
Abstract
Uterine myxoid smooth muscle tumors, including myxoid leiomyosarcoma, are rare and their genomic profile has not been fully characterized. With the discovery of uterine sarcomas with ZC3H7B-BCOR fusion and BCOR internal tandem duplications, the differential diagnosis of myxoid smooth muscle lesions is expanding to include molecularly-defined tumors. Thus, we aimed to explore the genomic landscape of myxoid smooth muscle tumor using comprehensive tools. We performed whole exome next-generation sequencing and a pan-sarcoma RNA fusion assay in tumoral paraffin-embedded tissue from nine well-characterized uterine myxoid smooth muscle tumors (seven myxoid leiomyosarcomas and two myxoid smooth muscle tumors of unknown malignant potential). By immunohistochemistry, all tumors were strongly positive for smooth muscle markers and negative for BCOR staining; 4/6 expressed PLAG1. None of the tumors harbored known fusions including ZC3H7B-BCOR, TRPS1-PLAG1, and RAD51B-PLAG1. None harbored exon 15 BCOR internal tandem duplications; however, four tumors contained BCOR internal tandem duplications of unknown significance (mostly intronic). Mutational burden was low (median 3.8 mutations/megabase). DNA damage repair pathway gene mutations, including TP53 and BRCA2, were found. Copy number variation load, inferred from sequencing data, was variable with genomic indexes ranging from 2.2 to 74.7 (median 25.7), with higher indexes in myxoid leiomyosarcomas than myxoid smooth muscle tumors of unknown malignant potential. The absence of clear driver mutations suggests myxoid smooth muscle tumors to be genetically heterogeneous group of tumours and that other genetic (eg., undiscovered translocation) or epigenetic events drive the pathogenesis of uterine myxoid smooth muscle neoplasia.
Similar content being viewed by others


Introduction
Myxoid leiomyosarcoma is a rare but well-recognized subset of uterine leiomyosarcoma characterized by abundant myxoid stroma separating smooth muscle fibres. Cumulative literature suggests that this tumor portends poor overall survival [1,2,3]. Given its rarity, myxoid leiomyosarcoma remains an elusive and diagnostically challenging lesion. Microscopic features indicative of malignancy in conventional smooth muscle tumors are frequently subtle or absent in myxoid leiomyosarcoma, and diagnosis relies on the presence of an infiltrative tumor border [1, 4].
The differential diagnosis of myxoid leiomyosarcoma includes other smooth muscle tumors such as conventional leiomyosarcoma and other myxoid neoplasms, mainly inflammatory myofibroblastic tumor and endometrial stromal sarcoma. Inflammatory myofibroblastic tumors are notable for a distinctly loose spindle cell population in myxoid stroma and ALK expression by immunohistochemistry, as a consequence of ALK rearrangements. The recent characterization of a novel subset of uterine sarcomas, categorized as high-grade endometrial stromal sarcoma harboring ZC3H7B-BCOR gene fusions, has expanded the list of mimickers of myxoid leiomyosarcoma [5]. The ZC3H7B-BCOR fusion-positive sarcoma also contains abundant myxoid stroma; in addition, it has a tongue-like permeative infiltration into the myometrium (akin to low grade endometrial stromal sarcoma) and diffuse CD10 expression. Interestingly, both myxoid leiomyosarcomas and BCOR-rearranged sarcomas both have documented aggressive behavior with widespread local and distant recurrences as well as poor survival.
The distinction between myxoid leiomyosarcoma and the above entities on morphologic and immunohistochemical grounds can be difficult. Mimickers of myxoid leiomyosarcoma have distinctive molecular aberrations that can be diagnostically useful: ZC3H7B-BCOR fusions and ALK rearrangements may be detectable by fluorescence in situ hybridization (FISH) and other gene fusion detection techniques [6, 7]. In contrast, the molecular landscape of myxoid leiomyosarcoma remains poorly characterized, and no mutations or structural variants specific for this entity have been documented yet. Aberrant (overexpressed or “null”) p53 and/or p16 immunohistochemistry patterns have been observed in ~50% of uterine myxoid leiomyosarcoma, correlating with TP53 and CDKN2A mutations [8]. Of note, a recent study documented TRPS1-PLAG1 and RAD51B-PLAG1 fusions in a subset of 4/15 tumors described as myxoid leiomyosarcomas [9].
In conventional smooth muscle tumors, the landscape of genetic alterations is diverse. TP53, ATRX, and MED12 mutations are some of the more frequently reported somatic mutations in conventional leiomyosarcoma, with alternative lengthening of telomeres phenotype being observed in those cases with loss of ATRX expression [10]. In addition, the burden of copy number variations (copy number variants) is heterogeneous, and it is associated with the tumor behavior. It has been shown that the genomic index, a derivative of the number copy number variations per chromosome, is significantly higher in conventional leiomyosarcoma compared to leiomyoma, and can stratify tumors of unknown malignant potential into low and high-risk groups [11, 12]. In comparison, the spectrum of genetic alterations of myxoid smooth muscle neoplasia in the uterus, particularly myxoid leiomyosarcoma, is largely unknown, and analysis restricted to this tumor category is needed.
The aim of this study is to characterize the genomic alterations of uterine myxoid smooth muscle neoplasia employing comprehensive sequencing and hybridization methods in a well-annotated sample. We also aimed to explore the presence of PLAG1 alterations in this family of tumors using sequencing data.
Materials and methods
Case selection and review
We identified patients with a diagnosis of myxoid smooth muscle tumor in our institutional database from July 2000 to June 2017. Cases with histologic material and at least one representative tumor block available were further selected. Archival material was reviewed and the diagnosis was confirmed by two gynecologic pathologists (CPH and MRN). For each case, demographic and pathologic variables were recorded including tumor size, mitotic count [per 10 high-power fields (HPFs)], and presence of necrosis. Inclusion required confirmation of smooth muscle morphology (fascicular growth, cigar-shaped nuclei) and presence of myxoid matrix highlighted by Alcian blue stain at pH 2.5. Further confirmation of the diagnosis was pursued with the use of immunohistochemistry (see section below). The diagnosis of myxoid leiomyosarcoma was based on the presence of: (1) infiltrative tumor borders plus any of the following three worrisome features: moderate to severe nuclear atypia, two or more mitoses in 10 high-power fields, and tumor cell necrosis, or (2) in the absence of infiltrative tumor borders, presence of two or three of the above features. The diagnosis of myxoid smooth muscle tumor of unknown malignant potential was made if: 1) tumor had infiltrative borders but no other worrisome features, or (2) tumor lacked infiltration and had one of the three worrisome features listed above. These definitions follow a previously published diagnostic algorithm [13].
Immunohistochemistry
Available formalin-fixed, paraffin-embedded material was stained for the following immunohistochemical markers: desmin (Ventana, DE-R-11), smooth muscle actin (Dako, 1A4), caldesmon (Dako, h-CD), ALK (Ventana, ALK01), CD10 (Ventana, SP67), estrogen receptor (Ventana, SP1), BCOR (Santacruz Biotechnology, SC-154576), and PLAG1 (Novus Biologicals, 3B7). Each was interpreted as negative (absent staining) or positive, the latter quantified in terms of distribution (1+ = 1–25% of positive tumor cells, 2+ = 26–50%, 3+ = >50%) and average staining intensity (1+ = weak, 2+ = moderate, 3+ = strong).
DNA extraction and whole-exome sequencing
Representative tumor areas were circled in the corresponding slide by one pathologist (CPH) and three cores, 1 mm in width and ~3 mm in depth, were manually punched out from the respective formalin-fixed, paraffin-embedded tissue blocks. All areas were entirely composed of tumor with a minimum tumor cellularity of 50%. Total genomic DNA (gDNA) from the tissue cores was extracted using the MagMAX formalin-fixed, paraffin-embedded DNA Isolation kit (ThermoFisher). The obtained gDNA samples were quantified using the TaqMan RNase P assay (ThermoFisher). The DNA samples were prepared using the Agilent SureSelect v5 Exome Capture kit as a paired-end library. Whole-exome sequencing was performed using an Illumina HiSeq 2500 to target coverage of ~100×.
Whole-exome sequencing data analysis
Alignment of sample reads against the GRCh38 reference genome assembly was performed using the Burrows–Wheeler Aligner (BWA, v0.7.9a Mem algorithm) [14]. Subsequent downstream alignment processing (e.g., sorting, duplicate read marking, base quality recalibration) was done using Picard v2.6.0 and the Genome Analysis Toolkit v3.6, respectively [15]. FASTQ and alignment metrics were performed using Picard v2.6.0 and FASTQC v0.11.5. At the completion of the alignment and post-processing stages, a final BAM file was produced with a quality report to identify key performance indicators for sequencing and alignment.
Somatic single nucleotide variants and small insertion/deletion events were identified using the VarScan v2.4.2 algorithm [16], and were annotated using ANNOVAR [17]. Intergenic region and intronic regions were excluded in the following analysis. The collection of base substitutions were filtered based on predicted impact on the gene function, as well as known allelic frequencies in control populations. The set of mutations were then further filtered for genes with known roles in various human cancers, as well as for genes found to be mutated in at least two of the cases. Because matched normal tissue sequence data were not available, we filtered out possible germline and single nucleotide polymorphisms by filtering out variants with minor allele frequency greater than 5% in the PopFreqMax database from ANNOVAR package [17], which consolidates allele frequencies from different databases, including the 1000 Genomes Project (October 2014 release, five ethnicity groups), Exome Aggregation Consortium, NHLBI-6500 Exomes (two ethnicity groups), and the Complete Genomics 46 genomes (CG46) database. All stop-gain and frameshift-causing in/del mutations were included in the subsequent analyses. For non-synonymous missense mutations, mutations predicted to be deleterious or possibly deleterious by either SIFT, PolyPhen2 or FATHMM were retained for the subsequent analyses. Extensive literature search in various databases (including COSMIC, ClinVar, My Cancer Genome) and primary research papers were then performed to generate a list of significantly mutated genes. Copy number variant analysis was performed using CopyWriteR, a copy number variant analysis algorithm suited for whole-exome data [18]. Adapting the methods used previously by Croce et al. to examine genomic index in conventional smooth muscle tumors [11], copy number variant burden was examined by calculating genomic index values. Genomic index values were calculated as A2/C (where A = total number of alterations (segmental gains and losses), and C = number of involved chromosomes).
While numerous duplications were identified by the algorithm, basic filtering criteria (i.e., in gene duplications, a minimum of two paired support reads and minimum mapping quality of 30) were used to limit the data to higher quality duplications. From the filtered single nucleotide variant data, the mutation burden was estimated based on the coding regions of the exome capture size of 38MB [19]. DeconstructSigs R package was used to examine for the trinucleotide mutational signatures previously described by Alexandrov et al. [20, 21].
gDNA-based gene fusion analysis
Sequencing data were surveyed for somatic structural rearrangements using DELLY as an initial tool [22], and manually explored duplications called from the DELLY structural rearrangement suite of tools. Manual surveying included search for ZC3H7B-BCOR, RPS1-PLAG1, and RAD51B-PLAG1 fusions, as well as BCOR internal tandem duplication, using IGV by viewing pairs of reads across the different gene boundaries.
RNA hybridization and BCOR RT-PCR
Total RNA was extracted and purified from paraffin-embedded tissue using the High Pure formalin-fixed, paraffin-embedded tissue RNA Isolation Kit from Roche (Roche Diagnostics, Indianapolis, IN). RNA quality and quantity was sufficient for downstream evaluations in six cases. The NanoString nCounter® Sarcoma Gene Fusion Panel assay (NanoString Technologies, Seattle, WA), a high-throughput hybridization technique, was utilized following the manufacturer’s protocol and analytical pipeline to interrogate for the presence of 174 unique sarcoma gene fusions previously detected in 25 sarcoma types [23]. Reverse transcription was carried out with the iScript cDNA Synthesis kit (Bio-Rad). Internal tandem duplications involving exon 15 of BCOR were interrogated with the following primers: F: CGGCAGGTTTCTGCAAGTCTC, R: ACTGTACATGGTGGGTCCA (expected amplicon size: 198 bp). PCR products were evaluated by ethidium bromide staining on a 1% agarose gel alongside 1 kb Plus DNA Ladder (Invitrogen). The PCR products were purified using 2 units of exonuclease (M0293S; New England Bio Labs) and 0.4 units of shrimp alkaline phosphatase (78390; Affymetrix) incubated for 18 min at 37 °C and 15 min at 80 °C. Purified products were Sanger-sequenced and manually reviewed and annotated in relationship to NM_001123385.1 reference sequence (corresponding to Ensembl ID ENST0000037844.8).
Results
Clinical features
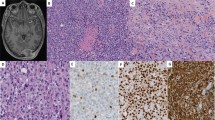
Nine patients met our inclusion criteria. The main clinical and pathologic characteristics of the sample are depicted in Table 1. Two patients had a diagnosis of myxoid smooth muscle tumor of unknown malignant potential and seven of myxoid leiomyosarcoma. Median age at presentation was 54 years (range 35–74). Median tumor size was 10 cm (range 5–23 cm). All tumors contained an abundant myxoid stroma representing ≥50% of the tumor volume and were positive for Alcian Blue at pH 2.5 (Fig. 1). We were able to assess the tumor borders in 8/9 cases, of which seven tumors had an infiltrative border (six myxoid leiomyosarcoma, one myxoid smooth muscle tumor of unknown malignant potential); one myxoid smooth muscle tumor of unknown malignant potential had a non-infiltrative border. 7/9 tumors had moderate or severe nuclear atypia (six myxoid leiomyosarcomas and the tumor of unknown malignant potential with a smooth border). Four cases had geographic tumor cell necrosis (all myxoid leiomyosarcomas). Mitotic count ranged from 1 to 30 mitotic figures per 10 HPFs (median 2).

Myxoid leiomyosarcoma is characterized by an irregular tumor border (a) and is composed of atypical hyperchromatic spindled and ovoid nuclei organized in bundles in a prominent pale myxoid matrix (b), which stains intensely blue with Alcian blue pH 2.5 stain (c). Myxoid leiomyosarcoma may show less prominent degrees of atypia (d) and low mitotic counts. This tumor was strongly and diffusely positive for desmin (e) and caldesmon (f) by immunohistochemistry
Immunohistochemistry
Immunohistochemical expression results are displayed in Table 2 and examples are depicted in Fig. 1. All nine tumors showed expression of at least two smooth muscle markers: Desmin was positive in all nine cases, and caldesmon was positive in 8/9 tumors. SMA was positive in all seven tumors tested for this marker. Importantly, smooth muscle marker expression was often strong and diffuse. CD10 was positive in 6/8 tumors tested, and ER expression was observed in 6/8 cases (strong and diffuse in all). ALK was negative in 8/8 cases. Similarly, BCOR expression was not detectable by immunohistochemistry in any cases (9/9). Diffuse, moderate to strong nuclear PLAG1 expression, was seen in 4/6 lesions with material available (3/5 myxoid leiomyosarcomas and 1/1 myxoid smooth muscle tumor of unknown malignant potential).
Sarcoma fusion analysis
Analysis of the whole-exome sequencing data showed no evidence of functional gene fusions predicted in any of the nine cases examined (DELLY [22]). This includes absence of ZC3H7B-BCOR, TRPS1-PLAG1, and RAD51B-PLAG1 fusions in all our cases. RNA hybridization testing using the NanoString nCounter platform was successful in six tumors (one myxoid smooth muscle tumor of unknown malignant potential and five myxoid leiomyosarcomas). No gene fusions were identified in any of the cases, including ALK rearrangements as well as ZC3H7B-BCOR, PLAG1-HAS2, or PLAG1-COL1A2 fusions.
Global single nucleotide and copy number variant analyses
Filtering for exonic, non-synonymous mutations, the total mutational burdens were low, with median total mutational burden being 3.8 mutations per MB (range 3.2–5.3) (Fig. 2a. Table 3). Total mutational burden values did not differ significantly when comparing by diagnosis (myxoid leiomyosarcoma vs. myxoid smooth muscle tumor of unknown malignant potential). We next examined the whole-exome sequencing data for mutational signatures, which examines the distribution of base substitutions at different trinucleotide sequences [21]. The most predominant mutational signature observed was signature 1, a signature that has been associated with aging (Supp.Fig. 1). Signature 16 (which is of unclear mechanistic association) and signature 26 (which may be seen with DNA mismatch repair) were seen as minor contributing signatures.

Global mutational burden in myxoid smooth muscle tumors. a Total mutational burden in myxoid smooth muscle neoplasms displayed as the number of exonic, non-synonymous single nucleotide variants per megabase. b copy number variant burden, displayed as genome index values based on whole exome sequencing data
We compared the copy number variant load using whole-exome sequencing-based genomic index values as a crude method to assess the copy number variant load in each case (Fig. 2b). Genomic index values based on whole-exome sequencing data were highly variable, ranging from 2240 to 74,705 (median 25,676). The genomic index values did not significantly differ when comparing based on mitotic count (>5 mitotic figures/10 HPFs vs. ≤5). Genomic index values tended to be higher in myxoid leiomyosarcomas compared to myxoid smooth muscle tumors of unknown malignant potential, although statistical significance was not reached (Table 3).
DNA damage repair pathways, epigenetic, and RNA metabolism pathways
Among the various genetic abnormalities identified, we identified mutations in PRSS3 (c.281_282insCC, p.R94fs, NM_001197098) and ZNF717 (c.2732delC, p.S911fs, NM_001128223) (Fig. 3). All the significant DNA changes are summarized in supplemental Table 1. We uncovered deleterious mutations in genes implicated in epigenetic regulation of gene expression, including CREBBP, EP300, KMT2C, KMT2D, ARID5B, and PPP4R1. Two tumors also harbored mutations in components of the SWI/SNF complexes, SMARCB1 and TTF2. Genes mutated with implication in RNA metabolism included PABPC1 (mutated in 5/9 cases), DROSHA, and DDX6.

Significantly mutated genes in myxoid leiomyosarcoma and myxoid smooth muscle tumor of unknown malignant potential. Shown is the breakdown of significant mutations uncovered through whole exome sequencing
Focusing on genes with known roles in the various DNA damage repair pathways, only one myxoid leiomyosarcoma (case 8) was found to harbor a deleterious TP53 mutation (p.Y31X). By copy number variant analysis, all nine cases were copy-neutral at 17p13, the region harboring TP53 (data not shown). Single cases harboring deleterious mutations in BRCA2, FANCM, and MEIOB were found.
Signaling pathways and cell cycle regulation
Examining for potentially targetable mutations in signaling pathways, mutations were found in MAML1, CSF1R, and WNT9A (Fig. 3). Downstream of various receptor tyrosine kinases, we found mutations in FRS3, SH2B3, SARM1, BLK, and FAM120A. No recurrent mutations in any Ras genes were found. Rather, we uncovered mutations in other Ras superfamily members, DOCK2, DOCK3, EPS8L2, MYO9A, and RGS21. Mutations in SMC2, INSC, ASPM, CEP350, and CEP120 were also found. No deleterious somatic CDKN2A mutations were detected, and by copy number variant analysis, all cases were copy-neutral for 9p21 (the region containing CDKN2A).
BCOR internal tandem duplications
Whole-exome sequencing identified internal tandem duplications in 4/9 cases (three myxoid leiomyosarcomas and one myxoid smooth muscle tumor of unknown malignant potential). Of these, three cases had internal tandem duplications in intronic regions, and one case (case 7) harbored an internal tandem duplication involving exon 15, in the 3’ untranslated region of BCOR, located 3’ to the BCOR internal tandem duplications previously identified in uterine sarcomas and clear cell sarcoma of the kidney [24, 25]. None of the genomic internal tandem duplications was expressed at the transcript level as per RT-PCR evaluations of BCOR exon 15 performed in seven cases.
Discussion
Given its rarity, the category of uterine myxoid smooth muscle neoplasia is still diagnostically challenging and poorly understood clinically and biologically. Our study is the first to comprehensively characterize the genomic landscape of myxoid smooth muscle neoplasia to date. Overall, we found low total mutational burdens (ranging from 3.2 to 5.3 mutations per MB), but a relatively high copy number variant burdens (genomic index values ranging from 2240 to 74,705). The genomic index was explored previously by Croce et al. in conventional smooth muscle tumors, showing accumulation of copy number variant burden to be a key event in leiomyosarcoma pathogenesis [10]. While it is difficult to compare the genomic index values between different platforms (comparative genomic hybridization used by Croce et al. vs. whole-exome sequencing), copy number variant accumulation appears to be also important in myxoid smooth muscle neoplasia pathogenesis and classification.
The absence of known sarcoma fusions in our series, including ZC3H7B-BCOR, by whole-exome and targeted RNA sequencing, as well as RPS1-PLAG1 and RAD51B-PLAG1 by whole-exome sequencing is an important finding. A major differential of myxoid smooth muscle neoplasia in the uterus is with other predominantly myxoid lesions, including fibromyxoid low-grade endometrial stromal sarcoma and the recently characterized uterine sarcoma harboring ZC3H7B-BCOR rearrangement. For this reason, and in order to verify the presence of smooth muscle differentiation in these lesions, we screened our cohort by careful histopathologic review, which included immunohistochemical analysis, as well as fusion analysis by DNA sequencing, RNA hybridization, and targeted BCOR RT-PCR. The nine myxoid smooth muscle tumors in our series had convincing smooth muscle morphologic and protein expression profiles; they all showed fascicular growth and nuclei with tapered ends, and they all were positive for smooth muscle markers, typically in a diffuse and strong fashion. On the other hand, CD10 was generally weak in intensity, and BCOR was consistently negative by immunohistochemistry. By DELLY method and manual inspection, we did not found evidence of PLAG1 fusions by whole exome sequencing in our case series. During the preparation of this manuscript, novel fusions involving the PLAG1 gene (TRPS1-PLAG1 and RAD51B-PLAG1) were reported in a four out of 15 tumors described as myxoid leiomyosarcoma [9]. Of note, the inclusion of cases in this publication was solely based on morphology, and expression of smooth muscle markers was not stated. Given the significant morphologic overlap between myxoid tumors of smooth muscle, endometrial stromal, myofibroblastic and other differentiation in the uterus, the true prevalence of PLAG1 fusions in myxoid leiomyosarcoma or other myxoid sarcomas of the uterus requires further exploration. Of importance, four out of six tumors in our series showed diffuse nuclear PLAG1 staining by immunohistochemistry despite the absence of PLAG1 fusions by whole-exome sequencing. This may be due to our testing methodology, since our RNA hybridization panel did not include the recently discovered TRPS1-PLAG1 and RAD51B-PLAG1 fusions. Alternatively, expression may be due to other PLAG1 alterations not yet unveiled. The specificity of this marker in the work-up of uterine myxoid tumors remains to be established.
BCOR internal tandem duplications have been recently described in high-grade uterine sarcomas with a morphologic spectrum ranging from endometrial stromal sarcoma to undifferentiated uterine sarcoma [5, 24, 26]. Internal tandem duplications involving the 3’ end of the BCOR gene have been formerly reported in 85–95% of clear cell sarcomas of the kidney, and subsequently in soft tissue undifferentiated round cell sarcomas of infancy. In these tumors, and emergently in high-grade uterine sarcomas, BCOR Internal tandem duplications are prevalent and appear to be mutually exclusive with YWHAE-NUTM2A/B fusions [27,28,29]. These alterations consistently lead to BCOR overexpression detectable by immunohistochemistry [26]. We did not identify exonic BCOR ITDs in our cohort. However, whole-exome sequencing detected BCOR internal tandem duplications in 4/9 (44%) myxoid smooth muscle tumors, in non-coding regions of BCOR (intronic in three tumors and 3’ untranslated region in another one). It is unknown whether these alterations result in transcriptional and functional alterations of BCOR, and therefore they are of unknown biological significance. However, the lack of detectable levels of BCOR expression by immunohistochemistry suggests that the mechanisms of BCOR dysregulation in myxoid smooth muscle tumor are different from those in uterine sarcomas with BCOR rearrangements or internal tandem duplications. Importantly, the spectrum of functional alterations of BCOR is wide and includes not only rearrangements and internal tandem duplications, but also exon deletions and mutations (frameshift, nonsense, or splicing site and copy number loss), which have been documented in myelodysplastic syndromes, T-cell lymphomas and small B-cell lymphomas [30,31,32], and suggest complex oncogenic and tumor-suppressor mechanisms that may result from heterogeneous loss or change of function genomic events.
We also report additional pertinent negative results. We had previously reported loss of p53 and p16 immunohistochemistry patterns to be frequent in conventional and myxoid leiomyosarcoma [8]. In the present cohort, however, TP53 mutations were found in only one case (case 8), and there was no loss of 17p13. CDKN2A (encoding p16) was also copy-neutral in all cases, with no deleterious exonic mutations. Thus, other mechanisms are likely playing a role in affecting p53 and p16 protein levels. No clear trunk driver mutations were identified in our sequencing data, suggesting that the copy number variant complexity observed in myxoid smooth muscle tumors may be driven by either structural variants not detected through our methods, or epigenetic events. Sequencing did not reveal any activating mutations or fusions involving receptor tyrosine kinases or common driver genes downstream of receptor tyrosine kinases, such as members of the Ras subfamily, in contrast to some other soft tissue sarcomas with myxoid features including inflammatory myofibroblastic tumor. Mutational burdens (in terms of single nucleotide variants) were similar between myxoid smooth muscle tumor of unknown malignant potential and myxoid leiomyosarcoma, thus suggesting a non-genetic mechanism driving the progression.
Our study is limited by the small sample size and the absence of sequencing of normal tissue for each tumor sample. Uterine myxoid leiomyosarcoma and myxoid smooth muscle tumor of unknown malignant potential are rare diagnoses, as evidenced by the fact that in a single tertiary-care institution, only nine cases with confirmed diagnosis and available material were successfully identified and included. We chose to concentrate resources in this, albeit small, well-characterized group of myxoid smooth muscle tumors diagnosed following strict criteria by two subspecialized pathologists. Matched normal tissue was not available for all cases. For this reason, we screened our data for possible germline and single nucleotide polymorphisms and filtered out variants with minor allele frequency greater than 5% in comprehensive public databases including ANNOVAR package, Exome Aggregation Consortium, NHLBI-6500 Exomes and the CG46 database. Confirmation of our findings requires independent and larger cohorts of patients.
In summary, comprehensive exome sequencing and RNA fusion analysis revealed that uterine myxoid smooth muscle neoplasia represents a genetically heterogeneous category. Myxoid smooth muscle tumors lack prevalent driver mutations and fusions, but often have significant copy number variant burdens reflected in high genomic indexes. Additional alterations, including those in DNA damage repair pathways, likely contribute to the complex genomic landscape of myxoid leiomyosarcoma. In our series of strictly characterized lesions, there was absence of BCOR structural aberrations, PLAG1 fusions and other sarcoma gene fusions. Nonetheless, frequent BCOR internal tandem duplications of unknown significance were discovered, which requires further investigation. Lastly, we believe that attribution of BCOR, PLAG1 and other novel and potentially useful genetic alterations to any established morphologic category of neoplasia requires rigorous tumor characterization and inclusion criteria. These molecular aberrations likely have a morphologic spectrum that goes beyond the currently defined tumor categories, and they may even redefine uterine sarcoma classification superseding phenotype, as has been the case in many other soft tissue neoplasms.
References
Parra-Herran C, Schoolmeester JK, Yuan L, Dal Cin P, Fletcher CD, Quade BJ, et al. Myxoid Leiomyosarcoma of the Uterus: a Clinicopathologic Analysis of 30 cases and review of the literature with reappraisal of its distinction from other uterine myxoid mesenchymal neoplasms. Am J Surg Pathol. 2016;40:285–301.
Wang WL, Soslow R, Hensley M, Asad H, Zannoni GF, de Nictolis M, et al. Histopathologic prognostic factors in stage I leiomyosarcoma of the uterus: a detailed analysis of 27 cases. Am J Surg Pathol. 2011;35:522–9.
Abeler VM, Røyne O, Thoresen S, Danielsen HE, Nesland JM, Kristensen GB. Uterine sarcomas in Norway. A histopathological and prognostic survey of a total population from 1970 to 2000 including 419 patients. Histopathology. 2009;54:355–64.
Burch DM, Tavassoli FA. Myxoid leiomyosarcoma of the uterus. Histopathology. 2011;59:1144–55.
Hoang LN, Aneja A, Conlon N, Delair DF, Middha S, Benayed R, et al. Novel high-grade endometrial stromal sarcoma: a morphologic mimicker of myxoid leiomyosarcoma. Am J Surg Pathol. 2017;41:12–24.
Parra-Herran C, Quick CM, Howitt BE, Dal Cin P, Quade BJ, Nucci MR. Inflammatory myofibroblastic tumor of the uterus: clinical and pathologic review of 10 cases including a subset with aggressive clinical course. Am J Surg Pathol. 2015;39:157–68.
Haimes JD, Stewart CJR, Kudlow BA, Culver BP, Meng B, Koay E, et al. Uterine inflammatory myofibroblastic tumors frequently harbor ALK fusions with IGFBP5 and THBS1. Am J Surg Pathol. 2017;41:773–80.
Schaefer I-M, Hornick JL, Sholl LM, Quade BJ, Nucci MR, Parra-Herran C. Abnormal p53 and p16 staining patterns distinguish uterine leiomyosarcoma from inflammatory myofibroblastic tumour. Histopathology. 2017;70:1138–46.
Arias-Stella JA 3rd, Benayed R, Oliva E, Young RH, Hoang LN, Lee CH, et al. Novel PLAG1 gene rearrangement distinguishes a subset of uterine myxoid leiomyosarcoma from other uterine myxoid mesenchymal tumors. Am J Surg Pathol. 2019;43:382–8.
Mäkinen N, Aavikko M, Heikkinen T, Taipale M, Taipale J, Koivisto-Korander R, et al. Exome sequencing of uterine leiomyosarcomas identifies frequent mutations in TP53, ATRX, and MED12. PLoS Genet. 2016;12:e1005850.
Croce S, Ribeiro A, Brulard C, Noel J-C, Amant F, Stoeckle E, et al. Uterine smooth muscle tumor analysis by comparative genomic hybridization: a useful diagnostic tool in challenging lesions. Mod Pathol. 2015;28:1001–10.
Croce S, Ducoulombier A, Ribeiro A, Lesluyes T, Noel JC, Amant F, et al. Genome profiling is an efficient tool to avoid the STUMP classification of uterine smooth muscle lesions: a comprehensive array-genomic hybridization analysis of 77 tumors. Mod Pathol. 2018;31:816–28.
Busca A, Parra-Herran C. Myxoid mesenchymal tumors of the uterus: an update on classification, definitions, and differential diagnosis. Adv Anat Pathol. 2017;24:354–61.
Li H, Durbin R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics. 2009;25:1754–60.
McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–303.
Koboldt DC, Zhang Q, Larson DE, Shen D, McLellan MD, Lin L, et al. VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 2012;22:568–76.
Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38:e164.
Kuilman T, Velds A, Kemper K, Ranzani M, Bombardelli L, Hoogstraat M, et al. CopywriteR: DNA copy number detection from off-target sequence data. Genome Biol. 2015;16:49.
Chalmers ZR, Connelly CF, Fabrizio D, Gay L, Ali SM, Ennis R, et al. Analysis of 100,000 human cancer genomes reveals the landscape of tumor mutational burden. Genome Med. 2017;9:34.
Rosenthal R, McGranahan N, Herrero J, Taylor BS, Swanton C. DeconstructSigs: delineating mutational processes in single tumors distinguishes DNA repair deficiencies and patterns of carcinoma evolution. Genome Biol. 2016;17:31.
Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SAJR, Behjati S, Biankin AV, et al. Signatures of mutational processes in human cancer. Nature. 2013;500:415–21.
Rausch T, Zichner T, Schlattl A, Stütz AM, Benes V, Korbel JO. DELLY: structural variant discovery by integrated paired-end and split-read analysis. Bioinformatics. 2012;28:i333–9.
Chang KTE, Goytain A, Tucker T, Karsan A, Lee CH, Nielsen TO, et al. Development and evaluation of a pan-sarcoma fusion gene detection assay using the NanoString nCounter platform. J Mol Diagn. 2018;20:63–77.
Marino-Enriquez A, Lauria A, Przybyl J, Ng TL, Kowalewska M, Debiec-Rychter M, et al. BCOR internal tandem duplication in high-grade uterine sarcomas. Am J Surg Pathol. 2018;42:335–41.
Ueno-Yokohata H, Okita H, Nakasato K, Akimoto S, Hata J-i, Koshinaga T, et al. Consistent in-frame internal tandem duplications of BCOR characterize clear cell sarcoma of the kidney. Nat Genet. 2015;47:861–3.
Chiang S, Lee C-H, Stewart CJR, Oliva E, Hoang LN, Ali RH, et al. BCOR is a robust diagnostic immunohistochemical marker of genetically diverse high-grade endometrial stromal sarcoma, including tumors exhibiting variant morphology. Mod Pathol. 2017;30:1251.
Kao YC, Sung YS, Zhang L, Huang SC, Argani P, Chung CT, et al. Recurrent BCOR internal tandem duplication and YWHAE-NUTM2B fusions in soft tissue undifferentiated round cell sarcoma of infancy: Overlapping genetic features with clear cell sarcoma of kidney. Am J Surg Pathol. 2016;40:1009–20.
Kenny C, Bausenwein S, Lazaro A, Furtwangler R, Gooskens SL, van den Heuvel Eibrink M, et al. Mutually exclusive BCOR internal tandem duplications and YWHAE-NUTM2 fusions in clear cell sarcoma of kidney: not the full story. J Pathol. 2016;238:617–20.
Karlsson J, Valind A, Gisselsson D. BCOR internal tandem duplication and YWHAE-NUTM2B/E fusion are mutually exclusive events in clear cell sarcoma of the kidney. Genes Chromosomes Cancer. 2016;55:120–3.
Tara S, Isshiki Y, Nakajima-Takagi Y, Oshima M, Aoyama K, Tanaka T, et al. Bcor insufficiency promotes initiation and progression of myelodysplastic syndrome. Blood. 2018;132:2470–83.
Damm F, Chesnais V, Nagata Y, Yoshida K, Scourzic L, Okuno Y, et al. BCOR and BCORL1 mutations in myelodysplastic syndromes and related disorders. Blood. 2013;122:3169–77.
Tanaka T, Nakajima-Takagi Y, Aoyama K, Tara S, Oshima M, Saraya A, et al. Internal deletion of BCOR reveals a tumor suppressor function for BCOR in T lymphocyte malignancies. J Exp Med. 2017;214:2901–13.
Acknowledgements
This work was supported in part by the SARC LMSARC research fund and the Brigham and Women’s Hospital Program in Precision Medicine (AME).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflicts of interest.
Ethical approval
This study was approved by the Research Ethics Board at Sunnybrook Health Sciences Centre, Toronto, ON, Canada.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Yoon, JY., Mariño-Enriquez, A., Stickle, N. et al. Myxoid smooth muscle neoplasia of the uterus: comprehensive analysis by next-generation sequencing and nucleic acid hybridization. Mod Pathol 32, 1688–1697 (2019). https://doi.org/10.1038/s41379-019-0299-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41379-019-0299-4
