
Abstract
Using a very high-resolution oligonucleotide array for copy number variant (CNV) screening of samples comprising schizophrenic patients, we detected a novel CNV within the critical region (NCBI36/hg18, Chr7: 158,630,410–158,719,410) previously shown to be associated with schizophrenia. We investigated the association between the novel CNV identified in the current study and schizophrenia. Three independent samples were used: (1) Screening set, 300 Japanese schizophrenic patients (53.28 ± 14.66 years); (2) Confirmation set, 531 schizophrenic patients (46.03 ± 12.15 years); and (3) 711 healthy controls (47.12 ± 11.03 years). All subjects enrolled in the study were Japanese. Chromosomal position was determined using fluorescence in situ hybridization. We identified a novel duplication within the region associated with schizophrenia identified on 7q36.3 that is adjacent to VIPR2 and is not associated with schizophrenia. In the Japanese population, the 35-kb region that harbors the common, novel CNV should be excluded from the region associated with schizophrenia on 7q36.3.
Similar content being viewed by others

Introduction
Schizophrenia is a chronic, debilitating illness characterized by impairments in cognition, affect and behaviour1. The Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition (DSM IV-TR)2 defines the essential features of schizophrenia as a mixture of characteristic signs and symptoms (both positive and negative) that have been present for a significant portion of time during a 1-month period (or for a shorter time if successfully treated), with some signs of the disorder persisting for at least 6 months. In this regard, positive refers to the presence of active symptoms including delusions and hallucinations. Negative symptoms refer to a loss, typically of emotions, speech, or motivation. Schizophrenic disorders exist on a continuum from mild to severe. The DSM IV-TR2 recognizes a number of different types, which include disorganized, catatonic, paranoid, schizophreniform, residual, schizoaffective, undifferentiated and not otherwise specified2. Schizophrenia is a relatively common disorder, with a lifetime prevalence of about 1%1. Although the overall sex ratio is almost equal, males tend to have an earlier onset than females, a finding accounted for by the later age of onset in those females who lack a family history of the disease3. Family history is the most important risk factor for schizophrenia, consistent with a genetic contribution to its etiology4. However, as with most mental disorders, the origins and mechanisms of schizophrenia are not fully understood.
Genetic factors influence human disorders by determining disease susceptibility or resistance5. Therefore, genetic studies can help pinpoint the exact molecular mechanism of a disease. Recent successes in the genetic mapping and molecular mechanism of the Mendelian traits have been remarkable, owning to the development of genome wide screening techniques6. As such, attention has been gradually shifting towards more complex, common, genetic disorders and traits that involve multiple genes and environmental effects, such as celiac disease7, diabetes8, rheumatoid arthritis9 and psychiatric disorders10. In this context, recurrent microdeletions at 1q21.111, 15q13.312 and 15q11.212, microduplications at 16p11.213 and copy number variations (CNVs) at other genomic loci14 have been shown to be associated with schizophrenia in large cohorts examined by CNV analyses and other molecular studies. Furthermore, duplication at chromosome 7q36.3, encompassing VIPR2, was implicated in schizophrenia for the first time in a recent report15. In a specific genome-wide association study of 8,290 patients with schizophrenia performed by Vacic et al.15, the authors found that 0.35% of these patients carry rare CNVs in the chromosomal locus 7q36.3. In contrast, these microduplications were much less frequent (0.03%) among the 7,431 healthy controls. All variants overlap with VIPR2 or lie within the noncoding subtelomeric region, <89 kb (NCBI36/hg18, Chr7: 158,630,410–158,719,410) from the transcriptional start site of VIPR2. This gene encodes the vasoactive intestinal peptide (VIP) receptor VPAC2, which is a G-protein-coupled receptor that is expressed in the suprachiasmatic nucleus, hippocampus, amygdala and hypothalamus16. VPAC2 binds VIP, activates cyclic AMP (cAMP)-signalling and PKA, regulates synaptic transmission in the hippocampus and promotes the proliferation of neural progenitor cells in the dentate gyrus17. Moreover, it has been shown that alteration in synaptic plasticity of hippocampal neurons may contribute to the symptoms observed in schizophrenic patients18. The aforementioned lines of evidence provide support for the role of VIPR2 as a candidate gene for schizophrenia from a biological point of view.
In the present study, by using a very high-resolution oligonucleotide array for CNV screening of samples from schizophrenic patients, we were able to detect CNV within the critical region (NCBI36/hg18, Chr7: 158,630,410–158,719,410) on 7q36.3 that was shown to be associated with schizophrenia by the Vacic et al. study15. Thus, the goal of the present study was to follow-up on the novel CNV that was previously detected in schizophrenic patients and further investigate any association between this CNV and schizophrenia.
Results
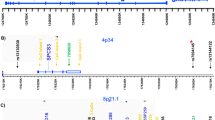
In the present study, we detected a smaller (35 kb) duplication (NCBI36/hg18, Chr7: 158,658,128–158,693,128) within the critical region identified by Vacic et al.15 (Figure 1). The observed frequency of the CNVs was ~2% and we did not detect any statistically significant difference between the patients and controls (Table 1). There was a 100% concordance rate between the custom NimbleGen 12 × 135,000 CGH arrays and the NimbleGen 3 × 720,000 CGH arrays or custom TaqMan copy number assay for the detection of the smaller (35 kb) duplication (NCBI36/hg18, Chr7: 158,658,128–158,693, 128) within the critical region (NCBI36/hg18, Chr7: 158,630,410–158,719,410) previously identified by Vacic et al.15.

High-resolution aCGH data.
Probe intensity ratios. The orange box (NCBI36/hg18, Chr7: 158,658,128–158,693,128) represents CNVs detected in the present study and the purple box (NCBI36/hg18, Chr7: 158,630,410–158,719,410) represents the region revealed by Vacic et al. to show an association peak in a schizophrenia group. Coordinates are based on the NCBI36 build.
In metaphase cells, all duplication-specific FISH signals localize to the subtelomeric region of 7q, confirming that the duplications lie adjacent to each other in the 7q36.3 region (Figure 2). In addition, NS102 exhibited two signals, one of which had a higher intensity compared to that of the other. This suggests that there is unilocus duplication in the VIPR2 promoter region. During the orientation analysis, an amplicon was detected by electrophoresis only in samples with duplication, which indicates that there is a head-to-tail orientation of the repeated DNA fragment (Supplementary Figure 1). Additionally, sequence analysis of the repeat junction revealed that all samples with duplication shared exactly the same sequence within the junction region (Supplementary Figure 2). Based on the panel of 8 SNPs in the CNV region that were detected in the current study, we did not observe the existence of any common haplotype (Supplementary Table 2).

Tandem duplications of 7q36.3 confirmed in two patients by fluorescence in situ hybridization (FISH).
7p-green (arrowheads) and 7q-red (arrows, CNV specific). Left NS102 (3 copies); Right NS004 (2 copies). Cytogenetic confirmation was obtained for two samples with and without duplication of VIPR2. Probes for duplicated region were produced by long range PCR. The subtelomeric probe, 7p-green (Abbott Molecular), was used as a reference. Hybridizations were performed according to the manufacturer's protocols.
Discussion
Using samples from only Japanese subjects, we identified a smaller 35-kb (NCBI36/hg18, Chr7: 158,658,128–158,693,128) common (>1%) duplication within the region that Vacic et al.15 previously showed was associated with schizophrenia (NCBI36/hg18, Chr7: 158,630,410–158,719,410). However, the common duplication that we detected in the present study was not found to be associated with schizophrenia. It is of note that these results may be specific to Japanese subjects and therefore further studies involving other population groups will need to be undertaken. We have experimentally confirmed that the common CNV detected in the current study was located adjacent to VIPR2. Our analysis of the breakpoint junctions at the sequence level showed there was no difference among the CNV carriers. On the basis of the 2-bp microhomology found at this junction (Supplementary Figure 2), we speculate that duplication formation occurs by the FoSTeS (fork stalling and template switching)/MMBIR (microhomology mediated break-induced replication) mechanism previously proposed by Lupski's research group23. In addition, we did not find any sequence motif that was characteristic for the breakpoint of recurrent rearrangements at the junction region. Although the junction sequence was exactly the same in all of the subjects in whom the 35-kb CNV was detected, we could not confirm that these subjects shared any common haplotype. Differences in the haplotype among the duplication carriers were likely due to the high recombination rate that occurs at the subtelomeric region24. Regarding the origin of the CNV that was identified in the current study, our observation of the same breakpoint junction sequence that was observed in the CNV carriers is highly suggestive of a common ancestral founder.
The main limitation of the current study was that we examined a much smaller number of samples as compared to the Vacic et al. study15. It is of note that the frequency of the common 35-kb CNV detected in the current study was 2% and thus with our current sample size of 300 schizophrenic patients, it was large enough to capture the variation. Regarding the individual with 4 copies, we do not have any data indicating whether the individual is a homozygote of duplication or is a carrier of triplication. This point should be considered as another limitation of the current study. The results of the current study do suggest that in case of a duplication event on 7q36.3, the relevant region is not the VIPR2 promoter (as has been suggested by Vacic et al.15), but rather suggest that it is the VIPR2 gene region. In addition, the 2-bp microhomology in the promoter region of VIPR2 may be associated with the relative meiotic instability of the region harboring the common CNV that is adjacent to the VIPR2 gene25. This in turn may give rise to the larger VIPR2 duplications that were shown to be associated with schizophrenia with an odds ratio of 4.014.
It is interesting that our findings demonstrated that CNV was detected in our study in contrast to the previous reports by both Vacic et al.15 and Beri et al.26. Moreover, CNV has not been listed in the database of genomic variants27. Although the CNV detected by our group may be specific to Japanese populations, further studies should be undertaken to ensure comprehensive characterization of the region surrounding the VIPR2 gene. In addition, to address the question regarding the origin of the CNV detected in the current study, it is necessary to perform family analysis of the carriers and determine whether CNV is a de novo event, or if it is transmitted from the parents. In conclusion, the 35-kb region that harbors the common CNV in the Japanese population should be excluded from the region of the association peak in the schizophrenia group reported in the Vacic et al. study15.
Methods
Three independent samples were used in the current study: (1) Screening set, 300 Japanese patients suffering from schizophrenia (53.28 ± 14.66 years); (2) Confirmation set, 531 patients suffering from schizophrenia (46.03 ± 12.15 years); and (3) 711 healthy control subjects (47.12 ± 11.03 years). All schizophrenic patients met the current DSM IV-TR criteria2, which was reflected by consensual diagnosis of two experienced psychiatrists. Prior to inclusion in the control set, subjects were screened on the basis of a brief diagnostic interview. Detailed characterization and psychiatric assessment of the subjects is available elsewhere19. All subjects enrolled in the study were Japanese and provided written informed consent prior to the study. Venous blood was drawn from each subject and genomic DNA was extracted according to the standard phenol/chloroform method. Comparative genomic hybridization of DNA was performed using the high-resolution NimbleGen (Roche NimbleGen, Inc., U.S.) CGH array (3 × 720,000 or 12 × 135,000). Labeling and hybridization of patient (test) and sex-matched commercial (Promega Corporation, U.S.) reference DNA was performed according to the manufacturer's protocols. Test and reference DNA were labeled by Cy3- and Cy5-labeled random primers, respectively and were combined and hybridized to the array for 40–72 h. Arrays were washed in four steps, as indicated in the protocol. Two-color scanning was performed using a NimbleGen MS 200 microarray scanner. Acquisition of the microarray images was performed with NimbleGen MS 200 software. Data extraction, analysis and visualization were done using NimbleScan version 2.4 software. CNV calling was performed using NEXUS software. The FASST2 Segmentation Algorithm, a Hidden Markov Model (HMM) based approach, was used to make copy number calls. The FASST2 algorithm, unlike other common HMM methods for copy number estimation, does not aim to estimate the copy number state at each probe, but uses many states to cover more possibilities, such as mosaic events. These state values are then used to make calls based on a log ratio threshold. The significance threshold for segmentation was set at 10−6 and also required a minimum of three probes per segment. The log ratio thresholds for single copy gain and single copy loss were set at 0.3 and −0.3, respectively. The log ratio thresholds for the gain of two or more copies and homozygous loss were set at 0.9 and −0.9 respectively.
Custom TaqMan copy number assay was specifically designed to interrogate a duplication region (NCBI36/hg18, Chr7: 158,630,410–158,719,410) without interspersed repeats, low complexity or a homologous DNA sequence. A TaqMan copy number assay for RNase P was used as a reference. Experiments were carried out on four technical replicates according to the manufacturer's protocol. CNV typing of the screening sample was performed using Roche NimbleGen, Inc. CGH array 3 × 720,000, while confirmation of the sample was performed using the TaqMan copy number assay. Sixteen randomly selected duplication events (both in the screening and confirmation samples) were validated using custom NimbleGen 12 × 135,000 CGH arrays (Roche NimbleGen, Inc., U.S.) covering the region (NCBI36/hg18, Chr7: 158,630,410–158,719,410) implicated in the Vacic et al. study15, with an average of one probe per 500 bp. P values derived from association analysis were based on Fisher's exact test.
We performed PCR based analysis to determine the orientation of the detected duplications. We designed forward and reversed primers to align with the region of the duplication junction (F: 5′-TGTGGATTCCCTTCAGAGGCGAC-3′, R: 5′-CATTCTTCAGCCCATGGAGTCATC-3′) (Supplementary Figure 1). Cytogenetic confirmation was obtained for two samples with and without duplication of VIPR2. Probes for duplicated region were produced by long range PCR (NCBI36/hg18, Chr7: 158,658,128–158,693,128). Subtelomeric probe, 7p-green (Abbott Molecular, U.S.), was used as a reference. Hybridizations were performed according to the manufacturer's protocols.
Haplotypes were estimated using the statistical software package PHASE version 3.4.1 (http://www.stat.washington.edu/stephens/)20,21,22. This program is based on a Bayesian statistical method using coalescent-based models that infers phases at loci from unphased genotype data for a sample of unrelated individuals20. The algorithm uses a flexible model for the decay of linkage disequilibrium with distance and explicitly incorporates an assumption about the recombination rate variation. PHASE uses Gibbs sampling, a Markov-Chain Monte Carlo algorithm for the estimation of the posterior distribution. Hence, the individual haplotype can be estimated from the posterior distribution by choosing the most likely haplotype reconstruction for each individual. Using the extension for unrelated individuals, we used the default settings to infer the haplotypes from the genotype data19 of the 8 SNPs (Supplementary Table 1) surrounding the duplication in sample that comprised 517 subjects (7 with a structural variant detected in the current study). Estimates of the sample haplotype frequencies together with their standard deviation, a list of the most likely pairs of haplotypes for each individual together with their probability and the estimates of recombination parameters in the region, were calculated using the same software.
References
van Os, J. & Kapur, S. Schizophrenia. Lancet 374, 635–45 (2009).
Author. Diagnostic and statistical manual of mental disorders (4th ed., text rev.). (American Psychiatric Association., Washington, DC, 2000).
Loranger, A. W. Sex difference in age at onset of schizophrenia. Arch Gen Psychiatry 41, 157–61 (1984).
Sullivan, P. F. The genetics of schizophrenia. PLoS Med 2, e212 (2005).
Manolio, T. A. et al. Finding the missing heritability of complex diseases. Nature 461, 747–53 (2009).
Chakravarti, A. Genomic contributions to Mendelian disease. Genome Res 21, 643–4 (2011).
Trynka, G. et al. Dense genotyping identifies and localizes multiple common and rare variant association signals in celiac disease. Nat Genet 43, 1193–201 (2011).
Todd, J. A. et al. Robust associations of four new chromosome regions from genome-wide analyses of type 1 diabetes. Nat Genet 39, 857–64 (2007).
Thomson, W. et al. Rheumatoid arthritis association at 6q23. Nat Genet 39, 1431–3 (2007).
Hamshere, M. L. et al. Genome-wide significant associations in schizophrenia to ITIH3/4, CACNA1C and SDCCAG8 and extensive replication of associations reported by the Schizophrenia PGC. Mol Psychiatry 18, 708–712 (2013).
Mefford, H. C. et al. A method for rapid, targeted CNV genotyping identifies rare variants associated with neurocognitive disease. Genome Res 19, 1579–85 (2009).
Stefansson, H. et al. Large recurrent microdeletions associated with schizophrenia. Nature 455, 232–6 (2008).
McCarthy, S. E. et al. Microduplications of 16p11.2 are associated with schizophrenia. Nat Genet 41, 1223–7 (2009).
Levinson, D. F. et al. Copy number variants in schizophrenia: confirmation of five previous findings and new evidence for 3q29 microdeletions and VIPR2 duplications. Am J Psychiatry 168, 302–16 (2011).
Vacic, V. et al. Duplications of the neuropeptide receptor gene VIPR2 confer significant risk for schizophrenia. Nature 471, 499–503 (2011).
Sheward, W. J., Lutz, E. M. & Harmar, A. J. The distribution of vasoactive intestinal peptide2 receptor messenger RNA in the rat brain and pituitary gland as assessed by in situ hybridization. Neuroscience 67, 409–18 (1995).
Zaben, M. et al. The neurotransmitter VIP expands the pool of symmetrically dividing postnatal dentate gyrus precursors via VPAC2 receptors or directs them toward a neuronal fate via VPAC1 receptors. Stem Cells 27, 2539–51 (2009).
Sanderson, T. M. et al. Alterations in hippocampal excitability, synaptic transmission and synaptic plasticity in a neurodevelopmental model of schizophrenia. Neuropharmacology 62, 1349–58 (2012).
Ikeda, M. et al. Genome-wide association study of schizophrenia in a Japanese population. Biol Psychiatry 69, 472–8 (2011).
Stephens, M. & Donnelly, P. A comparison of bayesian methods for haplotype reconstruction from population genotype data. Am J Hum Genet 73, 1162–9 (2003).
Stephens, M. & Scheet, P. Accounting for decay of linkage disequilibrium in haplotype inference and missing-data imputation. Am J Hum Genet 76, 449–62 (2005).
Stephens, M., Smith, N. J. & Donnelly, P. A new statistical method for haplotype reconstruction from population data. Am J Hum Genet 68, 978–89 (2001).
Hastings, P. J., Ira, G. & Lupski, J. R. A microhomology-mediated break-induced replication model for the origin of human copy number variation. PLoS Genet 5, e1000327 (2009).
Nachman, M. W. Variation in recombination rate across the genome: evidence and implications. Curr Opin Genet Dev 12, 657–63 (2002).
Warren, S. T. & Nelson, D. L. Trinucleotide repeat expansions in neurological disease. Curr Opin Neurobiol 3, 752–9 (1993).
Beri, S., Bonaglia, M. C. & Giorda, R. Low-copy repeats at the human VIPR2 gene predispose to recurrent and nonrecurrent rearrangements. Eur J Hum Genet 21, 757–61 (2013).
Zhang, J., Feuk, L., Duggan, G. E., Khaja, R. & Scherer, S. W. Development of bioinformatics resources for display and analysis of copy number and other structural variants in the human genome. Cytogenet Genome Res 115, 205–14 (2006).
Acknowledgements
Funding for this study was provided by research grants from the Ministry of Education, Culture, Sports, Science and Technology of Japan; the Ministry of Health, Labor and Welfare of Japan; Grant-in-Aid for “Integrated research on neuropsychiatric disorders” carried out under the Strategic Research Program for Brain Sciences by the Ministry of Education, Culture, Sports, Science and Technology of Japan; Grant-in-Aid for Scientific Research on Innovative Areas (Comprehensive Brain Science Network) from the Ministry of Education, Science, Sports and Culture of Japan; The Academic Frontier Project for Private Universities, Comparative Cognitive Science Institutes, Meijo University; the Core Research for Evolutional Science and Technology and SENSHIN Medical Research.
Author information
Authors and Affiliations
Contributions
B.A., I.K., T.O. and N.O. designed the study and wrote the protocol. B.A., I.K., T.O., N.I., H.K. and N.O. performed the literature review. B.A., I.K. and T.O. made and managed the sample database. B.A., I.K., T.O., M.I., S.K., Y.N., A.Y., T.K., S.I., H.K., N.I. and N.O. collected and managed the genome samples. B.A., I.K., T.O., M.I., S.K., Y.N., A.Y., T.K. and S.I. conducted the statistical analysis. B.A., I.K., T.O., M.I., S.K., Y.N., A.Y., T.K., S.I., H.K., N.I. and N.O. interpreted and discussed the results. B.A., I.K., T.O., M.I., H.K., N.I. and N.O. wrote the manuscript and edited the final manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Supplementary Information
Supplementary data
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Aleksic, B., Kushima, I., Ohye, T. et al. Definition and refinement of the 7q36.3 duplication region associated with schizophrenia. Sci Rep 3, 2587 (2013). https://doi.org/10.1038/srep02587
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep02587
This article is cited by
-
Role of cryptic rearrangements of human chromosomes in the aetiology of schizophrenia
Journal of Genetics (2023)
-
Genetic Variants Within Key Nodes of the Cascade of Antipsychotic Mechanisms: Effects on Antipsychotic Response and Schizophrenia Psychopathology in a Naturalistic Treatment Setting in Two Independent Korean and Italian Samples
Advances in Therapy (2017)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
