
Abstract
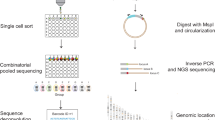
The influence of local chromatin context on gene expression can be explored by integrating a transcription reporter at different locations in the genome as a sensor. Here we provide a detailed protocol for analyzing thousands of reporters integrated in parallel (TRIP) at a genome-wide level. TRIP is based on tagging each reporter with a unique barcode, which is used for independent reporter expression analysis and integration site mapping. Compared with previous methods for studying position effects, TRIP offers a 100–1,000-fold higher throughput in a faster and less-labor-intensive manner. The entire experimental protocol takes ∼42 d to complete, with high-throughput sequencing and data analysis requiring an additional ∼11 d. TRIP was developed by using transcription reporters in mouse embryonic stem (mES) cells, but because of its flexibility the method can be used to probe the influence of chromatin context on a variety of molecular processes in any transfectable cell line.
This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 12 print issues and online access
$259.00 per year
only $21.58 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout





Similar content being viewed by others


Change history
16 January 2015
In the version of this article initially published, the final concentration of tamoxifen used in Step 53 of the Procedure was listed as 1 mM; it should be 1 µM. The error has been corrected in the HTML and PDF versions of the article.
References
Grewal, S.I. & Jia, S. Heterochromatin revisited. Nat. Rev. Genet. 8, 35–46 (2007).
Girton, J.R. & Johansen, K.M. Chromatin structure and the regulation of gene expression: the lessons of PEV in Drosophila. Adv. Genet. 61, 1–43 (2008).
Schoeftner, S. & Blasco, M.A. A 'higher order' of telomere regulation: telomere heterochromatin and telomeric RNAs. EMBO J. 28, 2323–2336 (2009).
Weber, F., de Villiers, J. & Schaffner, W. An SV40 'enhancer trap' incorporates exogenous enhancers or generates enhancers from its own sequences. Cell 36, 983–992 (1984).
Korzh, V. Transposons as tools for enhancer trap screens in vertebrates. Genome Biol. 8 (suppl. 1): S8 (2007).
Ruf, S. et al. Large-scale analysis of the regulatory architecture of the mouse genome with a transposon-associated sensor. Nat. Genet. 43, 379–386 (2011).
Sundaresan, V. et al. Patterns of gene action in plant development revealed by enhancer trap and gene trap transposable elements. Genes Dev. 9, 1797–1810 (1995).
Sun, F.L. et al. The fourth chromosome of Drosophila melanogaster: interspersed euchromatic and heterochromatic domains. Proc. Natl. Acad. Sci. USA 97, 5340–5345 (2000).
Gierman, H.J. et al. Domain-wide regulation of gene expression in the human genome. Genome Res. 17, 1286–1295 (2007).
Babenko, V.N. et al. Paucity and preferential suppression of transgenes in late replication domains of the D. melanogaster genome. BMC Genomics 11, 318 (2010).
Chen, M., Licon, K., Otsuka, R., Pillus, L. & Ideker, T. Decoupling epigenetic and genetic effects through systematic analysis of gene position. Cell Rep. 3, 128–137 (2013).
Akhtar, W. et al. Chromatin position effects assayed by thousands of reporters integrated in parallel. Cell 154, 914–927 (2013).
Pierce, S.E., Davis, R.W., Nislow, C. & Giaever, G. Genome-wide analysis of barcoded Saccharomyces cerevisiae gene-deletion mutants in pooled cultures. Nat. Protoc. 2, 2958–2974 (2007).
Gerlach, C. et al. One naive T cell, multiple fates in CD8+ T cell differentiation. J. Exp. Med. 207, 1235–1246 (2010).
Gerrits, A. et al. Cellular barcoding tool for clonal analysis in the hematopoietic system. Blood 115, 2610–2618 (2010).
Sharon, E. et al. Inferring gene regulatory logic from high-throughput measurements of thousands of systematically designed promoters. Nat. Biotechnol. 30, 521–530 (2012).
Masui, S. et al. An efficient system to establish multiple embryonic stem cell lines carrying an inducible expression unit. Nucleic Acids Res. 33, e43 (2005).
Ammar, I. et al. Retargeting transposon insertions by the adeno-associated virus Rep protein. Nucleic Acids Res. 40, 6693–6712 (2012).
Owens, J.B. et al. Chimeric piggyBac transposases for genomic targeting in human cells. Nucleic Acids Res. 40, 6978–6991 (2012).
Voigt, K. et al. Retargeting Sleeping Beauty transposon insertions by engineered zinc finger DNA-binding domains. Mol. Ther. 20, 1852–1862 (2012).
Huang, X. et al. Gene transfer efficiency and genome-wide integration profiling of Sleeping Beauty, Tol2, and piggyBac transposons in human primary T cells. Mol. Ther. 18, 1803–1813 (2010).
Santoni, F.A., Hartley, O. & Luban, J. Deciphering the code for retroviral integration target site selection. PLoS Comput. Biol. 6, e1001008 (2010).
Hui, E.K., Wang, P.C. & Lo, S.J. Strategies for cloning unknown cellular flanking DNA sequences from foreign integrants. Cell Mol. Life Sci. 54, 1403–1411 (1998).
Devon, R.S., Porteous, D.J. & Brookes, A.J. Splinkerettes–improved vectorettes for greater efficiency in PCR walking. Nucleic Acids Res. 23, 1644–1645 (1995).
Ochman, H., Gerber, A.S. & Hartl, D.L. Genetic applications of an inverse polymerase chain. Genetics 120, 621–623 (1988).
Saiki, R.K. et al. Primer-directed enzymatic amplification of DNA with a thermostable DNA polymerase. Science 239, 487–491 (1988).
Meyerhans, A., Vartanian, J.P. & Wain-Hobson, S. DNA recombination during PCR. Nucleic Acids Res. 18, 1687–1691 (1990).
McFarland, T.J. et al. Evaluation of a novel short polyadenylation signal as an alternative to the SV40 polyadenylation signal. Plasmid 56, 62–67 (2006).
Meir, Y.J. et al. Genome-wide target profiling of piggyBac and Tol2 in HEK 293: pros and cons for gene discovery and gene therapy. BMC Biotechnol. 11, 28 (2011).
Cadinanos, J. & Bradley, A. Generation of an inducible and optimized piggyBac transposon system. Nucleic Acids Res. 35, e87 (2007).
Langmead, B. & Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012).
Green, M.R. & Sambrook, J. Molecular Cloning: A Laboratory Manual 4th edn., Vol. 1 (Cold Spring Harbor Laboratory Press, 2012).
Acknowledgements
We thank the Netherlands Cancer Institute (NKI) Genomics Core Facility for sequencing support; the NKI Flow Cytometry Facility for fluorescence sorting support; G. Filion, A. Rosado and J.v. Arensbergen for their helpful suggestions; M. Amendola for providing the reference plasmid for IR copy number quantification; and members of our laboratories for their helpful discussions and critical reading of the manuscript. This work was supported by the Netherlands Consortium for Systems Biology (L.F.A.W., M.v.L. and B.v.S.); NWO-ALW open program grant (W.A. and M.v.L.); and EURYI, NWO-ALW VICI and European Research Council advanced grant no. 293662 (B.v.S.).
Author information
Authors and Affiliations
Contributions
W.A., A.V.P., M.v.L. and B.v.S. designed and developed the protocol. W.A. and A.V.P. wrote the manuscript. W.A., J.d.J. and L.P. developed the computational pipeline. J.t.H. developed the TRIP web page. M.v.L., B.v.S., L.F.A.W. and A.B. supervised the project and helped in writing.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Integrated supplementary information
Supplementary Figure 1 Potential artifacts during the preparation of mapping samples for Illumina high-throughput sequencing.
During iPCR-based mapping, intermolecular ligations between a barcoded fragment and an unrelated fragment of DNA or between two barcoded fragments can lead to the production of fragments with non-existing combinations of barcode and the genomic DNA.
Supplementary Figure 2 Determination of average copy number of IRs in a TRIP pool.
A scheme showing different amplicons present in TRIP pools and the reference plasmid, which are used for the estimation of the average copy number of IRs per cell (see also Box 2).
Supplementary Figure 3 Structure of DNA fragments prepared for Illumina high-throughput sequencing.
(a) The structure of fragments from normalization/expression samples. (b) The structure of fragments from mapping samples.
Supplementary information
Supplementary Figure 1
Potential artifacts during the preparation of mapping samples for Illumina high-throughput sequencing. (PDF 620 kb)
Supplementary Figure 2
Determination of average copy number of IRs in a TRIP pool. (PDF 193 kb)
Supplementary Figure 3
Structure of DNA fragments prepared for Illumina high-throughput sequencing. (PDF 188 kb)
Supplementary Table 1
(PDF 76 kb)
Supplementary Table 2
(PDF 90 kb)
Rights and permissions
About this article

Cite this article
Akhtar, W., Pindyurin, A., de Jong, J. et al. Using TRIP for genome-wide position effect analysis in cultured cells. Nat Protoc 9, 1255–1281 (2014). https://doi.org/10.1038/nprot.2014.072
Published:
Issue Date:
DOI: https://doi.org/10.1038/nprot.2014.072
This article is cited by
-
Selective sequestration of signalling proteins in a membraneless organelle reinforces the spatial regulation of asymmetry in Caulobacter crescentus
Nature Microbiology (2020)
-
Multiplexed Cas9 targeting reveals genomic location effects and gRNA-based staggered breaks influencing mutation efficiency
Nature Communications (2019)
-
The quest for mammalian Polycomb response elements: are we there yet?
Chromosoma (2016)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
