bp). Although truncated, the effect of HB-EGF was also evident with a 255-bp Cdx2 promoter (Figure 5b), thus indicating the probability of potent transcriptional responsive elements within the −220 to +34 region. In this regard, the presence of two potent transcription factors, AP-1 (−162 to −173) and NF-κB (−79 to −88) binding sites in the Cdx2 promoter region was revealed by promoter analysis software. From those results, we mutated AP-1 and NF-κB consensus sites using the wild-type pCdx2/255-luc backbone, and observed that introduction of either the AP-1 or NF-κB mutation reduced promoter activity, while the construct bearing a double mutation in both responsive sites completely diminished the effects of HB-EGF on the promoter construct (Figure 5c) transfected into HET1A cells. To know whether HB-EGF stimulation causes AP-1 and NF-kB activation, HET1A cells were transfected with these transcription factor binding reporter constructs, and then stimulated with HB-EGF. On HB-EGF stimulation, activity of these two transcription factors were increased significantly (Figures 5d and e). Taken together, these results firmly support the notion that HB-EGF-mediated Cdx2 activation occurs through AP-1 and NF-kB.

Recombinant HB-EGF-induced human Cdx2 promoter activity in esophageal squamous epithelial cells. (a) Schematic diagram showing the reporter vector containing the Cdx2 promoter. (b) HET1A-cells were plated into collagen type I coated 24-well plates, then various amounts of pCdx2-luc (0.1 μg per well) and pRL-TATA-luc (0.01 μg per well) vectors were transfected using Lipofectamine 2000. After 12 h of transfection, the cells were stimulated with HB-EGF (10 ng/ml) for 36 h and a dual-luciferase assay was performed with the cellular extracts. (c) Analysis of the responsive elements AP-1 and NF-κB after introducing point mutations in HB-EGF-treated pCdx2/255 transfected cells. (d and e) Reporter vectors, NF-kB (0.2 μg per well) containing five tandem repeats of the NF-κB consensus-binding sites, and AP-1 (0.2 μg per well) containing seven tandem repeats of AP-1-binding sites and with the internal control vector pRL-TATA-Luc (0.02 μg per well) were transfected to the corresponding wells by Lipofectamine 2000 (2.5 μl per well). After 36 h of HB-EGF (10 ng/ml) stimulation, dual-luciferase assay was carried out. Data are expressed as fold induction and compared with vehicle-treated cells. Error bars show s.e.m. values obtained from three independent experiments. *P<0.05 vs HB-EGF (−).
Profound Changes in Cellular Markers in HB-EGF-Treated Squamous Epithelial Cells
Changes in several cellular markers are important phenomena involved in the development of BE. To determine whether HB-EGF causes changes in the marker of normal esophageal epithelial cells, we treated cells with varying concentrations of recombinant HB-EGF, and assessed the expression of CK7 at the mRNA and protein levels. After 12 h of stimulation with HB-EGF at a dose of 1 ng/ml, CK7 mRNA was significantly upregulated (Figure 6a). Furthermore, intracellular CK7 protein was detected in HET1A cells after 5 days of incubation with recombinant HB-EGF (Figure 6b). In the recent studies along with the CK7, other cytokeratin markers in terms of the columnar phenotype are also implicated.24 Considering this, we aimed to screen some potent cytokeratin expression in HET1A cells at the mRNA level after stimulation with different doses of recombinant HB-EGF for 12 h. As shown in Figures 6c and d, we observed an upregulation of CK-8 and CK-18 gene expression in a dose-dependent manner. In addition, villin, an important columnar cell differentiation marker, was also significantly upregulated at the mRNA and protein levels (Figures 6e and f) when the cells were exposed to confined doses of HB-EGF. Collectively, these results imply that HB-EGF stimulates the expression genes of the columnar markers in squamous epithelial cells.

HB-EGF-induced several cytokeratins and villin expression in HET1A cells. (a) HET1A cells were treated with various concentrations of HB-EGF for 12 h, then CK7 mRNA was analyzed using real-time PCR. (b) HET1A cells were treated for 5 days with HB-EGF, then immunocytochemistry was performed using rhodamine-conjugated anti-human CK7 antibody, with nuclear staining by SYBR Green reagent. (c and d) CK8/18 expression at the mRNA level was checked by real-time PCR from HET1A cells treated with various concentrations of HB-EGF for 12 h. (e) Villin mRNA was assessed by real-time PCR using samples treated with various doses of HB-EGF for 12 h. (f) After treatment with HB-EGF, the expression of villin was determined from total protein using western blotting. β-Actin served as the loading control. Error bars show s.e.m. values obtained from three independent experiments. *P<0.05 vs HB-EGF (−).
HB-EGF Effects on Esophageal Epithelial Cells are Generated via EGFR
HB-EGF has been reported to exert its effects on different cell types by binding to EGFR.25 To know the status of EGFR expression and its phosphorylation, HET1A cells were stimulated with defined dose of HB-EGF and receptor phosphorylation was checked by immunoprecipitation using phospho-specific EGFR antibody. As shown in Figure 7a, cells treated with HB-EGF dramatically phosphorylates EGFR although the total EGFR contents remain same. The densitometric assay of the phosphorylated bands over that of non-HB-EGF-treated control clearly showed an upregulation of the phosphorylation status in HB-EGF-treated one (Figure 7b). Next, we used an EGFR inhibitor and carried out subsequent experiments to confirm whether HB-EGF induces changes in the transcription factor and cellular markers in HET1A cells mediated by EGFR. As shown in Figure 7c, the exogenous effects of HB-EGF on the expressions of Cdx2, CK7, and villin were considerably reduced because of the blocking of EGFR by its pharmacologic inhibitor, AG1478. Although AG1478 is commonly used as an inhibitor for EGFR, it may likely to have non-EGFR-specific effects. We, therefore, aimed to inhibit the endogenous expression of EGFR by using commercially available siRNAs specific for human EGFR mRNA. The efficiency of non-target-specific control as well as the EGFR-specific siRNAs was checked by its ability to abrogate target gene expression (data not shown). To elucidate EGFR-mediated recombinant HB-EGF effects, HET1A cells were transfected with siRNAs and then after stimulation with or without recombinant HB-EGF several gene expression statuses were evaluated. As shown in Figure 7d, cells treated with the EGFR-siRNAs dramatically reduced the effects of recombinant HB-EGF on Cdx-2, CK7, and villin gene expression, while those were found to be unaffected in non-target-specific control siRNA-treated conditions.

Role of EGFR in HB-EGF-mediated cellular effects. (a) HET1A cells were treated with HB-EGF for 30 min and then lysed and immunoprecipitated by anti-pEGFR from the immunoprecipitated samples. (b) Representative bars indicating the percentage of EGFR phosphorylation to total for the untreated vs treated samples as revealed by the densitometric analysis of the band intensities using Image J software. (c) HET1A cells were cultured with or without EGFR inhibitor at a dose of 100 nmol/l. After stimulation with HB-EGF, the cells were lysed, then immunoprecipitation for Cdx2 or western blotting for CK7 and villin was performed from cellular extracts using respective antibodies. β-Actin served as the loading control. (d) Effect of EGFR-siRNA on HET1A cells treated with recombinant HB-EGF. In brief, HET1A cells were plated into the 24-well plates, and then after 12 h, cells were transfected with human EGFR-specific siRNA (5–20 pM) or control siRNA (5–20 pM). After 12 h of transfection, the cells were stimulated with recombinant HB-EGF (10 ng/ml) for 24 h and then western blot for Cdx2, CK7, villin, and β-actin was checked from the total cellular protein extracts. (e and f) Effect of AG1478 or EGFR-siRNA on AP-1, and NF-κB activity in HB-EGF-treated HET1A cells. HET1A cells were plated into collagen type I coated 24-well plates, then co-transfected with reporter vectors (0.2 μg/ml each of AP-1 and NF-κB luciferase vectors) as well as siRNAs (5–20 pM each of EGFR or control siRNAs), and internal control renilla luciferase vector pRL-TATA-Luc (0.02 μg/ml) following the manufacturer protocol. After 12 h of transfection, cells were treated with recombinant HB-EGF (10 ng/ml) or AG1478 (100 nM/l) for 36 h and then dual-luciferase assay was carried out. Data are expressed as fold induction and compared with the respective vehicle control. Error bars show s.e.m. values obtained from three independent experiments. **P<0.01, *P<0.05 vs HB-EGF (−); #P<0.05 vs HB-EGF (+).
As, we previously showed that the recombinant HB-EGF induces the Cdx2 gene expression through activation of the transcription factors, AP-1 and NF-κB, as to read-out, we therefore, aimed to evaluate EGFR siRNAs effects and compared with that of AG1478 in terms of activation of these transcription factors. As shown in Figures 7e and f, we found to observe an inhibitory effect on AP-1 and NF-κB activation exerted by the EGFR-specific siRNAs transfection, which also co-related with the results with AG1478. Although the cells were stimulated with HB-EGF, we observed an insignificant decrease in luciferase values for both the AP-1 and NF-κB in negative control siRNA-treated conditions as compared with that of the no siRNA-transfected and HB-EGF-stimulated one, which may be due to the nonspecific effects of control siRNA oligonucleotides when transfected into the cell line. To authenticate our results, the statistical analysis was performed by comparing the results from control siRNA-transfected cells without HB-EGF-treatment. Depending on these handful evidences, we conclude that the acid pulse causes HB-EGF upregulation in fibroblast cells to promote Cdx2 induction in adjacent epithelia through EGFR and transcription factor AP-1 and NF-κB-mediated pathway (Figure 8).

Proposed model for HB-EGF-induced cellular changes in esophageal epithelial cells. An acid environment induces esophageal fibroblasts to secrete HB-EGF, a potent growth factor, which in turn triggers Cdx2 expression in adjacent squamous epithelial cells via EGFR through a paracrine mechanism.
DISCUSSION
An altered luminal acid environment induces several esophageal disorders by influencing numerous gene functions. In this study, we found a strong expression of HB-EGF in loosely adherent fibroblasts cells in the subepithelial area of broken mucosa exposed to the luminal surface in chronic reflux esophagitis patients. As chronic reflux esophagitis is the precursor condition of BE, we analyzed the role of HB-EGF in human esophageal squamous epithelial cells in the development of transdifferentiation observed in BE by evaluating several potential markers using an in vitro system. Although ample evidence indicates that stromal influences promote epithelial carcinogenesis in salivary glands, mammary glands, the urinary bladder, and skin,17, 18, 19 the interaction between cells of mesothelial origin and those in the outer epithelial layer of the esophagus is not well defined and new studies describing these events are of great interest.
Many malignancies arise from sites of infection, chronic irritation, and inflammation. In addition the tumor microenvironment, which is largely orchestrated by infiltrating inflammatory cells, growth factors, activated fibroblasts, and DNA-damage-promoting agents, promotes malignant progression by fostering proliferation, differentiation, survival, and migration. The basic concept of normal inflammation is associated with wound healing and engages epithelial and stromal cell types for reciprocal signaling dialog to facilitate the regenerative process.26 However, dysregulation of any of the converging pathways or factors can lead to abnormalities, which ultimately leads to neoplastic growth. Inflammatory bowel disease, Crohn's disease, and chronic ulcerative colitis are considered the best examples of pathologic inflammatory conditions in which the associated neoplasia turns into colorectal carcinoma.26 Apart from those, another frequently reported pathogenesis related to chronic inflammation is reflux esophagitis associated with BE, which ultimately develops into esophageal carcinoma.27 A number of studies have been conducted regarding the various esophageal aspects of reflux esophagitis, in which gastric acid is considered to be the most potent etiologic factor. In addition, we previously reported the effects of bile acid on esophageal epithelial cells.11
As the refluxant is a mixture of bile and physiologic acid, we assessed the effect of gastric acid alone on fibroblast cells in this study in an attempt to elucidate any potential paracrine factor that may induce columnar transdifferentiation. Exposure of OUMS-36T-6-cells to acidified (pH 5) medium for 20 min significantly upregulated the expression of known inflammatory chemokines, including interleukin 8, colony-stimulating factor 2, interleukin 1B, chemokine (C-X-C motif) ligand 5, tumor necrosis factor (ligand) super family member 12, and transforming growth factor beta 3, as well as the growth factors such as fibroblast growth factor 6 and 9, heparin-binding EGF-like factor, and platelet-derived growth factor alpha polypeptide and beta polypeptide. Our initial results of acid-treated fibroblast cells produced bulk amount of fibroblast growth factors even compared with HB-EGF or PDGF; however, in a preliminary study using recombinant FGF6 showed any significant effects on columnar phenotype initiated by Cdx2 (data not shown), thus suggesting its role in other events rather related to our hypothesis. Owing to these facts, from this array of upregulated genes, we selected HB-EGF as a potent paracrine factor for our subsequent experiments, after confirming its considerable expression in broken mucosal fibroblasts of chronic reflux esophagitis patients (Figure 3).
The normal esophageal squamous epithelial cell line, HET1A, has been widely used in several in vitro studies of BE.28 In this study, we evaluated the expressions of several columnar marker genes in HET1A cells treated with exogenous recombinant HB-EGF. Cdx2, an intestine-specific transcription factor and member of the caudal-related homeobox gene family, is important in early differentiation and maintenance of intestinal epithelial cells during gastrointestinal development.29 When expressed in the stomach, Cdx2 can induce differentiation of gastric epithelial cells to intestinal-type cells, thus a fundamental role in generating intestinal metaplasia has been suggested.30 In addition, increased Cdx2 expression has been reported in epithelium of BE and esophageal epithelium from subjects with esophagitis.30, 31 Our results showing increased Cdx2 expression in HB-EGF-treated squamous epithelial cells indicates its potency toward columnar transdifferentiation, which is a characteristic of BE. Moreover, cytokeratin subsets have been established as very useful organ-specific tumor markers.32 Recently, cytokeratin 7 expression was detected in esophagitis specimens as well as BE epithelium biopsy specimens.33 Consistent with those findings, we observed CK7 expression in squamous epithelial cells treated with HB-EGF at the same doses used for Cdx2 induction. In addition, we examined the effects of HB-EGF on squamous epithelial cell differentiation by detecting expression of the well-known marker villin, a cytoskeleton protein widely used as a marker of columnar cell differentiation, because of its localization in microvilli.34 The expression of villin in BE is tightly correlated with the presence of microvilli in that transformed condition.35 Taken together, our results strongly support a role for HB-EGF in the initiation of cellular transformation in BE.
Although we provided a maiden view of whether acid-induced fibroblast-derived factors can alter the potent transcription factors as well as several genes that may influence columnar phenotype, there are several points that we feel to address further. As fibroblast cells, herein we used a human embryo-derived OUMS-36T-6 cell line that are immortalized by hTRT. An interesting point may arise on the selection of embryonic vs adult fibroblast cell lines and their role in differential gene expression on acid stimulation. We speculate that depending on the differentiation status embryonic and adult fibroblast cells may show discrepancies on acid responsiveness in terms of gene expression, viability, proliferation, and apoptosis, and therefore further implicating the use of adult human fibroblast cells on this present in vitro model system. Similarly, we are also concerned regarding our findings of recombinant HB-EGF in terms of inducing several genes characteristics of columnar phenotypes by using the HET1A cells as a human primary squamous epithelial cell line that is immortalized by SV40 large Tag. SV40 is a potent DNA oncogenic polyomavirus, which is assumed to be associated with the primary viral gene product, large Tag, a protein responsible for steering the cells through transcriptional activation, repression, blockade of differentiation, stimulation of the cell cycle, and cellular transformation36 through complex interactions between the viral oncoproteins and various intracellular proteins.37 Apart from this, a correlation between promoter hypermethylation of several tumor suppressor genes (TSGs) and the presence of SV40 DNA in a heterogeneous group of leukemias and lymphomas are also evident to promote malignant transformation simultaneously by inactivating many TSGs, inducing telomerase activity, and activating several oncogenes and growth factors.38 As the HET1A cell line is transformed by SV40 large Tag, we cannot exclude some indirect effects because of the Tag-induced cellular transformation process. However, to exclude any possible artifacts because of gene methylation caused by SV40 Tag, we performed vehicle treatment instead of recombinant HB-EGF in every experiment as control. Moreover, we not only worked on only a single dose or time point, but also performed the dose- and time-dependent experiments to obtain reproducible data. In addition, we also provided the mechanistic data of how recombinant HB-EGF can modulate the genes of interest (Cdx2 and columnar markers) by focusing on its receptor and downstream pathways. Finally, we also selected another cell line OE33, which is not transformed by potent viral antigens and found concordance with the results of HET1A cells for recombinant HB-EGF-induced Cdx2 gene expression.
Conclusion and Future Directions
In this study, we gave a single pulse of acid stimulation to fibroblast cells to elucidate the nature of the stromal microenvironment and gene expression profile in reflux esophagitis. Although our findings only represent a snapshot of the condition, they indicate that chronic acid exposure might induce different panel of gene expression, which may subsequently activate diverse pathways for the initiation and progression of neoplastic changes. In our model, among several other upregulated genes, HB-EGF was given emphasis, because of its strong histological signals in broken mucosal fibroblasts from chronic reflux esophagitis patients. However, studies using other factors as described in Table 1 may also be prospective. Depending on our findings, we conclude that acid-induced HB-EGF in fibroblast cells may induce several columnar characteristic gene expressions in esophageal squamous epithelial cells.
Accession codes
References
Jankowski JA, Rebecca FH, Ian P, et al. Barrett's metaplasia. Lancet 2000;356:2079–2085.
Pera M, Cameron AJ, Trastek VF, et al. Increasing incidence of adenocarcinoma of the esophagus and esophagogastric junction. Gastroenterology 1993;104:510–513.
Lagergren J, Berqstrom R, Lindgren A, et al. Symptomatic gastroesophageal reflux as a risk factor for esophageal adenocarcinoma. N Engl J Med 1999;340:825–831.
Wild CP, Hardie LJ . Reflux, Barrett's oesophagus and adenocarcinoma: burning questions. Nat Rev Cancer 2003;3:676–684.
Fitzgerald RC, Omary MB, Triadafilopoulos G . Dynamic effects of acid on Barrett's esophagus. An ex vivo proliferation and differentiation model. J Clin Inves 1996;98:2120–2128.
Bremner RM, Crookes PF, DeMeester TR, et al. Concentration of refluxed acid and esophageal mucosal injury. Am J Surg 1992;164:522–526.
Stanciu C, Hoare RC, Bennett JR . Correlation between manometric and pH tests for gastro-esophageal reflux. Gut 1977;18:536–540.
Jang TJ, Min SK, Bae JD, et al. Expression of cyclooxygenase 2, microsomal prostaglandin E synthase 1, and EP receptors is increased in rat oesophageal squamous cell dysplasia and Barrett's metaplasia induced by duodenal contents reflux. Gut 2004;53:27–33.
Souza RF, Shewmake K, Terada LS, et al. Acid exposure activates the mitogen-activated protein kinase pathways in Barrett's Esophagus. Gastroenterology 2002;122:299–307.
Eda A, Osawa H, Satoh K, et al. Aberrant expression of CDX2 in Barrett's epithelium and inflamed esophageal mucosa. J Gastroenterology 2003;38:14–22.
Kazumori H, Ishihara S, Rumi MA, et al. Bile acids directly augment caudal related homeobox gene Cdx2 expression in oesophageal keratinocytes in Barrett's epithelium. Gut 2006;55:16–25.
Bissell MJ, Radisky D . Putting tumors in context. Nat Rev Cancer 2001;1:46–54.
Tlsty TD, Hein PW . Know thy neighbor: stromal cells can contribute oncogenic signals. Curr Opin Genet De 2001;11:5459.
Camps JL, Chang SM, Hsu TC, et al. Fibroblast-mediated acceleration of human epithelial tumor growth in vivo. Proc Natl Acad Sci USA 1990;87:75–79.
Sari A, Reidar G, Stina S . Fibroblasts can modulate the phenotype of malignant epithelial cells in vitro. Exp Cell Res 1997;235:180–187.
Okawa T, Michaylira CZ, Kalabis J, et al. The functional interplay between EGFR overexpression, hTERT activation, and p53 mutation in esophageal epithelial cells with activation of stromal fibroblasts induces tumor development, invasion, and differentiation. Genes Dev 2007;21:2788–2803.
Hodges GM, Hicks RM, Spacey GD . Epithelial-stromal interactions in normal and chemical carcinogen-treated adult bladder. Cancer Res 1977;37:3720–3730.
Sakakura T, Sakagami Y, Nishizuka Y . Accelerated mammary cancer development by fetal salivary mesenchyma isografted to adult mouse mammary epithelium. J Natl Cancer Ins 1981;66:953–959.
Smola H, Thiekötter G, Fusenig NE . Mutual induction of growth factor gene expression by epidermal-dermal cell interaction. J Cell Biol 1993;122:417–429.
Bhowmick NA, Neilson EG, Moses HL . Stromal fibroblast in cancer initiation and progression. Nature 2004;432:332–337.
Isomoto H, Wang A, Mizuta Y, et al. Elevated levels of chemokines in esophageal mucosa of patients with reflux esophagitis. Am J Gastroenterol 2003;98:551–556.
Isomoto H, Saenko VA, Kanazawa Y, et al. Enhanced expression of interleukin-8 and activation of nuclear factor kappa-B in endoscopy negative gastroesophageal reflux disease. Am J Gastroenterol 2004;99:589–597.
Rogler G, Gelbmann CM, Vogl D, et al. Differential activation of cytokines secretion in primary colonic fibroblast/myofibroblast cultures. Scand J Gastroenterol 2001;36:389–398.
Van Baal JW, Bozikas A, Pronk R, et al. Cytokeratin and CDX2 expression in Barrett's esophagus. Scand J Gastroenterol 2008;43:132–140.
El-Assal ON, Besner GE . HB-EGF enhances restitution after intestinal ischemia/reperfusion via PI3K/Akt and MEK/ERK1/2 activation. Gastroenterology 2005;129:609–625.
Coussens LM, Werb Z . Inflammation and cancer. Nature 2002;420:860–867.
Odze RD . Update on the diagnosis and treatment of Barrett esophagus and related neoplastic precursor lesions. Arch Pathol Lab Med 2008;132:1577–1585.
Kazumori H, Ishihara S, Kinoshita Y . Roles of caudal-related homeobox gene Cdx1 in oesophageal epithelial cells in Barrett's epithelium development. Gut 2009;58:620–628.
Silberg DG, Swain GP, Suh ER, et al. Cdx1 and Cdx2 expression during intestinal development. Gastroenterology 2000;119:961–971.
Eda A, Osawa H, Satoh K, et al. Aberrant expression of CDX2 in Barrett's epithelium and inflammatory esophageal mucosa. J Gastroenterol 2003;38:14–22.
Phillips RW, Frierson Jr HF, Moskaluk CA . Cdx2 as a marker of epithelial intestinal differentiation in the esophagus. Am J Surg Pathol 2003;27:1442–1447.
Moll R, Franke WW, Schiller DL, et al. The catalog of human cytokeratins: patterns of expression in normal epithelia, tumors and cultures cells. Cell 1982;31:11–24.
Ormsby AH, Goldblum JR, Rice TW, et al. Cytokeratin subsets can reliably distinguish Barrett esophagus from intestinal metaplasia of the stomach. Hum Pathol 1999;30:288–294.
Bretscher A, Weber K . Villin is a major protein of microvillus cytoskeleton which binds G and F actin in a calcium-dependent manner. Cell 1980;20:839–847.
Kumble S, Omary MB, Fajardo LF, et al. Multifocal heterogeneity in villin and Ep-cam expression in Barrett's esophagus. Int J Cancer 1996;66:48–54.
Moran E . DNA tumor virus transforming proteins and the cell cycle. Curr Opin Genet Dev 1993;3:63–70.
Ali SH, DeCaprio JA . Cellular transformation by SV40 large T antigen: interaction with host proteins. Semin Cancer Biol 2001;11:15–23.
Khaled A, Mounir T, Sonia Z, et al. Presence of simian virus 40 DNA sequences in diffuse large B-cell lymphomas in Tunisia correlates with aberrant promoter hypermethylation of multiple tumor suppressor genes. Int J Cancer 2007;121:2693–2702.
Acknowledgements
This work was supported in part by grants-in-aid for scientific research from the Ministry of Education, Science, Sports, and Culture of Japan. We thank Ms Keiko Masuzaki and Ms Rika Tohma for their technical support.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article
Cite this article
Rahman, F., Kadowaki, Y., Ishihara, S. et al. Fibroblast-derived HB-EGF promotes Cdx2 expression in esophageal squamous cells. Lab Invest 90, 1033–1048 (2010). https://doi.org/10.1038/labinvest.2010.71
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/labinvest.2010.71
Keywords
This article is cited by
-
Chronic exposure to submicromolar arsenite promotes the migration of human esophageal Het1A cells induced by heparin-binding EGF-like growth factor
Archives of Toxicology (2019)
-
The role of cancer-associated fibroblasts in esophageal cancer
Journal of Translational Medicine (2016)
-
Respond as reflux dials CDX 2
European Surgery (2010)
