
Abstract
Evolution of H1N1 influenza A outbreaks of the past 100 years is interesting and significantly complex and details of H1N1 genetic drift remains unknown. Here we investigated the clinical characteristics and immune cross-reactivity of significant historical H1N1 strains. We infected ferrets with H1N1 strains from 1943, 1947, 1977, 1986, 1999 and 2009 and showed each produced a unique clinical signature. We found significant cross-reactivity between viruses with similar HA sequences. Interestingly, A/FortMonmouth/1/1947 antisera cross-reacted with A/USSR/90/1977 virus, thought to be a 1947 resurfaced virus. Importantly, our immunological data that didn't show cross-reactivity can be extrapolated to failure of past H1N1 influenza vaccines, ie. 1947, 1986 and 2009. Together, our results help to elucidate H1N1 immuno-genetic alterations that occurred in the past 100 years and immune responses caused by H1N1 evolution. This work will facilitate development of future influenza therapeutics and prophylactics such as influenza vaccines.
Similar content being viewed by others


Introduction
The major challenge in regard to influenza surveillance and management is the propensity of the influenza virus to mutate, altering its immunogenic properties thereby allowing it to evade immune recognition and cause disease. Influenza A is classified according to the specific combination of its two surface molecules, hemagglutinin (HA) (H1 – H17) and neuraminidase (NA) (N1 – N9) isotypes1,2,3 and its diversity is attributed by two mechanisms: genetic mutation or by gene reassortment4,5. Typically genetic mutation is responsible for seasonal influenza outbreaks and the emergence of influenza pandemics is a consequence of gene reassortment among different strains and subtypes4,5,6. Since the extensively documented influenza pandemic in 1918–20, there have been a total of five influenza pandemics that have resulted in millions of deaths worldwide, where the fatality rate has reached more than 2.5% as in the case of 1918 pandemic5,7. In 1957, H2N2 surfaced and replaced H1N1 in the human population until 1977 when H1N1 resurfaced, leading to co-circulation of the 2 influenza subtypes. Compared to H3N28, the H1N1 subtype is believed to have a lower rate of antigenic drift which is associated with a smaller amount of mutations leading to amino acid changes9. Importantly the evolutionary behaviours of H1N1 and H3N2 are divergent leading to a dynamic and ever changing influenza climate in the human population where one virus subtype typically dominates over the other8,9. Throughout the 100 year history of H1N1, the specific clinical parameters and immunogenic response to the genetic drift of H1N1 remains to be clarified.
Subsequent to the 1918 H1N1 pandemic were several H1N1 epidemics occurring from the 1920's to the late 1950's9,10,11 which was followed by a 20 year disappearance and a re-emergence in the 1970's12. In 1947 a significant antigenic change transpired in the H1N1 virus creating strain distinct from previous 1943 viruses13. The new H1N1 virus termed “A-Prime” was relatively mild although widespread (a pseudopandemic) and hypothesized to be a reassortant from two distinct H1N1 strains9,13,14. An unusual H1N1 virus emerged in 1951 that was associated with severe disease: also thought to be a reassortant virus with genes from novel viruses and older H1N1 segments from the 1940's9. Succeeding a 20 year H1N1 disappearance, two H1N1 epidemics of interest came about, including the 1977 children's pandemic and a swine flu epidemic in 1976 that was feared to have pandemic potential and led to a massive public vaccination strategy14. The 1977 epidemic had limited infectivity to the immunologically naïve younger population (persons <25 years of age) which is thought to be due to the similar circulating H1N1 viruses of the 1950's14.
In 2009 a substantial change in the H1N1 virus occurred, unlike previous modifications, which allowed the virus to spread rapidly throughout the globe and prompted the WHO to declare a pandemic on 11 June 2009 15. Genomic analysis determined the virus was of swine origin and contained a triple reassortant of swine, human and avian influenza A genes16. Unlike previous contemporary seasonal influenza outbreaks, the 2009 H1N1pdm had age-related disparities in the frequency and severity of infection where the older age groups were less susceptible to the disease17,18. Furthermore, these differences in age-related severity are hypothesized to be due to previous exposure to older H1N1 viruses with similar antigenic epitopes17,18.
After influenza exposure, the body generates antibodies against the specific influenza strain it has encountered. Anti-HA production is often associated with immunity to the same or homologous influenza strains and some antibodies have virus neutralizing ability blocking viral entry to the host cell1,19. Importantly, previous exposure to influenza viruses influences how the body will respond to a subsequent influenza infection of the same or different genetic subtype20. To date, little is known about the immune and clinical response to H1N1 influenza viruses of the past 100 years and how the H1N1 subtype genetic drift and shift affected immune cross-reactivity. Previously Nelson et al., conducted a large scale genetic analysis of 71 H1N1 sequences to determine the evolutionary history of this virus since 1918 9. In our present study, we investigated the clinical characteristics and immune cross-reactivity of significant H1N1 influenza strains in the past 100 years in ferrets to determine the immunogenicity of important H1N1 viruses. We infected ferrets with historical H1N1 strains from 1943, 1947, 1977, 1986, 1999 and 2009 and monitored them for a 14-day time course to determine the clinical picture of each influenza strain infection and immune cross-reactivity. Our findings clarify the influenza clinical picture of historical H1N1 strains and the help to elucidate the antigenic alterations of H1N1 that occurred in the past 100 years. The work here will facilitate the understanding of the immune response toward H1N1 and with future work may aid the development of future influenza therapeutics and anti-viral treatments.
Results
Ferrets Infected with historical H1N1 strains show mild clinical symptoms
The ferret, Mustela putorius furo, is a superlative animal model for respiratory infections and the influenza ferret infectome has recently been published21,22,23,24,25,26,27,28,29. Ferrets show respiratory illness similar to humans and clinical features of disease are easily observed where fevers can persist days following infection of viruses such as 2009 H1N1pdm influenza21,23,30. As well as fever, nasal discharge and sneezing can also be observed in animals infected with influenza viruses21. Although the genetic evolution of H1N1 viruses between 1918 and 2008 has been studied9, the clinical features of these viruses have not been previously investigated and compared including the most recent 2009 H1N1 pandemic virus. Here we used ferrets to experimentally monitor and compare the clinical immune responses during infection with human historical H1N1 influenza strains.
We infected ferrets with six influenza A/H1N1 strains; A/AA/Marton/43 (Marton/43), A/FortMonmouth/1/1947 (FM/47), A/USSR/90/1977 (USSR/77), A/Taiwan/1/1986 (Taiwan/86), A/NewCaledonia/20/1999 (NCal/99) and A/NewYork/18/2009 (NY/09). These influenza A viruses were chosen due to their emergence and influence in H1N1 genetic history ( Fig. 1 , strains used in this study are marked with an asterisks) as covered in the introduction. Following infection, ferrets were monitored for body temperature, weight, inactivity level, sneezing and nasal discharge from each group were observed daily until Day 14 post-infection (pI). Infection by all strains produced an increase in temperature; the normal range for ferret temperature is indicated by the shaded area of each graph23 ( Supplementary Fig. S1a ). The pandemic H1N1 strain NY/09 induced the greatest fever on Day 2 to a temperature of 104% (of baseline). NCal/99 and Marton/43 infection also caused a high temperature of 103% from baseline, which peaked on Day 1 and Day 2 pI, respectively ( Table 1 ). USSR/77 and Taiwan/86 had moderate fevers and FM/47 had the smallest increase in temperature reaching only 101% above baseline ( Table 1 ).

History of H1N1 influenza epidemics and pandemics.
Analysis of weight loss showed that animals infected with of all viruses except pandemic NY/09 were able to recover to original weight or greater following infection ( Supplementary Fig. S1b and Table 1 ). NY/09 infected ferrets had the most significant weight loss compared to normal weight fluctuations (shaded area)23 which peaked at 91% of baseline weight on Day 6 and Day 7. USSR/77 and NCal/99 reached less than 95% and 95% of baseline weight, respectively, on Day 2 pI ( Table 1 ). Infection with Taiwan/86 produced the smallest amount of weight loss and animals infected with FM/47 did not lose any weight at all ( Table 1 ).
Secondary clinical signs were also measured and analysed for all infections, including nasal discharge, sneezing and inactivity level ( Supplementary Table S1 ). USSR/77 infected ferrets had the highest amount of nasal discharge and NY/09 ferrets had the greatest amount of sneezing and lethargy. Taken together, analysis of the complete clinical signs for each H1N1 strain infection suggested a unique clinical picture for each strain: NY/09 infection had the most severe with significant increase in temperature and an unrecovered weight loss compared to mildest strain, FM/47, which did not produce any weight loss and only a slight increase in temperature.
Analysis of HA immunogenicity in historical H1N1 strains
The HA protein, a homotrimeric protein that functions in viral entry into host cells31 and antibodies reactive toward HA have been associated with host resistance and a decrease in disease severity32,33,34. It has been shown that 60% of antibodies produced during an influenza infection are reactive toward the HA protein35. After determining the clinical signatures of ferrets infected with historical and contemporary H1N1 viruses, we went on to analyze the aspects of H1N1 HA immunogenicity.
The Hemagglutination Inhibition (HI) assay, is a common assay used to determine the reactivity and/or amount of antibody to the HA protein produced during a viral infection and the cross-reactivity of antibodies raised toward these specific historically important H1N1 viruses has not been investigated. To determine the cross-reactivity produced by infection among the historical and contemporary influenza strains, we used the harvested antisera from each ferret strain infection and perform HI assays with our panel of H1N1 viruses; PR/34 (A/PuertoRico/8/1934), Marton/43, FM/47, USSR/77, Taiwan/86, NCal/99, NY/09 and three antisera raised toward viruses from previous studies SI/06 (A/SolomonIslands/3/2006), Bris/07 (A/Brisbane/59/2007), Cal/09 (A/California/07/2009) in the analysis23 ( Supplementary Table S2 ).
Interestingly, ferret anti-sera toward the Marton/43 virus showed HI with its own virus and with the older PR/34 but not with its nearest chronological neighbour FM/47 ( Fig. 2a ). FM/47 and USSR/77 antisera recognized its respective virus and each other ( Fig. 2b and c ). Taiwan/86 and 2009 H1N1pdm (NY/09 or Cal/09) antisera both only reacted with their respective viruses ( Fig. 2d and h ). NCal/99 antisera only showed HI with its own NCal/99 virus ( Fig. 2e ). Unexpectedly, the SI/06 and Bris/07 antisera were able to inhibit the NCal/99 virus ( Fig. 2f and g ). Furthermore, SI/06 antisera also cross-reacted with the Bris/07 virus and the Bris/07 antisera recognized the SI/06 virus. Taken together, these results showed antigenic cross-reactivity to infection among historical and contemporary H1N1 influenza strains. This data suggested similarities of the host immune response between Marton/43 and PR/34, FM/47 and USSR/77 and NCal/99 with both SI/06 and Bris/07 but despairing responses in infection with Taiwan/86 and Cal/09.

Hemagglutination Inhibition (HI) analysis of H1N1 strain antisera shows cross-reactivity toward related strains.
Ferrets were infected with various H1N1 influenza strains Marton/43, FM/47, USSR/77, Taiwan/86, NCal/99, SI/06, Bris/07 and NY/09 and antisera was taken at 15 days pI. The antisera were used to measure HA specific antibody induction using hemagglutination inhibition assay against an H1N1 virus panel: PR/34, Marton/43, FM/47, USSR/77, Taiwan/86, NCal/99, SI/06, Bris/07 and NY/09 or Cal/09.
We also analysed the HI ability of WHO control antisera against our H1N1 virus panel ( Supplementary Table S3 ) and the WHO control viruses versus the H1N1 panel of antisera ( Supplementary Table S4 ). Expectedly, NCal/99 and SI/06 both reacted with the WHO seasonal H1N1 antisera and NY/09 reacted with the 2009 H1N1pdm antisera. As well, HI was seen for only the WHO control seasonal H1N1 virus with NCal/99, SI/06 and Bris/07 antisera and the WHO control 2009 H1N1pdm with NY/09 ( Supplementary Table S4 ). Importantly, no detection was observed for the Tawain/86 virus and earlier viruses with the WHO H1N1 seasonal control. Taken together, this work suggested that the WHO seasonal H1N1 antisera is only capable of detecting infection from recent seasonal H1N1 strains.
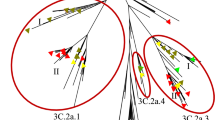
We investigated the phylogenetic relationship and sequence homology of the HA gene among the H1N1 viruses used in this study and included other strains implicated in important historical H1N1 outbreaks. The temporal structure of phylogenetic tree was comprised of 3 groups separated by long braches ( Fig. 3a ). The 1918 pandemic strains and 2009 pandemic isolates each formed their own distinct cluster. The middle cluster contained 10 epidemic isolates. Within this cluster, FM/47 was closer to USSR/77 than Marton/43 which was more similar to PR/34. The 3 latest strains of seasonal H1N1, NCal/99, SI/06 and Bris/07, were positioned as a clade which was separated from the early stains by a long branch and clustal alignment of these three viruses ( Supplementary Fig. S2 ) showed several amino acid changes from NCal/99 and conserved between SI/06 and Bris/07: A3V, T99K, Y111H, K157E, V182A, R224K, W268R, T283N and N489D. We calculated the protein homology by using the most variable region of the HA gene (HA135-295) which covers most of the viral surface and sequence change in this area has a strong impact in immunogenicity36. We found that 94% and higher homology scores led to HI cross-reactivity in all the cases. On the other hand, cross-reactivity with lower homologies of 91-93% may or may not take place ( Fig. 3b ). Taken together, our results suggested an important relationship and cross-reactivity between FM/47 and USSR/77 as well as the NCal/99, SI/06 and Bris/07 group that can be related to the genetic HA sequence. Further analysis of other influenza genes may also be important to the understanding of H1N1 evolution.

Phylogenetic relationship and homology of the influenza A H1N1 HA gene.
Phylogenetic relationship and homology among the hemagglutinin protein from different influenza strains are depicted. Evolutionary relationships among historical influenza H1N1 strains were conducted in MEGA5 using the Maximum Likelihood method and 500 bootstrap replications (a). Antibody cross-reactivity (determined by HI) among influenza strains is indicated by using the same colors. The numerical values displayed next to the branches indicate the percentage of bootstrap replicates in which the associated sequences clustered together. Hemagglutinin protein sequence homology among several influenza strains (b). Influenza strains were grouped according to the HI cross-reactivity observed in the serum from previously-infected ferrets (groups A through F). Alignments of protein sequences were performed using the highly variable HA135-295 region, which belongs to the HA-RBD area.
Discussion
The H1N1 influenza subtype is a significant viral agent affecting public health that has been responsible for 3 pandemics and several influenza epidemics globally. Here we investigated the clinical response and subsequent immunogenicity following infection with historically and contemporarily relevant H1N1 strains in ferrets. Importantly, we showed that infection with each strain produced a unique clinical signature which may be indicative of disease that affected humans. We also found significant cross-reactivity between viruses with similar HA aa sequences which can be extrapolated to protection or no protection following a subsequent infection with another H1N1 strain. These results are the first to compare the clinical signs and the antigenic relatedness of systematic infection of this panel of evolutionary important H1N1 viruses in a single study. These results add to the understanding of how the immune system responds to an evolving virus.
In the current study we analyzed the clinical parameters of evolutionary H1N1 strain infection in ferrets which has not been previously compared experimentally. Our results of the evolutionary H1N1 strains showed that infection with the FM/47 resulted in the mildest clinical disease. Interestingly, historical accounts of the 1947 H1N1 outbreak have indicated the virus to cause mild clinical symptoms and have a low death rate14. These records support our findings, where animals infected with FM/47 had the lowest percent nasal discharge, mildest fever and was the only virus infection which did not lead to weight loss. Furthermore, USSR/77 infection produced a moderate disease comparable to the reports of a mild clinical disease37. Our analysis of the clinical course of Taiwan/86 infection in ferrets showed a moderate disease consisting of a mild biphasic temperature increase and minimal weight loss. These observations were in agreement with clinical disease reported during the 1986 naval base outbreak of influenza which there was no deaths but patients described fever, non-productive cough, sore throat and myalgia38. Furthermore, in accordance with our NY/09 data which had the most significant temperature fluctuations and weight loss, analysis by Shrestha and colleagues demonstrated that H1N1pdm was more severe than the previous circulating seasonal influenza viruses39.
The influenza virus was first identified in 1932 and since then the epidemiology and phylogentic analysis of subsequent influenza epidemics and pandemics have been extensively investigated9,14,40. Importantly, our results build on the previous investigations as we report the immunogenic evolution of the HA protein of significant pandemic and epidemic strains. The epidemic isolates examined by our phylogenetic analysis showed the 1918 and H1N1pdm 2009 strains to comprise each their own cluster and the seasonal strains ranging from 1934 to 2007 to cluster together. As expected by chronological analysis, our results showed Marton/43 to cross-react with its predecessor PR/34 which was in agreement with the phylogenetic analysis that placed the HA of Marton/43 close to PR/34 in the tree.
Extensive phylogenetic analysis preformed by Nelson MI and colleagues suggested the 1947 strain to be a reassortant from two previous H1N1 viruses combining the PB1, NA and M segements from a 1943 virus with the PB2, PA, HA, NP and NS segments from an unknown H1N1 virus9. Furthermore, they showed that the HA1 region of the 1947 HA was significantly different from the HA of the earlier 1940's strains9. In accordance with this analysis, our results showed that the FM/47 antisera did not inhibit hemagglutination with the earlier PR/34 and Marton/43 viruses and the Marton/43 antisera did not cross-react with the FM/47 virus. The inability for immunity induced by Marton/43 virus infection to target the FM/47 virus compliments the influenza vaccine performance in the human population of the 1940's as well as previously published studies41. Specifically, a related 1943 H1N1 virus strain (A/weiss/43), as well as A/PR/8/34 and B/Lee/40 were used in the influenza vaccine preparation which was completely ineffective during the ’47 influenza outbreak14,41. Although FM/47 antisera did not recognize the earlier H1N1 strains, it did cross-react with the HA molecule of the USSR/77 virus as did the USSR/77 antisera with the FM/47 virus. In support of these findings, the 1977 epidemic was thought to be the resurfacing of the FM/47 strain9. The USSR/77 outbreak was known as ‘The Children's Pandemic' since only persons younger than 26 years old were affected14. During this time, older individuals were considered primed for the 1977 outbreak strain by previous infections from the 40's and 50's37 which is supported by our immunological evidence. Together, our results, the phylogenetic analysis and the epidemiological records suggest the triggering of a unique immune response following infection with Marton/43 and FM/47 and a similar response from infection with the related strains FM/47 and USSR/77.
In the fall of 1986 a significant outbreak of influenza occurred on a US Naval Base which was found to be caused by strains that had been circulating in Asia since the spring of that year38,42,43,44. Taiwan/86 was isolated from this outbreak and genomic analysis of its HA have suggested that the Taiwan/86 virus had evolved from viruses circulating in the early 1980‘s in Hong Kong45. Furthermore, it has also been suggested that this virus was the product of a reassortment event that had occurred between two H1N1 viruses9. Our results showed that the Taiwan/86 strain shared lower homology to 99, 06 and 07 viruses seen on our phylogenetic tree. The immunogenic findings showed antisera produced from ferret infection with Taiwan/86 was not able to inhibit hemagglutination with the other viruses on our virus panel, which compliments the literature describing the 1986 strain. The influenza vaccine in 1986 included the A/Chile/1/83 influenza strain38. Interestingly, during the 1986 US Naval Base outbreak, the soldiers who were vaccinated were poorly protected from infection with Taiwan/198638. Furthermore, it was also shown that antibodies produced by vaccination with the A/Chile/1/83 vaccine preparation were not able to provide hemagglutination inhibition toward the newly arisen Taiwan/86 and Taiwan/86-like viruses44. From this evidence, it was then recommended that the Taiwan/86 strain be included in the subsequent influenza monovalent vaccine preparation42,44. Our findings together with the previous reports on the Taiwan/86 outbreak suggest that the 1986 viruses were an antigenically unique group of strains that induced a specific immune response important when considering the evolution of H1N1 immunity.
It can be argued that the H1N1 virus underwent minor genetic drift in the 10 year span from 1999 to 2009 which can be evidenced by the consistent inclusion of the NCal/99 in the influenza vaccine preparation9,46. Our genetic analysis showed the three contemporary H1N1 seasonal viruses, NCal/99, Bris/07 and SI/06, to be proximal to each other on the phylogenetic tree with a long separation from the previous Taiwan/86 virus and in a distinct cluster from the 2009 H1N1 pandemic viruses. Moreover, Bris/07 and SI/06 were closer to each other than they were to NCal/99. Interestingly, the antisera generated by infection with NCal/99 did not cross-react with the other two contemporary seasonal H1N1 viruses although the SI/06 and Bris/07 antisera recognized the NCal/09 virus. These results suggested there to be an amino acid change in Bris/07 and SI/06 from NCal/99 that may have resulted in a new antigenic site that was not originally present in NCal/99 strain. To investigate this contention we generated a clustal alignment of these viruses which showed numerous amino acid changes between Bris/07 and SI/06 sequences with NCal/99. These results may indicate the gradual yet still present drift of the H1N1 virus in this time period. As well, the immunogenic evolution seen in our analysis may question the consistent usage of the NCal/99 virus in influenza vaccine preparations from 2000–2001 to 2006–2007 9,47.
The most significant emergent in H1N1 history since 1918 ensued in late spring 2009 and was declared a pandemic shortly after15. The HA from newly emerged H1N1 virus was identified to be similar to the HA of viruses isolated from North American swine which had recent and historical precedence48. This virus was designated 2009 H1N1 pandemic (H1N1pdm) and was shown to be a triple reassortant virus containing genes from swine, human and avian influenza A viruses16. In our phylogenetic analysis it was clearly evident that the triple reassortant viruses clustered far from the previously circulating seasonal H1N1 viruses in agreement with similar alignments. Furthermore, HI assays performed using ferret antisera produced raised against the NY/09 H1N1pdm antisera was not able to recognize the HA of any other viruses. These results are supported by previous published findings that determined the cross-protection initiated following infection with H1N1pdm which was preceded by infection with contemporary seasonal H1N1 did not occur through an HA targeted antibody response20. Interestingly, although an H1N1 component had been included in the seasonal influenza vaccine preparations since 1977 49, antigen directed pre-existing immunity to 2009 H1N1pdm was only detected in the elderly suggesting the majority of the current human population was naïve to the this novel virus50,51. Cross-reactive antibodies found in the elderly population suggested that protective immunity was induced from exposure to an older H1N1 virus with antigenic similarities to 2009 H1N1pdm25,50,51,52. Our study directly tested the ability of infection with older H1N1 strains from the years 1943 to 1947 to bind the HA molecule of the 2009 H1N1pdm virus. Against current hypotheses, we found that the immune production of antisera from infection with viruses originating from the 1940's were not able to recognize the HA of the novel pandemic viruses. Likewise, the H1N1pdm antisera did not detect the panel of viruses ranging from 1934 to 2007. Recently, the effect of priming ferrets with historical H1N1 viruses on the immune response of secondary infection the H1N1pdm was described53,54. These studies addressed distinct immunological questions as our study utilizing a different set of H1N1 viruses. These priming studies complimented our study as the historical H1N1 strains used were not the same as ours. The purpose of the previous studies was to determine protection from priming infection/infections whereas we queried immune detection, cross-reactivity and clinical analysis. Importantly, a significant issue that remains is the affect of age on the outcome of infection/sequential infection with the historical and contemporary viruses. Historical accounts mention clinical pathology among age groups but do not have a comprehensive analysis of the comparative severity14,38,49,55. As well, it is not know how the aging immune system responds to the evolving influenza virus. Although a protective response was seen in priming studies in adult animals, it is not know if protection of the same magnitude would remain during age related immunosenescence or if the clinical response in the aged would be similar as we have shown in this study.
Influenza A H1N1 and Influenza A H3N2 have varying patterns of antigenic and genetic evolution where specifically H1N1 has a lower rate of acquired mutations8,9. These evolutionary behaviours are divergent leading to a dynamic and ever changing influenza climate in the human population where one Influenza A subtype typically dominates over the other. Importantly, understanding the interplay between the H1N1 and H3N2 infection evolution remains a significant hurdle in influenza reasearch8,9. Nelson and colleagues investigated the genetic evolution of the H1N1 virus by analyzing the evolution of the whole virus as well as the individual viral segments from strains since 1918 9. In this study the authors were able to identify instances where divergent clades co-circulated as well as events of intra-H1N1 subtype reassortment9. Previously, Smith and colleagues compared the antigenic and genetic evolution of the H3N2 virus from strains originating in 1968 to 2003 and found that antigenic and genetic evolution showed differing patterns8. Antigenic evolution was punctuated where the strains formed clusters and genetic change was more even and gradual as time increased8. Here we sought to elucidate the antigenic evolution of the H1N1 virus. As with the H3N2 virus8, we found the antigenic change did not vary evenly by year but instead corresponded to genetic changes in the HA molecule. These studies are significant for elucidating the intricacies of influenza evolution as the continuous change of the influenza virus is the greatest challenge to the management of disease and containment of further outbreaks. A better understanding of the evolution of the influenza virus and the interplay between the H3N2 and H1N1 strains will improve the design of prophylactics such as by guiding vaccine strain selection for yearly trivalent influenza vaccine or may even give insight to the development of a universal influenza vaccine. More work is needed to understand the true nature of influenza virus dynamics and evolution.
Animal models remain the most appropriate mode for the methodical scientific investigation of human influenza virus pathogenesis and the testing of influenza prophylactics and therapeutics. Currently, the ferret is thought to be the most suitable animal model for respiratory influenza virus infection56 since ferrets are physiologically susceptible to wild type influenza viruses due to a similar respiratory tract and are able to transmit the virus once infected. As mentioned previously, ferrets display similar clinical disease as humans following influenza infection which include fever, weight loss and sneezing and these positive features have been well discussed previously21,24,26,30,56. As well, the ferret has been shown to have similar features as human in regard to IFN pathway function and is potential model for the study of human IFN-gamma signalling24,57. Although the ferret is an appropriate model, it is imperative to be mindful of the disparities between animal models and the nature human infection to have an accurate assessment of research findings especially from ferret-influenza studies. During experimental influenza infections in animal models, a specific and known amount of virus is administered to the animal in a controlled manor, usually by intranasal inoculation. Modeling the infectious behaviour of a virus is more predictable when the infection route and dose are controlled. This is in contrast to natural infection where humans are infected with an unknown amount of virus in an unknown route, which must be considered when extrapolating results from animal model studies. Most significantly, ferrets as well as other animal models used in influenza studies are typically ‘Specific Pathogen Free' (SPF). In this case, the ferrets are determined to be influenza free prior to study initiation. Unlike humans who have been previously exposed to various subtypes of influenza viruses, ferrets used in influenza studies are completely naïve to the virus. The nuances of the immune response subsequent to multiple influenza infections, such as the phenomenon of original antigenic sin58, has only started to be elucidated in the ferret model53 and much work is still required to have a full understanding of the effect of sequential influenza infection on the specific immune response mounted. Furthermore, since the influenza virus is subject to both genetic shift and genetic drift35, the clinical manifestations of sequential virus infections by a virus that undergone multiple genetic drifts would differ from a response of a virus that had undergone a significant genetic shift as that of the H1N1 2009 pandemic variant.
Influenza virus infection has been thought to plague the human population for hundreds of years, where symptoms of influenza can be traced back in the writings of the early Greek cultures to 412 BC40. As many questions still exist in relation to evolution and emergence of influenza strains, we analyzed the immunological evolution of past and contemporary influenza viruses. Our results shed light on the cross-protective immune response toward the HA molecule and suggest that unique immunity is induced dependent on the HA sequence. Further work is needed to determine the evolutionary placement of the 1918 and 1934 virus immunogenicity, as well as the placement of newly emerging influenza strains. Our results describing the antigenic evolution of the H1N1 influenza subtype together with previous work will add to the understanding of the transcendence of influenza viruses and have implications on the design of future influenza vaccines.
Methods
Ethics statement
All animal work was conducted in strict accordance with the Canadian Council of Animal Care (CCAC) guidelines. The University Health Network (UHN) has certification with the Animals for Research Act (Permit Number: #0045 and #0085 of the Ontario Ministry of Agriculture, Food and Rural Affairs) and follows NIH guidelines (OLAW #A5408-01). The animal use protocol was approved by the Animal Care Committee (ACC) of the UHN. Infections and subsequent sample collection were performed under 5% isofluorane anesthesia in an effort to minimize suffering.
Viruses
All viruses, A/PuertoRico/8/1934 (PR/34), A/AA/Marton/43 (Marton/43), A/FortMonmouth/1/1947 (FM/47), A/USSR/90/1977 (USSR/77), A/Taiwan/1/1986 (Taiwan/86), A/NewCaledonia/20/1999 (NCal/99) and A/NewYork/18/2009 (NY/09), A/SolomonIslands/3/2006 (SI/06), A/Brisbane/59/2007 (Bris/07), A/California/07/2009 (Cal/09) are provided by Center for Disease Control and Prevention ((CDC), Atlanta, GA, USA) or American Type Culture Collection ((ATCC), Manassas, VA, USA). All virus work was performed in BSL-2 facility.
Infection and ferret monitoring
Maintenance and monitoring of infected ferrets has been previously described23. Briefly, male ferrets 4–6 months old were bred in an on-site SPF ferret colony (University Health Network, Toronto, ON, Canada). Prior to infection, all ferrets were screened for influenza and shown to be seronegative by HI assay against circulating influenza A and B strains (2010-2011 WHO Influenza Reagent Kit for Identification of Influenza Isolates (WHO collaborating center for surveillance, epidemiology and control for influenza infection division)). The kits contain the circulating influenza strains for the particular year. Prior to infection, ferrets were randomly selected and pair-housed in cages contained in bioclean portable laminar-flow clean-room enclosures (Lab Products, Seaford, DE) in the BSL-2 animal holding area. Baseline body temperature and weight were measured on Day 0 for each animal. Temperatures were measured by using a subcutaneous implantable temperature transponder (BioMedic Data Systems, Inc., Seaford, DE). Upon infection, ferrets were anesthetized and infected 1mL of virus preparation for each ferret (0.5mL in each nostril). Ferrets were infected with Marton/43 (N = 12), FM/47 (N = 10), USSR/77 (N = 12), Taiwan/86 (N = 10), NCal/99 (N = 14) and NY/09 (N = 12). All viruses were used at 105 EID50 except for FM/47 which was infected at 106 EID50 since it was reported to be a milder virus14. Clinical signs (body temperature, body weight, level of activity, nasal discharge and sneezing) were observed daily for 14 days pI. We examined animals at the same time each day for consistency. Nasal discharge includes crusty nose, mucous and transparent exudates/fluids. The scores were calculated from the total animals displaying any nasal discharge symptom over the total number of animals. The sneezing scores were calculated from the total animals found sneezing over the total number of animals. Scores were calculated daily for 14 days and only the peak values for each infection are summarized. The inactivity scoring system is based on the reference Reuman et al., 1989 59 to assess the inactivity level: 0, alert and playful; 0.5, alert but playful only when stimulated; 1, alert but not playful when stimulated; 2, neither alert nor playful when stimulated. A relative inactivity index was calculated as follows: Σ(day 1 to day 14)[score+1]n/Σ(day 1 to day 14)n, where n equals the total number of observations. A value of 1 was added to each observation unit score so that a score of 0 could be divided by a denominator, resulting in an index value of 1.0 as the minimum value.
Determination of influenza specific antibody responses
Influenza specific antibody responses from the uninfected or infected ferrets were measured by HI as previously described23. Briefly, receptor destroying enzyme ([RDE], Accurate Chemical & Scientific Corp., Westbury, NY, USA) treated ferret anti-sera was serially diluted and HI titers were determined by the highest dilution that completely inhibited influenza hemagglutination (4HAU) of turkey erythrocytes.
Comparative analysis of hemagglutinin sequences
Phylogenetic analysis of influenza hemagglutinin DNA sequences were conducted in MEGA560 using the Maximum Likelihood method based on the Tamura-Nei model61, 500 bootstrap repetitions were performed. To analyze the protein similarity of influenza hemagglutinin from different H1N1 strains, the aminoacid sequences from the protein region HA135-295 were aligned using ClustalW262. This region of the HA protein is part of the hemagglutinin receptor binding domain (HA-RBD), which is located in the external surface of the virus and it concentrates most of the antigenic potential of the HA protein36. Amino Acid HA GenBank accession numbers for Clustal alignment ( Supplementary Fig. S2 ): A/NewCaledonia/20/1999(H1N1) (AAP34324.1); A/SolomonIslands/3/2006(H1N1) (ABU99109.1); A/Brisbane/59/2007(H1N1) (ADE28750.1). Amino Acid HA GenBank accession numbers for homology table ( Fig. 3b ): A/BrevigMission/1/1918 (AAD17218); A/PuertoRico/8/1934 (CAA24272); A/AA/Marton/1943-(ABO38054); A/FortMonmouth/1/1947 (ABD77807); A/USSR/90/1977 (ABD95350); A/Taiwan/1/1986 (ABF21274); A/NewCaledonia/20/1999 (ABF21272); A/SolomonIslands/3/2006 (ABU99109); A/Brisbane/59/2007 (ACA28844); California/07/2009 (ACP41953); A/NewYork/18/2009 (ACR67196). GenBank accession numbers for the phylogenetic analysis of HA DNA sequence ( Fig. 3a ): A/PR/8/1934 (V01088); A/BrevigMission/1/1918 (AF116575); A/South Carolina/1/1918 (AF117241); A/AA/Marton/1943 (CY02028); A/FortMonmouth/1/1947 (CY009612); A/USSR/90/1977 (CY010372); A/Taiwan/01/1986 (DQ508873); A/NewCaledonia/20/1999 (DQ508857); A/SolomonIslands/3/2006 (EU124177); A/Brisbane/59/2007 (CY030230); A/California/07/2009 (FJ966974); A/NewYork/18/2009 (GQ232064).
References
Sym, D., Patel, P. N. & El Chaar, G. M. Seasonal, avian and novel H1N1 influenza: prevention and treatment modalities. Ann. Pharmacother. 43, 2001–2011 (2009).
Fouchier, R. A. et al. Characterization of a novel influenza A virus hemagglutinin subtype (H16) obtained from black-headed gulls. J. Virol. 79, 2814–2822 (2005).
Tong, S. et al. A distinct lineage of influenza A virus from bats. Proc. Natl. Acad. Sci. U. S. A 109, 4269–4274 (2012).
Holmes, E. C. RNA virus genomics: a world of possibilities. J. Clin. Invest 119, 2488–2495 (2009).
Girard, M. P., Tam, J. S., Assossou, O. M. & Kieny, M. P. The 2009 A (H1N1) influenza virus pandemic: A review. Vaccine 28, 4895–4902 (2010).
World Health Organization (WHO). Influenza (Seasonal) http://www.who.int/mediacentre/factsheets/fs211/en/index.html. Ref Type: Generic Accessed: 2012.
Watanabe, T. & Kawaoka, Y. Pathogenesis of the 1918 pandemic influenza virus. PLoS Pathog. 7, e1001218 (2011).
Smith, D. J. et al. Mapping the antigenic and genetic evolution of influenza virus. Science 305, 371–376 (2004).
Nelson, M. I. et al. Multiple reassortment events in the evolutionary history of H1N1 influenza A virus since 1918. PLoS Pathog. 4, e1000012 (2008).
Logan, W. P. & MacKay, D. G. Development of influenza epidemics. Lancet 1, 264–265 (1951).
Collins, S. D. & Lehmann, J. Trends and epidemics of influenza and pneumonia: 1918–1951. Public Health Rep. 66, 1487–1516 (1951).
Scholtissek, C., Rohde, W., Von, H. V. & Rott, R. On the origin of the human influenza virus subtypes H2N2 and H3N2. Virology 87, 13–20 (1978).
Salk, J. E. & Suriano, P. C. Importance of antigenic composition of influenza virus vaccine in protecting against the natural disease; observations during the winter of 1947–1948. Am J Public Health Nations. Health 39, 345–355 (1949).
Kilbourne, E. D. Influenza pandemics of the 20th century. Emerg. Infect. Dis. 12, 9–14 (2006).
World Health Organization (WHO) New Influenza A(H1N1) virus: global epidemiological situation, June 2009. Weekly Epidemiological Record 25, 249–260 (2009).
Shinde, V. et al. Triple-reassortant swine influenza A (H1) in humans in the United States, 2005-2009. N. Engl. J Med 360, 2616–2625 (2009).
Manicassamy, B. et al. Protection of mice against lethal challenge with 2009 H1N1 influenza A virus by 1918-like and classical swine H1N1 based vaccines. PLoS Pathog. 6, e1000745 (2010).
Greenberg, M. E. et al. Response to a monovalent 2009 influenza A (H1N1) vaccine. N. Engl. J Med 361, 2405–2413 (2009).
World Health Organization (WHO). WHO Manual on Animal InfluenzaDiagnosis and Surveillance. http://www.who.int/vaccine_research/diseases/influenza/WHO_manual_on_animal-diagnosis_and_surveillance_2002_5.pdf. 2002. Ref Type: Generic Accessed: 2012.
Fang, Y. et al. Seasonal H1N1 Influenza Virus Infection Induces Cross-Protective Pandemic H1N1 Virus Immunity through a CD8-Independent, B Cell-Dependent Mechanism 25. J. Virol. 86, 2229–2238 (2012).
Rowe, T. et al. Modeling host responses in ferrets during A/California/07/2009 influenza infection. Virology 401, 257–265 (2010).
Huang, S. S. et al. Differential Pathological and Immune Responses in Newly Weaned Ferrets are associated with Mild Clinical Outcome of Pandemic 2009 H1N1 Infection. J Virol. (2012). Reviewer Comment 3.
Huang, S. S. et al. Comparative analyses of pandemic H1N1 and seasonal H1N1, H3N2 and influenza B infections depict distinct clinical pictures in ferrets. PLoS One 6, e27512 (2011).
Kelvin, A. A. et al. Ferret TNF-a and IFN-g Immunoassays in Trends in Immunolabelled and Related Techniques (ed. Dr. Eltayb Abuelzein) (InTech, 2012). Reviewer Comment 3.
Maines, T. R. et al. Transmission and pathogenesis of swine-origin 2009 A(H1N1) influenza viruses in ferrets and mice. Science 325, 484–487 (2009).
Banner, D. & Kelvin, A. A. The current state of H5N1 vaccines and the use of the ferret model for influenza therapeutic and prophylactic development. J Infect. Dev. Ctries. 6, 465–469 (2012).
Cameron, M. J. et al. Lack of Innate Interferon Responses during SARS Coronavirus Infection in a Vaccination and Reinfection Ferret Model. PLoS One 7, e45842 (2012).
Leon, A. J. et al. Sequencing, annotation and characterization of the influenza ferret infectome. J Virol. 87, 1957–1966 (2013).
Danesh, A. et al. Early gene expression events in ferrets in response to SARS coronavirus infection versus direct interferon-alpha2b stimulation. Virology 409, 102–112 (2011).
Zitzow, L. A. et al. Pathogenesis of avian influenza A (H5N1) viruses in ferrets. J. Virol. 76, 4420–4429 (2002).
Rossman, J. S. & Lamb, R. A. Influenza virus assembly and budding. Virology 411, 229–236 (2011).
Dowdle, W. R., Coleman, M. T., Mostow, S. R., Kaye, H. S. & Schoenbaum, S. C. Inactivated influenza vaccines. 2. Laboratory indices of protection. Postgrad. Med J 49, 159–163 (1973).
Kilbourne, E. D., Christenson, W. N. & Sande, M. Antibody response in man to influenza virus neuraminidase following influenza. J Virol. 2, 761–762 (1968).
Potter, C. W. & Oxford, J. S. Determinants of immunity to influenza infection in man. Br. Med Bull. 35, 69–75 (1979).
Chen, J. & Deng, Y. M. Influenza virus antigenic variation, host antibody production and new approach to control epidemics. Virol. J 6, 30 (2009).
Aguilar-Yanez, J. M. et al. An influenza A/H1N1/2009 hemagglutinin vaccine produced in Escherichia coli. PLoS One 5, e11694 (2010).
Glezen, W. P. et al. Age distribution of patients with medically-attended illnesses caused by sequential variants of influenza A/H1N1: comparison to age-specific infection rates, 1978–1989. Am J Epidemiol. 133, 296–304 (1991).
Klontz, K. C. et al. An outbreak of influenza A/Taiwan/1/86 (H1N1) infections at a naval base and its association with airplane travel. Am J Epidemiol. 129, 341–348 (1989).
Shrestha, S. S. et al. Estimating the burden of 2009 pandemic influenza A (H1N1) in the United States (April 2009-April 2010). Clin. Infect. Dis. 52 (Suppl 1), S75–S82 (2011).
Potter, C. W. A history of influenza. J Appl. Microbiol. 91, 572–579 (2001).
Kilbourne, E. D. et al. The total influenza vaccine failure of 1947 revisited: major intrasubtypic antigenic change can explain failure of vaccine in a post-World War II epidemic. Proc. Natl. Acad. Sci. U. S. A 99, 10748–10752 (2002).
CDC Update: influenza activity-worldwide. MMWR 35, 433–434 (1986).
CDC Antigenic variation of recent influenza A(H1N1) viruses. MMWR 35, 685–687 (1986).
CDC Recommendation of the Immunization Practices Advisory Committee Monovalent Influenza A(H1N1) Vaccine, 1986–1987. MMWR 35, 517–521 (1986).
Robertson, J. S. Sequence analysis of the haemagglutinin of A/Taiwan/1/86, a new variant of human influenza A(H1N1) virus. J Gen. Virol. 68 (Pt 4), 1205–1208 (1987).
Hay, A. J., Gregory, V., Douglas, A. R. & Lin, Y. P. The evolution of human influenza viruses. Philos. Trans. R. Soc. Lond B Biol Sci. 356, 1861–1870 (2001).
CDC Update: influenza activity - United States and worldwide, 2006 - 2007 and composition of the 2007 - 2008 influenza vaccine. MMWR 56, 789–794 (2007).
Garten, R. J. et al. Antigenic and genetic characteristics of swine-origin 2009 A(H1N1) influenza viruses circulating in humans. Science 325, 197–201 (2009).
Couch, R. B. et al. Prior infections with seasonal influenza A/H1N1 virus reduced the illness severity and epidemic intensity of pandemic H1N1 influenza in healthy adults. Clin. Infect. Dis. 54, 311–317 (2012).
Hancock, K. et al. Cross-reactive antibody responses to the 2009 pandemic H1N1 influenza virus. N. Engl. J Med 361, 1945–1952 (2009).
Miller, E. et al. Incidence of 2009 pandemic influenza A H1N1 infection in England: a cross-sectional serological study. Lancet 375, 1100–1108 (2010).
Itoh, Y. et al. In vitro and in vivo characterization of new swine-origin H1N1 influenza viruses. Nature 460, 1021–1025 (2009).
Carter, D. M. et al. Sequential Seasonal H1N1 Influenza Virus Infections Protect Ferrets Against Novel 2009 H1N1 Influenza. J Virol. (2012).
O'Donnell, C. D. et al. The effect of priming with H1N1 influenza viruses of variable antigenic distance on challenge with 2009 pandemic H1N1 virus. J Virol. (2012).
Teare, E. L. et al. Failure of influenza vaccine to prevent two successive outbreaks of influenza A H1N1 in a school community. Br. J Gen. Pract. 40, 10–12 (1990).
Belser, J. A., Katz, J. M. & Tumpey, T. M. The ferret as a model organism to study influenza A virus infection. Dis. Model. Mech. 4, 575–579 (2011).
Danesh, A. et al. Cloning, expression and characterization of ferret CXCL10. Mol. Immunol. 45, 1288–1297 (2008).
Pan, K. Understanding original antigenic sin in influenza with a dynamical system. PLoS One 6, e23910 (2011).
Reuman, P. D., Keely, S. & Schiff, G. M. Assessment of signs of influenza illness in the ferret model. J. Virol. Methods 24, 27–34 (1989).
Tamura, K. et al. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance and maximum parsimony methods. Mol. Biol Evol. 28, 2731–2739 (2011).
Tamura, K. & Nei, M. Estimation of the number of nucleotide substitutions in the control region of mitochondrial DNA in humans and chimpanzees. Mol. Biol Evol. 10, 512–526 (1993).
Larkin, M. A. et al. Clustal W and Clustal X version 2.0. Bioinformatics. 23, 2947–2948 (2007).
Acknowledgements
All influenza strains, A/PuertoRico/8/1934, A/AA/Marton/43, A/FortMonmouth/1/1947, A/USSR/90/1977, A/Taiwan/1/1986, A/NewCaledonia/20/1999, A/SolomonIslands/3/2006, A/Brisbane/59/2007 and A/NewYork/18/2009 were obtained through the Influenza Reagent Resource, Influenza Division, WHO Collaborating Center for Surveillance, Epidemiology and Control of Influenza, Centers for Disease Control and Prevention, Atlanta, GA, USA. We thank the Li Ka-Shing Foundation of Canada, Immune Diagnostics & Research and Shantou University Medical College for the support of conducting this study. A/Mexico/4108/2009 virus was obtained through the Influenza Reagent Resource, Influenza Division, WHO Collaborating Center for Surveillance, Epidemiology and Control of Influenza, Centers for Disease Control and Prevention, Atlanta, GA, USA.
Author information
Authors and Affiliations
Contributions
S.S.H.H. and Z.L. conducted the majority of the experimental and technical work and assisted with figure preparation. D.B. ran and supervised all ferret maintenance and experiments. A.J.L. performed the comparative analysis of hemagglutinin sequences. S.G.P. provided technical assistance with animal studies. B.R., S.R. and Y.G. assisted with data analysis, data interpretation and manuscript preparation. D.J.K. and A.A.K. designed the experiments and supervised the project. A.A.K. analyzed the data and wrote the paper.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Supplementary Information
Supplemental Material
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Huang, S., Lin, Z., Banner, D. et al. Immunity toward H1N1 influenza hemagglutinin of historical and contemporary strains suggests protection and vaccine failure. Sci Rep 3, 1698 (2013). https://doi.org/10.1038/srep01698
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep01698
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
