


















« Prev Next »
Homo sapiens are just one of millions of Earth's species. With only about 10% (1.8 million) of all eukaryotic organisms described to date, most such organisms are yet to be discovered (Hawksworth & Kalin-Arroyo, 1995). To tackle this immense diversity, scientists often prioritize their efforts by sampling "biodiversity hotspots" (Figure 1), or areas that harbor unique and diverse organisms that are often threatened by extinction. Cataloging biodiversity through species inventory projects is a first step toward developing an understanding of how various organisms interact with their environment, which is a key factor in establishing these organisms' roles in the ecosystem and their potential utility to humankind.
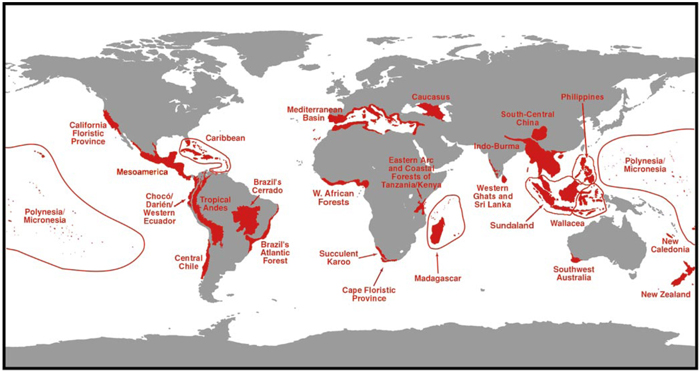
Figure 1: Prominent biodiversity hotspots.
As many as 44% of all species of vascular plants and 35% of all species in four vertebrate groups are confined to 25 hotspots comprising only 1.4% of the land surface of the Earth.
© 2000 Nature Publishing Group Myers, N. et al. Biodiversity hotspots for conservation priorities. Nature 403, 853 (2000). All rights reserved. 
Full Genome Sequences
It would be ideal if we could have a giant database of full genome sequences of all living species. For now, however, scientists are overwhelmed with the data generated from sequencing only a few dozen genomes. Although this data provides the virtual map of an organism, some questions can be answered using fairly short gene sequences. The race is currently on to make genome sequencing faster, cheaper, and much more efficient. Complete sequences of a growing number of genomes (e.g., mouse, fruit fly, cat, dog, ape, and human) allow us to compare the DNA of closely related species in order to establish and analyze their genetic differences, a field known as comparative genomics.
For example, after full genome sequences of humans and several apes became available, scientists were eager to find out what genes set us apart from our closest living relative, the bonobo. Researchers thus compared the two sequences to establish the amount and type of genetic variation. It turns out that the human and ape genomes are remarkably similar; in fact, a recent comparison of the two found a nucleotide divergence of only 1.23% (Mikkelsen et al., 2005). To better appreciate the scale of this variation, consider the fact that African and European human populations exhibit 0.08% nucleotide divergence (Yu et al., 2002). Knowing that all genes do not evolve at the same rate, researchers were also able to isolate those human alleles that have undergone the most change since the human and ape lineages split about 2 million years ago. This knowledge was then used to determine our rate of evolutionary divergence from the great apes, thus helping calibrate the molecular clock used to build the primate family tree.
To further fine-tune this clock, efforts are now underway to sequence the full genome of our closest extinct relative, the Neanderthal. This is no easy task: fossils are often contaminated by bacteria and human samples, thus causing the authentic DNA to become fragmented and degraded. However, persistence and a few well-preserved bones have taken us one million base pairs closer to the goal of obtaining the full nuclear genome of the Neanderthal (Green et al., 2006). One of the first major discoveries to come out of working with the Neanderthal genome fragments is the presence in Neanderthal DNA of the modern human FOXP2 gene, which is known to play a role in speech development. FOXP2 was also found in the genes of the common ancestor of humans and Neanderthals, which may mean that language has been around for much longer than we initially supposed (Krause et al., 2007). Major milestones in this field are achieved quite rapidly; at the time this article was authored, scientists unveiled the full mitochondrial genome sequence of a 38,000-year old Neanderthal bone fossil. The full nuclear genome of our last extinct relative is to follow shortly thereafter (Green, et. al., 2008). Such discoveries hold great potential for identifying the key genes that helped spawn human civilization.
Some argue that given the rate of current advances in cloning technology, a full genome sequence can be viewed as species' "insurance policy" that will guarantee that it doesn't become extinct. But even as we advance our cloning and sequencing methods, the availability of a genetic sequence is no guarantee of long-term species survival, especially for an organism that inhabits a unique and threatened habitat, or a narrow trophic niche.
Partial Gene Sequences
The method of comparative genomics can be applied not just to full genome sequences, but also to single genes and gene fragments to study their function and help establish relationships among species. Indeed, a species' place on an evolutionary tree is a valuable predictor of the structure and function of neighboring taxa.
The current convention of describing (defining) organisms new to science and establishing their evolutionary relationships is based on total evidence; in other words, the organisms' genetic, morphological, and ecological characters are described and analyzed against other sets of data. Taken together, these techniques can be very informative, having thus far provided us with a detailed road map of Earth's biota. But for systematics - the study of biological diversity and common ancestry - rapid technological advances in the field of comparative genomics are both a blessing and a curse. Consider, for example, the technique called DNA bar coding, which is based on using short fragments of mitochondrial gene CO1 to uniquely identify and document animal species (Savolainen, 2005). This technique has applications across all living organisms, but the precise genetic methodology is still being developed. In addition, the debate among scientists regarding the use and the utility of DNA bar coding has been quite vociferous. On one hand, this technique brings the promise of instant species identification to a much wider community with minimal biological training. Indeed, it is hypothetically possible to carry a hand-held device out in the field and input species sequences into a rapidly expanding database; all for a fraction of the price, knowledge, and effort associated with the conventional manual method or with human-curated taxonomic identification. So what's the catch?
One major problem with DNA bar coding is that it operates on the assumption that species have evolved in perfect percentile distances of genetic diversion. Thus, with this technique, in order for any two organisms to be deemed the same species, they must share 88-98% of genetic code at the chosen CO1 mitochondrial gene fragment (Savolainen, 2005). The exact suggested threshold has to be characterized for each group, and neither the threshold nor the groups have been clearly defined for most taxa. Thus, DNA bar coding has been called a "quick fix" and an oversimplification of systematics. Indeed, wide variation in the CO1 gene is found not only among species, but also within them, and even between the cells of an individual organism - a phenomenon known as mitochondrial heteroplasmy (Kmiec & Woloszynska, 2006). Furthermore, there is a broad overlap of inter- and intraspecific genetic distances among closely related species (Goldstein et al., 2000).
These issues come into focus when you consider the devastating malaria epidemic that kills one to three million people worldwide every year. The pathogens that cause malaria are protozoan parasites from the genus Plasmodium that are transmitted through the bite of mosquitoes of the genus Anopheles. Both of these animal genera contain hundreds of species, although only a few are involved in transmitting malaria in humans. Recent genetic studies of the symbiotic bacteria in the midgut of the Anopheles stephensi mosquitoes have yielded promising results: Enterobacter agglomerans bacteria were genetically engineered to display two anti-Plasmodium effector molecules that kill the parasite before it is transmitted to humans (Riehle et al., 2007). Now consider the genetic and physiological differences between the wild-type and genetically modified Anopheles stephensi mosquitoes: they are still the same species by all major standards of species definition, yet what a difference it would make for humankind if the Plasmodium-resistant genetically modified strain were dominant. This example highlights the importance of studying genomes and biological associations of the narrowest niches of life. It also underlines the vital potential for the unpredictable outcomes of genome sequencing-major advances are often made using information generated for completely unrelated reasons.
Metagenomic Studies
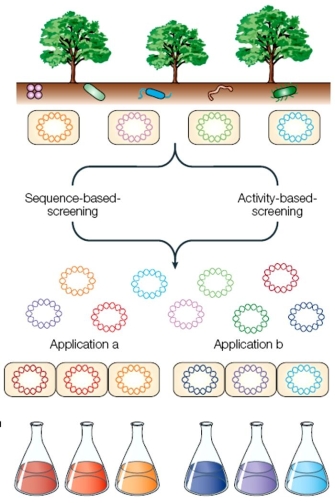
Figure 2: Industrial enzymes - from the metagenome to the applications and processes.
A library of cloned DNA is produced from the metagenome. Primary screening, including both sequence-based screening and activity-based screening, produces enzyme libraries that serve as platforms for subsequent development. Secondary screening of the enzyme library identifies process-specific properties such as substrate specificity, activity, and stability. Subsets of cloned enzymes are then used in scale-up applications or process testing to identify suitable enzyme candidates.
© 2005 Nature Publishing Group Lorenz, P. et al. Metagenomics and industrial applications. Nature Reviews Microbiology 3, 512 (2005). All rights reserved.
Of course, other bacterial biomes are far greater than our body, and consequently, they are not investigated as extensively. The vast majority of microorganisms from the oceanic strata, for example, remain virtually unknown. Scientists have taken a "shotgun" approach to this dilemma by sequencing genetic material found in the ocean waters. This method of obtaining DNA directly from non-lab samples is called metagenomics, and it is revamping the field of microbial oceanography by tapping a rich source of genetic diversity.
In addition to focusing on the complete genome sequences obtained by metagenomic sampling, functional inventories often skip a step by going directly for the gene products, rather than getting to know the organisms that produce them (Figure 2). Such studies focus on investigating the functional aspects of the environment, allowing scientists to directly infer habitat-specific metabolic demands by targeting the proteins encoded by a community. In a study that analyzed soil and environment samples from 3 deep-ocean whale skeletons, Tringe, et. al. (2005) highlighted significant differences in energy production and population density among the three communities. Despite these intriguing findings, large proportions of the gene fragments discovered in the course of this and similar studies remains unidentified. Often times, however, the sequence data alone is sufficient to predict factors like energy sources and pollution levels of a given environment. The potential of metagenomic studies is therefore truly astonishing, and the commercial incentive is equally great. Gene fragments obtained in the course of metagenomic research can be expressed in E. coli, for example, to produce molecular materials that can then be used by the biotechnology and pharmaceutical industries in product and drug development (Handelsman, 2004).
As a result of this ever-expanding amount of genomic data and the availability of progressively more efficient sampling, sequencing, and data-storage technologies, we are seeing rapid advances in the discovery of new genes, gene products and organisms. Genome sequencing is more affordable than ever; in fact, we are nearing the benchmark of a "$1,000 genome" (Wolinsky, 2007).
References and Recommended Reading
Allen, E., & Banfield, J. Community genomics in microbial ecology and evolution. Nature Review Microbiology 3, 489–498 (2005) doi:10.1038/nrmicro1157 (link to article)
Blow, N. Technology feature: Metagenomics-Exploring unseen communities. Nature 453, 687–690 (2008) doi:10.1038/453687a (link to article)
Falush, D., et. al., Traces of human migrations in Helicobacter pylori populations, Science 299, 1582-1585 (2003)
Goldstein, P. Z., et al. Conservation genetics at the species boundary. Conservation Biology 14, 120–131 (2000) doi:10.1046/j.1523-1739.2000.98122.x
Green, R. E., et al. Analysis of one million base pairs of Neanderthal DNA. Nature 444, 330–336 (2006) doi:10.1038/nature05336 (link to article)
———. A complete Neandertal mitochondrial genome sequence determined by high-throughput sequencing. Cell 134(3): 416–428 (2008) doi:10.1016/j.cell.2008.06.021
Handelsman, J. Metagenomics: Application of genomics to uncultured organisms. Microbiology and Molecular Biology Reviews 68, 669–685 (2004) doi:10.1128/MBR.68.4.669-685.2004
Hawksworth, D. L., & Kalin-Arroyo, M. T. Magnitude and distribution of biodiversity. In Global Biodiversity Assessment, ed. V. H. Heywood (Cambridge, Cambridge University Press, 1995)
Kmiec, B., et al. Heteroplasmy as a common state of mitochondrial genetic information in plants and animals. Current Genetics 50, 149–159 (2006)
Krause, J., et al. The derived FOXP2 variant of modern humans was shared with Neanderthals. Current Biology 17, 1908–1912 (2007)
Lorenz, P., & Jurgen, E. Metagenomics and industrial applications, Nature Reviews Microbiology 3, 510-516 (2005), doi:10.1038/nrmicro1161 (link to article)
Mikkelsen, T. S., et al. Initial sequence of the chimpanzee genome and comparison with the human genome. Nature 437, 69–87 (2005) doi:10.1038/nature04072 (link to article)
Morgan, J. First complete Neanderthal genome sequenced. Nature News: August 7 (2008) doi:10.1038/news.2008.1026
Myers, N., et al. Biodiversity hotspots for conservation priorities. Nature 403, 853–858 (2000) doi:10.1038/35002501 (link to article)
Riehle, M. A., et al. Using bacteria to express and display anti-Plasmodium molecules in the mosquito midgut. International Journal of Parasitology 37, 595–603 (2007)
Savolainen, V., et al. Towards writing the encyclopaedia of life: An introduction to DNA bar coding. Philosophical Transactions of the Royal Society 360, 1805–1811 (2005)
Tringe, S. G. et al. Comparative metagenomics of microbial communities. Science 308, 554-557 (2005)
Wolinsky, H. The thousand-dollar genome. European Molecular Biology Organization Reports 8, 900–903 (2007) doi:10.1038/sj.embor.7401070.pdf (link to article)
Yu, N., et al. Larger genetic differences within Africans than between Africans and Eurasians. Genetics 161, 269–274 (2002)










