
Abstract
Although there seems to be a common stem cell for the two epithelial cell types in the breast, the vast majority of breast cancers exhibit a luminal phenotype. Pure myoepithelial carcinomas are rare. We report our findings of genetic alterations in these tumors. We have analyzed 10 cases of pure myoepithelial cell carcinomas using laser capture microdissection and comparative genomic hybridization. The mean number of changes was 2.1 (range 0–4), compared with a mean of 8.6 (range 3.6–13.8) in unselected ductal carcinomas. Common alterations included loss at 16q (3/10 cases), 17p (3/10), 11q (2/10), and 16p (2/10), regions also commonly deleted in ductal carcinomas. The single case in which both pure myoepithelial carcinoma and invasive ductal carcinoma was present showed 2 alterations in the myoepithelial tumor (losses at 17p and 17q), whereas the invasive ductal component showed 15 alterations (5 gains and 9 losses), including loss at 17p. The sharing of 17p loss in myoepithelial and ductal carcinoma is consistent with a common stem cell model in the breast. The relatively few genetic alterations in otherwise aggressive neoplasms suggests that myoepithelial tumors may be a good model for the delineation of genes important in breast tumorigenesis.
Similar content being viewed by others

Introduction
Myoepithelial cells are normally located between the continuous luminal epithelial cells and the basal lamina of acini and ducts of the breast (Ahmed, 1974). They are found mostly in the acinar component of sweat and salivary glands (Foschini et al, in press). These flat, elongated cells are immunoreactive with smooth muscle actin (Bussolati et al, 1980), smooth muscle myosin heavy chain, cytokeratin 14, s-100 protein (Martinez-Madrigal and Micheau, 1991), calponin, caldesmon (Wang et al, 1997), and GFAP antibodies (Dardick, 1995, Viale et al, 1991). Because of their contractile properties, the principal function is to serve as a pump for discharging secretory material into the lumen. Paradoxically, they are often inconspicuous in the lactating breast.
Myoepithelial cells can be prominent in sclerosing adenosis and in papillomata of the breast. Tumors with myoepithelial cell component have been recognized for some time within the salivary gland and the breast (Cameron et al, 1974; Crissman et al, 1977; Hamperl, 1970; Sciubba and Brannon, 1982). However myoepithelial cell differentiation in the neoplastic population of “ordinary” ductal and lobular carcinomas of the breast is rare. In the last two decades, tumors showing myoepithelial cell differentiation have been reported more frequently (Foschini and Eusebi, 1998). These include adenoid cystic carcinoma, low grade adenosquamous carcinoma, adenomyoepitheliomas, pure myoepithelial cell carcinoma, and poorly differentiated myoepithelial rich carcinoma (Damiani et al, 1997; Foschini and Eusebi, 1998; Makek and von Hochstetter, 1980; Rosen and Ernsbergr, 1987; Tavassoli, 1991). All of these tumors are relatively rare in clinical practice, and references in the literature are generally composed of single case reports (Accurso et al, 1990; Desautels, 1990; Erlandson and Rosen, 1982; Eusebi et al, 1987; Lakhani et al, 1995; Thorner et al, 1986).
It is becoming increasingly clear that while the multistep model of carcinogenesis provides a simple model for the transformation of normal cells into cancer cells, the behavior of neoplastic cells is under a complex control system and includes paracrine influences from neighboring non-neoplastic cells. Evidence is accumulating that the myoepithelial cell may play a critical paracrine role in preventing the progression of in situ carcinoma to frankly invasive tumor (Kedeshian et al, 1998; Sternlicht and Barsky, 1997). This type of regulation of tumor cell behavior is likely to be of as great a significance as the accumulation of genetic mutations in oncogenes within the neoplastic luminal epithelial cells.
As part of our investigation of genetic alterations in preinvasive breast disease, we have demonstrated that loss of heterozygosity (LOH) identified in invasive carcinoma is already present in “normal” luminal and myoepithelial cells independently (Lakhani et al, 1999). This argues for a common stem cell that must have acquired the genetic alteration prior to differentiation into the two epithelial cell types. It is intriguing, therefore, that tumors showing myoepithelial differentiation are so rare within the breast. The morphology of tumors showing myoepithelial differentiation is generally different from tumors derived from luminal cells. Pure myoepithelial carcinomas tend to resemble sarcomas by having a predominantly spindle cell pattern of growth (Foschini and Eusebi, 1998; Lakhani et al, 1995; Schurch et al, 1985). Although there are only a few reports in the literature, it seems that no fewer than 50% of the published cases had an aggressive behavior (Foschini and Eusebi, 1998).
To date, no attempt has been made to evaluate the molecular events involved in the pathogenesis of these tumors. As a first step in trying to understand why myoepithelial cells transform so rarely, despite sharing a probable common origin with luminal tumors, we have investigated whether the differences in morphology are reflected at the molecular cytogenetic level. We have therefore investigated 10 pure myoepithelial carcinomas using comparative genomic hybridization and compared the data to the literature on invasive breast carcinomas showing luminal cell differentiation.
Results
The clinico-pathological data are summarized in Table 1. All patients were female, with an age range of 47 to 72 years. The size of the tumors ranged from 0.8 cm to 12 cm in maximum dimension. Three out of ten patients had lymph node metastases. Follow-up data were available for seven of the ten patients, four of whom died a few months to 6 years after diagnosis.
A summary of the comparative genomic hybridization (CGH) data on the 10 myoepithelial carcinomas is depicted in Figure 1. A total of 21 DNA copy number changes was seen (2 gains and 19 losses). Table 2 illustrates the alterations in each of the cases. The number of alterations (gains or losses) varied from zero to four. The most common alterations were loss of 16q and 17p (3/10 cases).

Summary karyogram showing regions of DNA copy number change for the 10 cases of myoepithelial carcinoma analyzed by comparative genomic hybridization (CGH). Gains and losses are shown by the solid bars to the right and left of the chromosome, respectively.
In Case 2, classic invasive ductal carcinoma of no special type and myoepithelial carcinoma were present within the same tissue block, and these were microdissected and analyzed separately. CGH results from the two lesions from the same case are shown in Figure 2. The myoepithelial carcinoma showed just two alterations, loss at 17p and 17q, while the invasive ductal carcinoma showed nine regions of loss and five regions of gain in chromosomal material.

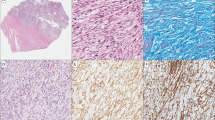
a, Photomicrograph of myoepithelial carcinoma from Case 2, haematoxylin and eosin stain, magnification × 400. Genetic alterations detected by CGH in this lesion were losses at 17p and 17q. b, Photomicrograph of invasive ductal carcinoma in the same tissue block from Case 2, haematoxylin and eosin stain, magnification × 400. Genetic alterations detected by CGH in this lesion were losses at 1p, 6q, 8p, 11p, 16q, 17p, 19p, 20p, and 22q; and gains at 7q, 8q, 15q, 17q, and 20q.
Discussion
We have demonstrated that LOH identified in invasive carcinoma is present independently in both the luminal and myoepithelial cell types suggesting the presence of a common stem cell within the breast lobule (Lakhani et al, 1999). Because the alterations must have occurred within the stem cell, it is surprising that tumors with myoepithelial differentiation are rare compared with tumors having luminal cell differentiation. This suggests that myoepithelial cells in the breast are resistant to transformation, and this is reflected in their rarity. We were therefore interested in investigating the genetic profile of pure myoepithelial carcinomas compared with carcinomas with luminal phenotype. CGH analysis was carried out in 10 pure myoepithelial carcinomas and compared with data from ductal carcinomas of no special type, which was derived from the literature.
The most striking observation from this study is the paucity of alterations identified in myoepithelial carcinomas (mean 2.1) compared with “ordinary” breast carcinomas having luminal differentiation. The mean number of alterations reported in the literature in Grade I invasive ductal carcinomas of no special type is 5.4 (range of means is 3.6–8.0), and 11.7 in Grade III tumors (range of means 8.4–13.8) (Buerger et al, 1999; Nishizaki et al, 1997; Roylance et al, 1999; Schwendel et al, 1998; Tirkkonen et al, 1998). The data in myoepithelial tumors are surprising in view of their aggressive morphology and behavior. Seven of ten tumors were larger than 2 cm in size, and four of seven patients for whom follow-up information was available had died within 6 years of diagnosis. Due to the relatively small number of cases, it is not possible to analyze the association between genetic alterations and specific clinico-pathological features. This type of analysis would require a large number of cases; however, these tumors are extremely rare in clinical practice.
None of the alterations identified in myoepithelial carcinomas are unique, and they have been previously described in invasive breast carcinomas with luminal phenotype (Buerger et al, 1999; Nishizaki et al, 1997; Roylance et al, 1999; Schwendel et al, 1998; Tirkkonen et al, 1998). The most common alterations identified in myoepithelial carcinoma (loss of 16q and 17p) are also regions commonly deleted in ductal carcinomas of no special type. Gains of chromosomal material were less commonly seen in myoepithelial tumors; however, these were also at loci associated with ductal carcinomas.
Although there was considerable overlap in the genetic profile of the two distinct epithelial tumor types, some of the most common alterations described in ductal carcinomas, such as gains of 1q, 8q, and 20q, and losses of 1p, 8p, and 13q (Buerger et al, 1999; Nishizaki et al, 1997; Roylance et al, 1999; Schwendel et al, 1998; Tirkkonen et al, 1998), were not identified in myoepithelial carcinomas. On the assumption that the two cell types are derived from a common stem cell (Lakhani et al, 1999), genetic alterations that overlap between luminal and myoepithelial tumors must have occurred within stem cells before differentiation into the two epithelial cell types. Those alterations that do not overlap would have to occur after stem cell division. In one of our cases, we had the opportunity to investigate this, because ductal carcinoma and myoepithelial carcinoma were present within the same tissue block. Loss at 17p was seen in both tumor types. Only 1 other alteration was seen in the myoepithelial tumor, while the ductal carcinoma showed 13 other alterations. We would therefore hypothesize that loss at 17p occurred within a stem cell, while alterations in other loci occurred after differentiation into the two epithelial cell types. We cannot completely exclude the possibility that the myoepithelial cell with 17p loss was derived from the luminal cell, as has been suggested recently (Petersen et al, 1998). A possible candidate gene at 17p is the TP53 gene, but TP53 mutations are generally believed to be late events in breast carcinogenesis because p53 positivity using immunohistochemistry is not seen in benign breast disease (Allred et al, 1994). We are currently in the process of narrowing this region of loss to determine whether this or another putative gene may be implicated at this early stage in tumorigenesis.
Because invasive ductal carcinomas exhibit a large number of alterations, it has been difficult to differentiate between pathogenic mutations and nonspecific mutations caused by genomic instability. Myoepithelial carcinomas, in contrast, seem resistant to transformation and exhibit few genetic alterations. Because the alterations identified in myoepithelial carcinomas are also those seen in ductal carcinomas, these alterations are likely to be pathogenetically significant in breast carcinogenesis. The presence of relatively few genetic alterations in otherwise aggressive neoplasms suggests that myoepithelial tumors may be a good model for the delineation of genes important in breast tumorigenesis.
Materials and Methods
Patients
The cases studied were gathered from consult material at University College London, or from the Department of Histopathology, Bologna. The clinical details of the patients are listed in Table 1.
The diagnosis of myoepithelial carcinoma in all cases was initially made on the characteristic morphology on haematoxylin and eosin (H&E)-stained sections that showed polygonal or spindle cells. The nuclei of neoplastic cells were pleomorphic, and atypical mitoses were frequent. The cytoplasm was occasionally abundant, eosinophilic, and filamentous. All tumors were composed of at least 80% neoplastic myoepithelial cells. These were immunoreactive with antibodies to vimentin, S100 protein, smooth muscle actin, smooth muscle myosin, calponin, caldesmone, and cytokeratin 14. One case (Case 2) was a compound tumor being constituted by areas of actin- and calponin-rich spindle and polygonal elements which were adjacent to areas of poorly differentiated invasive ductal carcinoma of no special type (Scarpellini et al 1997).
Microdissection and DNA Extraction
Microdissection was carried out using the PixCell II Laser Capture Microdissection system (Arcturus, Mountain View, California) using 5-μm-thick sections cut from formalin-fixed, paraffin-embedded tissue. Normal lymph nodes from the axillary dissection were used as control constitutional DNA. Lesions were microdissected and the DNA extracted overnight in a humidified chamber at 55° C in 20 μl extraction buffer (0.5 μg/μl proteinase K in 50 mm KCl; 10 mm Tris-HCl, pH 8.0; 2.5 mm MgCl2; 0.1 mg/ml gelatin; 0.45% NP40; 0.45% Tween 20). Before PCR, proteinase K was inactivated at 95° C for 10 minutes. In one case, classic invasive ductal carcinoma of no special type and myoepithelial carcinoma were present within the same tissue block, and these were dissected and analyzed separately.
Comparative Genomic Hybridization Analysis
Amplification and fluorescent labeling of the DNA from microdissected tissue was carried out by DOP-PCR in two rounds as previously published (Wells et al, 1999), using 2 to 5 μl of the total volume of extracted DNA. Normal male metaphase spreads (Vysis UK Ltd, Richmond, England) were denatured at 75° C for 5 minutes in 70% formamide, 2 × SSC, and dehydrated through a series of alcohols. Test (fluoroscein-labeled, FluoroGreen; Amersham Life Sciences Ltd., Buckinghamshire, England) and reference (rhodamine-labeled, FluoroRed, Amersham Life Sciences Ltd.) DNA samples from the DOP-PCR reaction were co-precipitated with 60 μg human Cot-1 DNA (Life Technologies Ltd., Paisley, Scotland) and 10 μg salmon sperm DNA, and resuspended in 10 μl of hybridization buffer (50% deionized formamide; 20% w/v dextran sulfate; 2 × SSC; 0.1 mm EDTA, pH 8.0; 0.2 mm Tris-HCl, pH 7.6). The denatured probes were then hybridized to the metaphases under a coverslip for 2 to 3 days at 37° C. After hybridization, the slides were washed in 50% formamide, 2 × SSC (3 × 10 minutes at 45° C), 2 × SSC (2 × 10 minutes at 45° C, 1 × 10 minutes at room temperature), TNT buffer (10 mm Tris-HCl, pH 8.0; 0.15 M NaCl; 0.05% Tween; 10 minutes) and ddH2O (10 minutes), before dehydration through the alcohol series. Finally, the slides were mounted in anti-fade containing 4,6-diamino-2-phenyllindole (DAPI) as a counterstain. Metaphase chromosome preparations were captured using a Zeiss Axioskop microscope (Photometrics, Munich, Germany), KAF1400 CCD camera, and Vysis SmartCapture software. Image analysis was performed using Vysis Quips CGH software. Between five and ten representative images of high quality hybridizations were analyzed and the results combined to produce an average fluorescence ratio for each chromosome. Control experiments were carried out using normal:normal (microdissected normal lymph node) co-hybridizations, whose average red:green ratio levels and 95% confidence intervals were used to set the lower and upper limits for scoring losses and gains of genetic material as 0.80 to 1.20.
References
Accurso A, Donofrio V, Insabato L, and Mosella G (1990). Adenomyoepithelioma of the breast. A case report. Tumori 76:606–610.
Ahmed A (1974). The myoepithelium in human breast carcinoma. J Pathol 113:129–135.
Allred DC, O’Connell P, Fuqua SA, and Osborne CK (1994). Immunohistochemical studies of early breast cancer evolution. Breast Cancer Res Treat 32:13–18.
Buerger H, Otterbach F, Simon R, Schäfer KL, Poremba C, Diallo R, Brinkschmidt C, Dockhorn-Dworniczak B, and Boecker W (1999). Different genetic pathways in the evolution of invasive breast cancer are associated with distinct morphological subtypes. J Pathol 189:521–526.
Bussolati G, Alfani V, Weber K, and Osborn M (1980). Immunocytochemical detection of actin on fixed and embedded tissues: Its potential use in routine pathology. J Histochem Cytochem 28:169–173.
Cameron HM, Hamperl H, and Warambo W (1974). Leiomyosarcoma of the breast originating from myothelium (myoepithelium). J Pathol 114:89–92.
Crissman JD, Wirman JA, and Harris A (1977). Malignant myoepithelioma of the parotid gland. Cancer 40:3042–3049.
Damiani S, Riccioni L, Pasquinelli G, and Eusebi V (1997). Poorly differentiated myoepithelial cell rich carcinoma of the breast. Histopathology 30:542–548.
Dardick I (1995). Myoepithelioma: Definitions and criteria. Ultrastruct Pathol 19:335–345.
Desautels JE (1990). Malignant myoepithelioma of the breast: A case report. Can Assoc Radiol J 41:387–388.
Erlandson RA and Rosen PP (1982). Infiltrating myoepithelioma of the breast. Am J Surg Pathol 6:785–793.
Eusebi V, Casadei GP, Bussolati G, and Azzopardi JG (1987). Adenomyoepithelioma of the breast with a distinctive type of apocrine adenosis. Histopathology 11:305–315.
Foschini MP and Eusebi V (1998). Carcinomas of the breast showing myoepithelial cell differentiation. Virchows Arch 432:303–310.
Foschini MP, Scarpellini F, Gown AM, and Eusebi V (2000). Different expression of myoepithelial markers in salivary, sweat and mammary gland. Int J Surg Pathol 8:29–37.
Hamperl H (1970). The myothelia (myoepithelial cells). Normal state; regressive changes; hyperplasia; tumors. Curr Top Pathol 53:161–213.
Kedeshian P, Sternlicht MD, Nguyen M, Shao ZM, and Barsky SH (1998). Humatrix, a novel myoepithelial matrical gel with unique biochemical and biological properties. Cancer Lett 123:215–226.
Lakhani SR, Chaggar R, Davies S, Jones C, Collins N, Odel C, Stratton MR, and O’Hare M (1999). Genetic alterations in “normal” luminal and myoepthelial cells of the breast. J Pathol 189:496–503.
Lakhani SR, O’Hare MJ, Monaghan P, Winehouse J, Gazet JC, and Sloane JP (1995). Malignant myoepithelioma (myoepithelial carcinoma) of the breast: A detailed cytokeratin study. J Clin Pathol 48:164–167.
Makek M and von Hochstetter AR (1980). Pleomorphic adenoma of the human breast. J Surg Oncol 14:281–286.
Martinez-Madrigal F and Micheau F (1991). Major salivary glands. In: Sternberg SS (editor) Histology for pathologists. New York:Raven Press, 405–432.
Nishizaki T, DeVries S, Chew K, Goodson WH 3rd, Ljung BM, Thor A, and Waldman FM (1997). Genetic alterations in primary breast cancers and their metastases: Direct comparison using modified comparative genomic hybridization. Genes Chromosomes Cancer 19:267–272.
Petersen OW, Ronnov-Jessen L, Weaver VM, and Bissell MJ (1998). Differentiation and cancer in the mammary gland: Shedding light on an old dichotomy. Adv Cancer Res 75:135–161.
Rosen PP and Ernsbergr D (1987). Low-grade adenosquamous carcinoma. A variant of metaplastic mammary carcinoma. Am J Surg Pathol 11:351–358.
Roylance R, Gorman P, Harris W, Liebmann R, Barnes D, Hanby A, and Sheer D (1999). Comparative genomic hybridization of breast tumors stratified by histological grade reveals new insights into the biological progression of breast cancer. Cancer Res 59:1433–1436.
Scarpellini F, Usellini S, and Foschini MP (1997). Mioepitelioma maligno associato a carcinoma duttale in situ ed invasivo NAS. Descrizione di un caso e revisione della letteratura. Pathologica 89:420–424.
Schurch W, Potvin C, and Seemayer TA (1985). Malignant myoepithelioma (myoepithelial carcinoma) of the breast: An ultrastructural and immunocytochemical study. Ultrastruct Pathol 8:1–11.
Schwendel A, Richard F, Langreck H, Kaufmann O, Lage H, Winzer KJ, Petersen I, and Dietel M (1998). Chromosome alterations in breast carcinomas: Frequent involvement of DNA losses including chromosomes 4q and 21q. Br J Cancer 78:806–811.
Sciubba JJ and Brannon RB (1982). Myoepithelioma of salivary glands: Report of 23 cases. Cancer 49:562–572.
Sternlicht MD and Barsky SH (1997). The myoepithelial defense: A host defense against cancer. Med Hypotheses 48:37–46.
Tavassoli FA (1991). Myoepithelial lesions of the breast. Myoepitheliosis, adenomyoepithelioma, and myoepithelial carcinoma. Am J Surg Pathol 15:554–568.
Thorner PS, Kahn HJ, Baumal R, Lee K, and Moffatt W (1986). Malignant myoepithelioma of the breast. An immunohistochemical study by light and electron microscopy. Cancer 57:745–750.
Tirkkonen M, Tanner M, Karhu R, Kallioniemi A, Isola J, and Kallioniemi OP (1998). Molecular cytogenetics of primary breast cancer by CGH. Genes Chromosomes Cancer 21:177–184.
Viale G, Gambacorta M, Coggi G, Dell’Orto P, Milani M, and Doglioni C (1991). Glial fibrillary acidic protein immunoreactivity in normal and diseased human breast. Virchows Arch 418:339–348.
Wang NP, Wan BC, Skelly M, Frid MG, Glukhova MA, Koteliansky VE, and Gown AM (1997). Antibodies to novel myoepithelium associated proteins distinguished benign lesions and carcinoma in situ from invasive carcinoma of the breast. Appl Immunohistochem 5:141–151.
Wells D, Sherlock JK, Handyside AH, and Delhanty JDA (1999). Detailed chromosomal and molecular genetic analysis of single cells by whole genome amplification and comparative genomic hybridization. Nucleic Acids Res 27:1214–1218.
Acknowledgements
We would like to thank Dr. Andy Hanby, Guys Hospital, London; Dr. Margaret Jeffreys, St Mary’s Hospital, Portsmouth; Dr. Su Ramachandra, Whittington Hospital, London; and Dr. Reuben Tooze, Addenbrookes Hospital, Cambridge, for allowing us to include their cases in this study. We are grateful to Professor Joy Delhanty and Professor A. Munro Neville for encouragement and support.
Supported in part by the Ludwig Institute for Cancer Research and the Sydney and Phyllis Goldberg Memorial Charitable Trust and MURST (Rome). CJ is supported by the UCL and Middlesex Special Trustees. DW is supported by the Wellcome Trust (Grant No. 046416), which also funded the image capture equipment (Grant No. 039938).
Author information
Authors and Affiliations
Corresponding author
Additional information
Y-JL is supported by Breakthrough Breast Cancer.
Rights and permissions
About this article
Cite this article
Jones, C., Foschini, M., Chaggar, R. et al. Comparative Genomic Hybridization Analysis of Myoepithelial Carcinoma of the Breast. Lab Invest 80, 831–836 (2000). https://doi.org/10.1038/labinvest.3780087
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1038/labinvest.3780087
This article is cited by
-
K5/K14-positive cells contribute to salivary gland-like breast tumors with myoepithelial differentiation
Modern Pathology (2013)
-
Primary myoepithelial carcinoma of palate
World Journal of Surgical Oncology (2011)
-
Myoepithelial carcinoma of the breast: a clinicopathological and immunohistochemical study of 15 diagnostically challenging cases
Virchows Archiv (2010)
-
Molecular and morphological analysis of adenoid cystic carcinoma of the breast with synchronous tubular adenosis
Virchows Archiv (2009)
-
Mechanisms of Disease: breast tumor pathogenesis and the role of the myoepithelial cell
Nature Clinical Practice Oncology (2006)
