






Advertisement
-
-
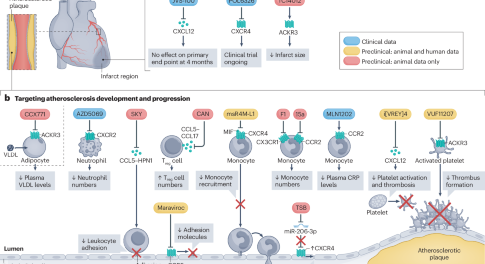
Targeting immune cell recruitment in atherosclerosis
In this Review, the authors discuss the receptors, ligands and interactors that regulate immune cell recruitment in atherosclerosis, describe mechanisms that promote the resolution of inflammation in atherosclerotic lesions, and highlight potential strategies to target these pathways for the treatment of atherosclerotic cardiovascular disease.
-
-








Anticoagulants
Anticoagulant drugs are used to prevent and treat thrombotic disorders in millions of patients worldwide. This Milestone plots the history of anticoagulant drugs, starting with the discovery and clinical trials of heparin and warfarin.
Advertisement
Trending - Altmetric
Advertisement
