



 Figure 1
Figure 1















« Prev Next »
To understand the relationship between the genome and the functioning of cells, scientists have sought to study the products of the genome, namely proteins and expressed RNAs, such as tRNA and rRNA. Proteomics is the study of the set of proteins in a cell or tissue, and it includes details on protein quantity and diversity. However, the proteome may not tell a cell's entire story. After all, proteins are dynamic and interacting molecules, and their changeability can make proteomic snapshots difficult at best. Furthermore, there are many technical challenges in characterizing molecules that cannot be easily amplified and have several post-translational modifications. Thankfully, measuring the intermediate step between genes and proteins—in other words, transcripts of messenger RNA—bridges the gap between the genetic code and the functional molecules that run cells.
Transcriptomes Are Indicative of Gene Activity
In multicellular organisms, nearly every cell contains the same genome and thus the same genes. However, not every gene is transcriptionally active in every cell — in other words, different cells show different patterns of gene expression. These variations underlie the wide range of physical, biochemical, and developmental differences seen among various cells and tissues and may play a role in the difference between health and disease. Thus, by collecting and comparing transcriptomes of different types of cells or tissues, researchers can gain a deeper understanding of what constitutes a specific cell type and how changes in transcriptional activity may reflect or contribute to disease.
A transcriptome represents that small percentage of the genetic code that is transcribed into RNA molecules — estimated to be less than 5% of the genome in humans (Frith et al., 2005). The proportion of transcribed sequences that are non-protein-coding appears to be greater in more complex organisms. In addition, each gene may produce more than one variant of mRNA because of alternative splicing, RNA editing, or alternative transcription initiation and termination sites. Therefore, the transcriptome captures a level of complexity that the simple genome sequence does not (Figure 1).
By studying transcriptomes, researchers hope to determine when and where genes are turned on or off in various types of cells and tissues. The number of transcripts can be quantified to get some idea of the amount of gene activity or expression in a cell. For example, transcript information may help reveal what genes give stem cells their unique properties of developmental plasticity and continuous growth in culture, or which particular gene expression changes are associated with cancer. Furthermore, by considering the transcriptome, it is possible to generate a comprehensive picture of what genes are active at various stages of development.
Expressed Sequences and cDNA Libraries
All nucleated cells share the same genetic material; what differentiates these cells is the specific genes that are expressed in each cell at specific times. The genes involved in tissue-specific or developmental processes traditionally have been studied by making libraries of all expressed genes for an organ or developmental stage. Complementary DNA (cDNA) libraries give a snapshot of actively expressed genes by capitalizing on the fact that during the transcription of mRNA in eukaryotes, a poly(A) tail (consisting of a long sequence of adenine nucleotides) is added. This poly(A) tail distinguishes mRNA from other expressed RNAs and can therefore be used as a primer site for reverse transcription.
To make a library of transcribed sequences, scientists isolate all the RNA from their cells of interest and use a single-stranded primer complementary to the unique poly(A) tail, as well as a viral enzyme called reverse transcriptase. Because they are produced from transcribed mRNA found in the nucleus, cDNA libraries contain primarily the protein-encoding regions of the genome. Once a cDNA has been at least partially sequenced, unique polymerase chain reaction (PCR) primer pairs that identify short stretches of each cDNA can be designed. These regions, called expressed sequence tags or ESTs, can then be used to produce probes to determine the presence or absence of similar transcripts in other tissues. The identification of ESTs has proceeded rapidly, with approximately 52 million ESTs now available in public databases (e.g., GenBank). Moreover, current methods allow expressed RNAs to be made into cDNA or cRNA and sequenced en masse using pyrosequencing, which promises to accelerate the rate at which new EST data is added to these databases.
Microarrays, SAGE, and Transcriptome Resources
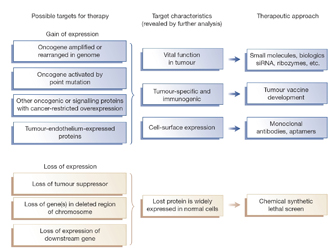
© 2004 Nature Publishing Group Strausberg, R. L. et al. Oncogenomics and the development of new cancer therapies. Nature 429, 470. All rights reserved. 
The sheer volume of expression and annotation data being produced necessitates sophisticated computational methods for the sake of analysis. For example, bioinformatic approaches attempt to establish functional linkages between fully sequenced genomes and their expressed RNA products. The information generated by these approaches can be used to infer which genetic circuitry is needed for all cells, versus which gene networks give cancer, liver, or stem cells their unique properties. These linkages can also be applied to understanding evolutionary relationships among species (phylogeny) and to the study of the function of homologous proteins in model organisms (Kalia & Gupta, 2005). For example, the U.S. National Human Genome Research Institute (NHGRI) is participating in two projects — the Mammalian Gene Collection and the Mouse Transcriptome Project — that will create transcriptome resources that will be made available to researchers around the world.
Already, large-scale transcript sequencing projects in the United States and Brazil have resulted in the establishment and presentation of data in the Cancer Genome Anatomy Project, a key reference for the definition of human gene expression in normal tissues and tumors (Strausberg et al., 2004). The findings from this public collaborative project will support new approaches to drug discovery in cancer (Figure 2). Similarly, the FANTOM consortium based in Japan reported the transcriptome of the mouse in 2002 (Okazaki, et al.). This consortium's analysis of 61,000 full-length cDNA sequences derived from over 200 tissue samples was by far the most extensive sampling of the mouse transcriptome so far. Among the highlights of this project was the suggestion, supported by the genomic sequencing data, that a significant class of genes whose ultimate products are not proteins, but rather RNAs with novel functions, may exist (Paigen, et al., 2003).
Indeed, Claverie (2005) writes: "The notion that transcription is limited to protein-coding genes is being challenged. The intergenic, intronic, and antisense transcribed sequences that were once deemed artifactual are now a testimony to our collective refusal to depart from an oversimplified gene model. Perhaps it's time to go back to the cDNA sequence databases and reevaluate the numerous unexpected objects they contain. Transcription will never be simple again, but how complex will it get?"
References and Recommended Reading
Claverie, J. M. Fewer genes, more noncoding RNA. Science 309, 1529–1530 (2005) doi:10.1126/science.1116800
Frith, M. C., et al. Genomics: The amazing complexity of the human transcriptome. European Journal
of Human Genetics 13, 894–897 (2005) doi:10.1038/sj.ejhg.5201459 (link to article)
Gimelbrant, A., et al. Widespread monoallelic expression on human autosomes. Science 318, 1136–1140 (2007) doi:10.1126/science.1148910
Kalia, A., & Gupta, R. P. Proteomics: A paradigm shift. Critical Reviews in Biotechnology 25, 173–198 (2005)
Ma, Y., et al. Prevalence of off-target effects in Drosophila RNA interference screens. Nature 443, 359–363 (2006) doi:10.1038/nature05179 (link to article)
Newman, J. R. S., et al. Single-cell proteomic analysis of S. cerevisiae reveals the architecture of biological noise. Nature 441, 840–846 (2006) doi:10.1038/nature04785 (link to article)
Paigen, K. One hundred years of mouse genetics: An intellectual history. Part II: The molecular revolution (1981-2002). Genetics 163, 1227–1235 (2003)
Okazaki, Y., et al. Analysis of the mouse transcriptome based on functional annotation of 60,770 full-length cDNAs. Nature 420, 563–573 (2002) doi:10.1038/nature01266 (link to article)
Strausberg, R. L., et al. Oncogenomics and the development of new cancer therapies. Nature 429, 469–474 (2004) doi:10.1038/nature02627 (link to article)










