
Abstract
Detection of somatic point substitutions is a key step in characterizing the cancer genome. However, existing methods typically miss low-allelic-fraction mutations that occur in only a subset of the sequenced cells owing to either tumor heterogeneity or contamination by normal cells. Here we present MuTect, a method that applies a Bayesian classifier to detect somatic mutations with very low allele fractions, requiring only a few supporting reads, followed by carefully tuned filters that ensure high specificity. We also describe benchmarking approaches that use real, rather than simulated, sequencing data to evaluate the sensitivity and specificity as a function of sequencing depth, base quality and allelic fraction. Compared with other methods, MuTect has higher sensitivity with similar specificity, especially for mutations with allelic fractions as low as 0.1 and below, making MuTect particularly useful for studying cancer subclones and their evolution in standard exome and genome sequencing data.
Similar content being viewed by others



Main
Somatic single-nucleotide substitutions are an important and common mechanism for altering gene function in cancer. Yet they are difficult to identify. First, they occur at a very low frequency in the genome, ranging from 0.1 to 100 mutations per megabase (Mb), depending on tumor type1,2,3,4,5,6,7. Second, the alterations may be present only in a small fraction of the DNA molecules originating from the specific genomic locus for reasons including contaminating normal cells in the analyzed sample, local copy-number variation in the cancer genome and presence of a mutation only in a subpopulation of the tumor cells8,9,10,11 ('subclonality'). The fraction of DNA molecules harboring an alteration ('allelic fraction') has been reported to be as low as 0.05 for highly impure tumors8. The study of the subclonal structure of tumors is not only critical to understanding tumor evolution both in disease progression and response to treatment12 but also for developing reliable clinical diagnostic tools for personalized cancer therapy13.
Recent reports on subclonal events in cancer have used three different nonstandard experimental strategies: (i) analysis of clonal mutations present in several, but not all, of the metastases from the same patient, which suggested that these mutations were subclonal in the primary tumor14; (ii) detection of subclonal mutations by ultra-deep sequencing11; or (iii) sequencing of very small numbers of single cells15,16,17. In contrast, tens of thousands of tumors are being sequenced at standard depths of 100–150× for exomes and 30–60× for whole genomes as part of large-scale cancer genome projects, such as The Cancer Genome Atlas1,2,7 and the International Cancer Genome Consortium18. To detect clonal and subclonal mutations present in these samples, one needs a highly sensitive and specific mutation-calling method. Although specificity can be controlled through subsequent experimental validation, this is an expensive and time-consuming step that is impractical for general application.
The sensitivity and specificity of any somatic mutation–calling method varies along the genome and depends on several factors, including the depth of sequence coverage in the tumor and a patient-matched normal sample, the local sequencing error rate, the allelic fraction of the mutation and the evidence thresholds used to declare a mutation. Characterizing how sensitivity and specificity depend on these factors is necessary for designing experiments with adequate power to detect mutations at a given allelic fraction, as well as for inferring the mutation frequency along the genome, which is a key parameter for understanding mutational processes and significance analysis19,20.
To meet these critical needs of high sensitivity and specificity, which are not adequately addressed by available methods21,22,23, we developed a caller of somatic point mutations, MuTect. During its development, MuTect was used in many collaborative studies1,2,3,4,7,19,24,25,26,27,28,29,30,31,32,33,34,35. Here we describe the publicly available version of MuTect, including the rationale behind its different components. We also estimate its performance as a function of the aforementioned factors using benchmarking approaches that, to our knowledge, have not been described before. The performance of the method is also supported by independent experimental validation in previous studies3,4,7,19,24,25,26,27,28,29,30 as well as by its application to data sets analyzed in other publications36. We demonstrate that our method is several times more sensitive than other methods for low-allelic-fraction events while remaining highly specific, allowing for deeper exploration of the mutational landscape of highly impure tumor samples and of the subclonal evolution of tumors.
MuTect is freely available for noncommercial use at http://www.broadinstitute.org/cancer/cga/mutect (Supplementary Data).
Results
Benchmarks for assessing mutation callers
Many mutation-detection methods have been developed, but there are few systematic approaches for benchmarking their performance on real sequencing data. Previous publications have described simulation methods ranging from fully synthetic models21 to ones that better capture real sequencing errors11. However, none of these methods model the full diversity of nonrandom sequencing errors of both the reference and alternate alleles at the genomic site. To better evaluate the performance of mutation-detection methods, we used two benchmarking approaches, downsampling and 'virtual tumors'.
Downsampling uses subsets of reads from primary sequencing data of validated somatic mutations to measure the sensitivity with which a mutation caller identifies the known mutations. Subsets are generated by randomly excluding reads from the experimentally derived data set until a desired depth of coverage is reached. Notably, downsampling preserves the expected allelic fraction of the original mutation because reads are removed regardless of whether or not they contain the mutant allele. The downsampling approach is limited in four respects: (i) the number of validated events is typically small, resulting in larger error for the sensitivity estimate; (ii) because allele fractions are preserved, only previously validated allele fractions can be explored; (iii) the analysis excludes any mutations that were not originally detected and hence may overestimate the true sensitivity; and (iv) specificity cannot be measured.
To address the problems with downsampling, we developed a benchmarking procedure that involves creating 'virtual tumors' in which we know all true mutations with certainty (Online Methods and Supplementary Fig. 1). To measure specificity, we created virtual tumor and normal data sets, at controlled depths, from sequencing data generated in two different sequencing experiments of the same normal sample (designated sample A). All mutations identified are necessarily false positives. To measure sensitivity, we simulated somatic mutations at controlled allele fractions by replacing selected reads in the virtual-tumor data set with reads from a second sample (designated sample B) at loci where sample A is the reference and sample B harbors a high-confidence germ-line heterozygous event. We then assessed the ability of an algorithm to detect these simulated somatic mutations. In this manner, we can measure sensitivity using real sequencing data at a desired depth of coverage and allelic fraction.
The two benchmarking approaches are complementary. Downsampling uses real somatic mutations but is limited in the parameter regimes it can be used to explore, and it cannot be used to measure specificity directly. In contrast, the virtual-tumor approach does not have these limitations. However, it simulates somatic mutations using germ-line events, which differ from somatic mutations in their nucleotide substitution frequencies and context. As recalibrated base qualities vary for the different bases (owing to biases in machine errors), there is variable sensitivity in the detection of different substitutions (Supplementary Fig. 2). Because the difference in sensitivity is minimal, we chose to use all the germ-line events. However, with the virtual-tumor approach it is possible to simulate the mutation spectrum of a specific tumor type by reweighting the germ-line events to match the expected mutation spectrum of the tumor.
Detection of somatic mutations with MuTect
MuTect takes as input sequence data from matched tumor and normal DNA after alignment of the reads to a reference genome and standard preprocessing steps38,39,40, which include marking of duplicate reads, recalibration of base quality scores and local realignment. The method operates on each genomic locus independently and consists of four key steps (Fig. 1): (i) removal of low-quality sequence data (Supplementary Methods); (ii) variant detection in the tumor sample using a Bayesian classifier; (iii) filtering to remove false positives resulting from correlated sequencing artifacts that are not captured by the error model; and (iv) designation of the variants as somatic or germ-line by a second Bayesian classifier.

MuTect takes as input next-generation sequencing data from tumor and normal samples and, after removing low-quality reads (Supplementary Methods), determines whether there is evidence for a variant beyond the expected random sequencing errors. Candidate variant sites are then passed through six filters to remove artifacts (Table 1). Next, a panel of normal samples (PON) filter is used to screen out remaining false positives caused by rare error modes only detectable in additional samples. Finally, the somatic or germ-line status of passing variants is determined using the matched normal sample. STD, standard; HC, high confidence.
Variant detection
Variants in the tumor data are identified by analyzing the data at each site under two alternate models: (i) a reference model, M0, which assumes there is no variant at the site and any observed nonreference bases are due to random sequencing errors, and (ii) a variant model,  , which assumes the site contains a true variant allele m at allele fraction f in addition to sequencing errors. The allele fraction f is unknown and is estimated as the fraction of tumor-sample reads that support m. This explicit modeling of f, instead of assuming a heterozygous, diploid event, makes our method more sensitive than other methods21,22. We declare m to be a candidate variant if the log-likelihood ratio of the data under the variant and reference models (that is, the log odds (LOD) score) exceeds a predefined decision threshold that depends on the expected mutation frequency and the desired false positive rate (Online Methods). The choice of decision threshold can be used to control the tradeoff between specificity and sensitivity, as described by a receiver operating characteristic (ROC) curve (Fig. 2a). We used a fixed threshold of 6.3 for all results presented here unless indicated otherwise. This threshold corresponds to a 106.3:1 odds ratio in favor of the reference model, which is reasonable because the frequency of mutations in many tumors is only 1–10 per Mb and thus the a priori odds of a site harboring a mutation may be as low as 1:105 or 1:106.
, which assumes the site contains a true variant allele m at allele fraction f in addition to sequencing errors. The allele fraction f is unknown and is estimated as the fraction of tumor-sample reads that support m. This explicit modeling of f, instead of assuming a heterozygous, diploid event, makes our method more sensitive than other methods21,22. We declare m to be a candidate variant if the log-likelihood ratio of the data under the variant and reference models (that is, the log odds (LOD) score) exceeds a predefined decision threshold that depends on the expected mutation frequency and the desired false positive rate (Online Methods). The choice of decision threshold can be used to control the tradeoff between specificity and sensitivity, as described by a receiver operating characteristic (ROC) curve (Fig. 2a). We used a fixed threshold of 6.3 for all results presented here unless indicated otherwise. This threshold corresponds to a 106.3:1 odds ratio in favor of the reference model, which is reasonable because the frequency of mutations in many tumors is only 1–10 per Mb and thus the a priori odds of a site harboring a mutation may be as low as 1:105 or 1:106.

(a) Sensitivity and specificity of MuTect for mutations with an allele fraction of 0.2, tumor sample sequencing depth of 30× and normal sample sequencing depth of 30× using various values of the LOD threshold (θ T) (0.1 ≤ θ T ≤ 100). Calculated sensitivity and false positive rate using a model of independent sequencing errors with uniform Q35 base quality scores and accurate read placement (Calculation) are shown as well as results from the virtual-tumor approach for the standard (MuTect STD) and high-confidence (MuTect HC) configurations. A typical setting of θ T = 6.3 is marked with black circles. (b) Sensitivity as a function of tumor sample sequencing depth and allele fraction (f ) using θ T = 6.3. Calculated sensitivity as in a is shown as well as results from the virtual-tumor approach and the downsampling of validated colorectal mutations7. Error bars, 95% confidence intervals (typically smaller than marks).
The LOD score is useful as a threshold for detection, as observed in the concordance of predicted sensitivity and measured sensitivity from the virtual-tumor approach (Fig. 2). Nonetheless, the LOD score cannot be immediately translated into the probability that a variant is due to true mutation rather than to sequencing error because the LOD score is calculated under an assumption of independent sequencing errors and accurate read placement. As we discuss below, these assumptions are incorrect, and as a result, although direct application of the LOD score accurately estimates the sensitivity to detect a mutation, it substantially underestimates the false positive rate.
Variant filtering
To eliminate these additional false positives due to inaccurate read placement and non-independent sequencing errors, we developed six filters (Fig. 1 and Table 1). In addition, we used a panel of normal samples as controls to eliminate both germ-line events and artifacts (Online Methods). Subsets of these filters define several versions of the method (Fig. 1): (i) standard (STD), which applies no filters and thus includes all detected variants; (ii) high-confidence (HC), which applies the six filters and (iii) high-confidence plus panel of normal samples (HC + PON), which additionally applies the 'panel of normal samples' (PON) filter.
We tested the utility of these filters by applying them to the virtual-tumors benchmark and recomparing the results with the calculations (Fig. 2a). The sensitivity estimated for both with (HC) and without (STD) filters was similar, indicating that the model is accurate with respect to detection and that the filters do not adversely impact sensitivity. However, after applying the filters (HC), specificity increased and closely followed the calculations, suggesting that the filters largely eliminate systematic false positives (Fig. 2a and Supplementary Fig. 3).
Variant classification
Finally, each variant detected in the tumor sample is designated as somatic (not present in the matched normal sample), germ-line (present in the matched normal sample) or variant (present in the tumor sample but indeterminate status in the matched normal sample as a result of insufficient data). To perform this classification, we used a LOD score that compares the likelihood of the data under models in which the variant is present as a heterozygote or absent in the matched normal sample (Online Methods). We declare that there are insufficient data for classification if the power to make a germ-line classification is less than 95%. We also used public germ-line variation databases41 as a prior probability of an event being germ-line.
Sensitivity
We applied several benchmarking methods to evaluate the sensitivity of our method to detect mutations as a function of sequencing depth and allelic fraction (Fig. 2b). First, we calculated the sensitivity under a model of independent sequencing errors and accurate read placement using our statistical test given an allelic fraction and tumor sample sequencing depth, and assuming that all bases have a fixed base quality score of Q35 (approximate mean base quality score in simulation data; Online Methods and Supplementary Fig. 4).
Next, to apply the downsampling benchmark, we used 3,753 validated somatic mutations, stratified by allele fraction (median = 0.28, range = 0.07–0.94), in colorectal cancer7 with deep-coverage (≥100×), exome-capture sequencing data downloaded from the database of Genotypes and Phenotypes (dbGAP; phs000178). Finally, to apply the virtual-tumor benchmark, we used deep-coverage data from two high-coverage, whole-genome samples (Coriell individuals NA12878 and NA12981) sequenced on Illumina HiSeq instruments as part of the 1000 Genomes Project42 and another previous study43, across 1 Gb of genomic territory. Note that we cannot use the PON filter (HC + PON) in the virtual-tumor sensitivity benchmark because it discards common germ-line sites.
Sensitivity estimates based on these three approaches were highly consistent with each other (median coefficients of variation for each depth of 3.1%). This suggests that the benchmarking approaches accurately estimate the sensitivity of mutation-calling methods and also that the calculated sensitivity is robust across a large range of parameter values, enabling us to confidently extrapolate to higher sequencing depths and lower allele fractions (Supplementary Table 1).
Based on this analysis, we observed that MuTect is a highly sensitive detection method. It detected mutations at a site with 30× depth in the tumor data (typical of whole-genome sequencing) and an allele fraction of 0.2 with 95.6% sensitivity. The sensitivity increased to 99.9% with deeper sequencing (50×) and dropped to 58.9% for detecting mutations with allelic fraction of 0.1 (at 30× sequencing; Fig. 2b and Supplementary Table 1). With 150× sequencing depth (typical of exome sequencing) we observed 66.4% sensitivity for 3% allele fraction events. It is this sensitivity to detect low-allele-fraction events that uniquely positions MuTect to analyze samples with low purity or with complex subclonal structure.
This detailed understanding of the factors determining sensitivity is critical for targeting the appropriate depth of sequencing. Because the allelic fraction of a mutation depends on the tumor purity, local copy number and clonality8, one can calculate the sequencing depth required for a desired sensitivity on a tumor-specific basis. Also, given a sequencing data set we can calculate the sensitivity to have detected a mutation with a particular allelic fraction for each base along the genome. This allows us to assert the absence of a mutation (with a specified allele fraction), which is particularly important in a clinical setting.
Specificity
It is trivial to create an extremely sensitive method to detect somatic mutations by identifying any site with a single nonreference read as a candidate mutation. Clearly, such an approach would result in an enormous false positive rate. Therefore in evaluating the performance of a mutation-detection method, it is critical to thoroughly characterize its specificity. There are two sources of false positives: (i) overcalling events for the tumor data and (ii) undercalling true germ-line events in the matched normal data. Overcalling in the tumor data is typically due to sequencing errors and inaccurate read placements, whereas undercalling of true germ-line events in the matched normal sample is often due to low sequencing depth in the normal sample.
To measure the false positive rate owing to overcalling for tumor data, we used the virtual-tumor approach across 1 Gb of NA12878 sequence data at various depths in the virtual tumor and at 30× in the virtual normal sample. All detected events are false positives, but to eliminate from consideration those resulting from undercalling germ-line events, we excluded all known germ-line variant sites. Using no filters (STD), the false positive rate increased with depth (from 6.7 Mb−1 at 5× coverage to 20.1 Mb−1 at 30× coverage; Fig. 3a). This was due to the increased power to call mutations with lower allele fractions, which are enriched with false positives (Fig. 3b). The HC filters reduced the false positive rate by an order of magnitude (1.00 Mb−1 at 30× coverage). The PON filter (HC + PON) then filtered out most of the remaining rare, but recurrent, artifacts (0.51 Mb−1 at 30× coverage). Certain filters, such as the 'poor mapping' filter, had the biggest effect at low depths, whereas other filters were more invariant with changes in sequencing depth, such as the 'proximal gap' filter (Fig. 3c). The 'clustered position' filter rejects the most sites exclusively. However, the majority of false positives are rejected by several filters.

(a) Somatic miscall error rate for true reference sites as a function of tumor sample sequencing depth for the STD, HC and HC + PON configurations of MuTect. Dashed line, desired false positive rate. Error bars, 95% confidence intervals. (b) Distribution of allele fraction for all miscalls as a function of tumor sample sequencing depth. (c) Fraction of events rejected by each filter; hashed regions indicate events rejected exclusively by each filter. (d) Somatic miscall error rate for true germ-line heterozygous single-nucleotide polymorphism sites by sequencing depth in the normal sample when the site is known to be variant in the population (in dbSNP) and previously unknown (not in dbSNP). Error bars, 95% confidence intervals. (e,f) Power as a function of sequencing depth in the normal sample to have classified these events as germ-line or somatic at previously unknown (e) and known (f) germ-line variant sites.
We then studied the errors owing to undercalling of true germ-line events in the matched normal sample with the same approach but instead using the ∼1 million germ-line variant loci in the same territory (Fig. 3d–f). In classifying an event as germ-line or somatic, MuTect uses different prior probabilities at sites of common germ-line variation versus the rest of the genome, and therefore we report the false positive rates separately for these two scenarios (Fig. 3d) along with the power to have classified such events (Fig. 3e,f). We observed that with ≤ 7 reads in the normal data at previously unknown germ-line variation sites (Fig. 3e) or with ≤18 reads at sites of known germ-line variation (Fig. 3f), there was insufficient data to classify a variant as being somatic or germ-line, and hence we kept such sites as 'variant' and never made false positive somatic calls in these cases. Once there was sufficient data to make a classification, the error rate dropped rapidly from 2.4 × 10−3 at 8× coverage in the normal sample to below 0.2 × 10−3 at 12× coverage, which corresponds to less than one misclassified germ-line event in the entire exome (∼30 Mb in the exome × 50 previously unknown germ-line variants Mb−1 × 0.2 × 10−3 error rate).
Finally, we have used MuTect in several recent studies and found a consistent validation rate of ∼95% in coding regions based on multiple orthogonal validation technologies3,4,7,19,24,25,26,27,28,29,30 (Table 2). These studies had used earlier versions of MuTect, which were less sensitive, but in a publication13 using the version of MuTect described in this paper, mutations present at 7% allelic fraction (8 of 102 reads) were detected and subsequently validated by ultra-deep sequencing (∼6,000× coverage). In fact, the validation rate is not the best measure for comparing false positive rates across studies because it depends on the ratio of false positive to true mutations, which varies across tumor types. We therefore also report the false positive rate itself (Table 2). We observed a median false positive rate of 0.16 Mb−1, which is lower than the rate we reported using whole-genome data (Fig. 3) but is consistent with the rate measured when restricting the analysis to coding regions (Supplementary Fig. 5), indicating that coding regions are less prone to sequencing and alignment errors.
Comparison to other methods
We used the downsampling and virtual-tumor benchmarking approaches to compare MuTect with other commonly used methods: SomaticSniper21, JointSNVMix22 and Strelka23. We tested each method in two configurations, standard (STD) and high confidence (HC), with thresholds chosen to produce similar false positive rates across the methods. For SomaticSniper (v1.0.0), we used the published configurations. For JointSNVMix (v0.7.5), we used a detection threshold of P(somatic) ≥ 0.95 for STD and P(somatic) ≥ 0.9998 for HC. For Strelka (version 0.4.7), we used the recommended configuration with a quality score ≥ 15 for HC and quality score ≥ 1 for STD.
We evaluated the sensitivity of the methods with regard to allele fraction and tumor sample sequencing depth using virtual-tumor (Fig. 4a) and downsampling (Supplementary Fig. 6) approaches, and observed a sharp distinction in sensitivity, particularly at lower allele fractions. We analyzed data for 30× sequence coverage. In the standard configurations, all methods showed ≥ 99.3% sensitivity for mutations at an allele fraction of 0.4. However, in the HC configurations, MuTect, JointSNVMix and Strelka remained sensitive (98.8%, 96.6% and 98.5%, respectively), whereas SomaticSniper sensitivity dropped to 91.5%. At an allele fraction of 0.1, MuTect HC detected more than half of the mutations (53.2%), whereas Strelka HC, JointSNVMix HC and SomaticSniper HC detected 29.7%, 16.8% and 7.4% of the mutations, respectively. At an even lower allele fraction of 0.05, MuTect HC had 16.0% sensitivity but this increased to 51.9% with 60× coverage. By contrast, JointSNVMix HC and SomaticSniper HC had a sensitivity of ≤ 2.0%, and the sensitivity did not increase appreciably with tumor sample sequencing depth. Strelka HC detected just 4.6% of the events at 30× coverage and only increased to 20.8% at 60× coverage. Sensitivity for such low-allelic-fraction events is critical for characterizing impure tumors or subclonal mutations in heterogeneous tumors, and MuTect was much more sensitive in this regime.

(a) Sensitivity as a function of tumor sample sequencing depth and mutation allele fraction (f ) for the indicated mutation-detection methods and configurations. (b) Somatic miscall error rate for true germ-line sites as a function of sequencing depth in the normal sample. (c) Somatic miscall error rate for true reference sites as a function of tumor sample sequencing depth. Dashed line, desired false positive rate. (d) Sensitivity as a function of specificity for mutations with an allele fraction of 0.1, tumor sample sequencing depth of 30× and normal sample sequencing depth of 30× for indicated methods and configurations. Black dashed lines indicate change in sensitivity and specificity between STD and HC configurations for a method. Gray solid lines are the MuTect results of virtual-tumor approach from Supplementary Figure 3. Error bars, 95% confidence intervals (a–c).
As a more sensitive method may also be less specific, we also compared the performance of the methods with regard to the two kinds of false positives. We observed a very low false positive rate owing to miscalled germ-line sites for all methods given sufficient depth (≥15×) in the matched normal sample (Fig. 4b). The false positive rates per megabase owing to miscalled reference sites (Fig. 4c) are comparable above 20× coverage in both the STD configuration (median = 10.2, range = 0.7–20.1) and the HC configuration (median = 1.0, range = 0.2–3.1).
We can summarize the tradeoff between sensitivity and specificity for each of the methods using a ROC curve, which depends on the sequencing depths in the tumor and normal samples and the mutation allele fraction. In Figure 4d we give an example using tumor sample sequencing depth of 30×, normal sample sequencing depth of 30× and allele fraction of 0.1, showing that MuTect is a generally more sensitive for a given specificity and also has a much smaller decrease in sensitivity for a similar increase in specificity gained by the HC configuration.
We also compared the sensitivity of the methods using previously reported sequencing data and validated mutations in the COLO-829 melanoma cell line37 (Supplementary Table 2). Although MuTect is slightly more sensitive than the other methods, this data set represents a pure cell line with easily detectable high-allelic-fraction events (median = 0.55) and thus does not expose differences between methods. By running MuTect and the other mutation callers we found additional mutations not originally reported (Supplementary Tables 3 and 4), underscoring that comparisons to mutations reported in the literature typically underestimate the sensitivity as the complete ground truth set of somatic mutations is often unknown.
Discussion
As new somatic-mutation callers are developed, the cancer genomics community will greatly benefit from a systematic measurement of performance using the approaches described here across the entire parameter space of tumor and normal samples at various sequencing depths and mutation allele fraction. Our method as well as the tools we developed to benchmark mutation-detection methods are available, and we encourage developers to report the characteristics of their method using these metrics. The approaches described here can be extended to other alterations such as insertion-deletions (indels) or rearrangements.
Our data suggest that MuTect has an advantage over other methods in terms of its tradeoff between specificity and sensitivity (Fig. 4). The advantage in sensitivity of MuTect is derived from the variant-detection statistical test, which includes an estimation of the allele fraction of the event and the working point chosen along the ROC curve. SomaticSniper and JointSNVMix use a model based on a clonal mutation in a pure, diploid tumor (and thus assume a fixed 50% allele fraction). This assumption reduces sensitivity for lower allele fraction events. In contrast, Strelka specifically considers allele fraction and thus in the STD configuration has similar sensitivity to MuTect. However, when running in the recommended HC configurations to control false positives, MuTect has only a minor drop in sensitivity compared with the other methods. This is likely because the filters in MuTect were carefully tuned to reject true false positive calls without sacrificing sensitivity.
We showed that MuTect is much more sensitive at a given specificity than competing methods, allowing us to more comprehensively characterize the landscape of somatic mutations, particularly those present in a small fraction of cancer cells. Moreover, this can be done with standard sequencing depths, enabling analysis of the large data sets that are being generated worldwide. Analysis of subclonal mutations and changes in the fractions of cancer cells that harbor them is a powerful way to study the evolution of subclones as they progress during treatment, metastasis and relapse11,12,44,45. In particular, we demonstrated that the presence of subclonal mutations in genes involved in driving chronic lymphocytic leukemia is an independent prognostic factor beyond the currently used clinical parameters13. Using standard exome sequencing data, we detected mutations present in as low as 10% of cancer cells, representing an expected allele fraction of 0.05 (assuming heterozygous mutations in a diploid region) even before accounting for stromal contamination, and found that these mutations appear to have an effect on time to first therapy13.
Because other methods are not as sensitive to low-allelic-fraction events, they may miss important subclonal drivers of progression or resistance. Therefore, the sensitivity of MuTect in detecting subclonal mutations with low allele fractions is a substantial advance, essential to future discoveries regarding the subclonal architecture of cancer and the translation of those discoveries into clinical diagnostics affecting cancer patient treatment and outcomes.
Methods
Virtual-tumor benchmarking approach.
The virtual-tumor approach begins with deep-coverage data from a high-coverage, whole-genome sample (NA12878) sequenced on Illumina HiSeq instruments: two libraries42, 'Solexa-18483' and 'Solexa-18484', at 30× each and one library43, 'Solexa-23661', at 30×. These data are publicly available; details are available in Supplementary Table 5.
First, we randomly divided the sequencing data into several partitions. We created six partitions from each of the three libraries (18 partitions total), therefore creating data partitions with ∼5× coverage each. We accomplished this by sorting the BAM39 by name using SortSam from the Picard (http://picard.sourceforge.net/) tools to effectively give the reads random ordering. We then randomly allocated each read to one of the partitions and wrote it to a partition-specific BAM file.
To measure specificity, we can designate certain partitions as the 'tumor' and others as the 'normal', and process them through MuTect (or any other method). Somatic mutations identified in this process are false positives as they are either germ-line events that are undercalled in the normal or erroneous variants resulting from sequencing noise overcalled in the partitions designated as tumor. We drew reads from libraries Solexa-18483 and Solexa-23661 for the tumor sample and from the library Solexa-18484 for the normal sample.
To measure sensitivity, we turned to additional sequencing data on a second individual (Supplementary Table 5). In this case we chose NA12891, which was also sequenced to 60× as part of the 1000 Genomes Project. Using the published high-confidence single-nucleotide polymorphism (SNP) genotypes for those samples from the 1000 Genomes Project, we identified a set of sites that are heterozygous in NA12891 and homozygous for the reference in NA12878. We then used a second utility, SomaticSpike, which is part of the MuTect software package, to perform a mixing experiment in silico. At each of the selected sites, this utility attempts to replace a number of reads determined by a binomial distribution using a specified allelic fraction in the NA12878 data with reads from the NA12891 data, therefore simulating a somatic mutation of known location, type and expected allele fraction. If there are not enough reads in NA12891 to replace the desired reads in NA12878, the site is skipped. The output of this process is a virtual tumor BAM with the in silico variants and a set of locations of those variants. Sensitivity is then estimated by attempting to detect mutations at these sites.
Variant detection.
For each site we denote the reference allele as r ∈{A,C,G,T} and denote by bi and ei the called base of read i (i = 1...d) that covers the site and the probability of error of that base call (each base has an associated Phred-like quality score qi where 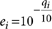 ). To call a variant in the tumor we try to explain the data using two models: (i) model M0 in which there is no variant at the site and all nonreference bases are explained by sequencing noise, and (ii) model
). To call a variant in the tumor we try to explain the data using two models: (i) model M0 in which there is no variant at the site and all nonreference bases are explained by sequencing noise, and (ii) model 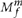 in which a variant allele m truly exists at the site with an allele fraction f and, as in M0, reads are also subject to sequencing noise. Note that M0 is equivalent to
in which a variant allele m truly exists at the site with an allele fraction f and, as in M0, reads are also subject to sequencing noise. Note that M0 is equivalent to 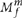 with f = 0.
with f = 0.
The likelihood of the model  is given by
is given by

assuming the sequencing errors are independent across reads. If all substitution errors are equally likely, that is, occur with probability ei/3, we obtain
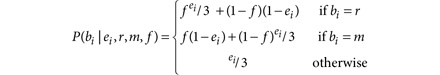
Variant detection is performed by comparing the likelihood of both models and if their ratio, that is, the LOD score, exceeds a decision threshold (log10 δT) we declare m as a candidate variant at the site. We calculate
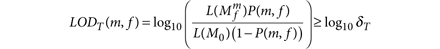
and set δT to 2 to ensure that we are at least twice as confident that the site is variant as compared to noise. We can then also rewrite LODT as

To determine P(m,f), we first assume that P(m) and P(f) are statistically independent and that P(f) is uniformly distributed (that is, P(f) = 1) and P(m) is one-third of the expected mutation frequency for the studied tumor type (representing equal prior for all substitutions). In practice, we used a typical mutation frequency of 3 × 10−6, which yields θT = 6.3.
We find the maximum LODT across all three values of m and to set the unknown allelic fraction parameter f, we could use maximum likelihood estimation, that is, find f that maximizes LODT. However, for computational efficiency, we instead estimated 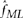 as
as
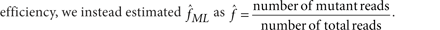
A common source of false positive mutation calls is contamination of the tumor DNA with DNA from other individuals. Germ-line SNPs in the contaminating DNA appear as somatic mutations. We have previously demonstrated that such contamination can yield many false positives and developed a tool, ContEst46, to estimate the contamination level, fcont, in sequencing data. Low-level contamination of DNA is a common phenomenon, and even 2% contamination can give rise to 166 false positive calls per megabase and 10 false positive calls per megabase when excluding known SNP sites46. To protect against this type of false positives and enable analysis of contaminated samples, we replaced the reference model with a variant model, 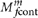 . This guarantees that variants are called only when they are highly unlikely to be explained by contamination.
. This guarantees that variants are called only when they are highly unlikely to be explained by contamination.
Variant filters: panel of normal samples.
To reduce false positives and miscalled germ-line events, we used a panel of normal samples as a filter. To create this filter, we ran MuTect on a set of normal samples as if they were tumor samples without a matched normal sample in STD mode. From these data, a VCF file is created for the sites that were identified as variant by MuTect in more than one normal sample.
This VCF is then supplied to the caller, which rejects these sites. However, if the site was present in the supplied VCF of known mutations it is retained because these sites could represent known recurrent somatic mutations that have been detected in the panel of normal samples when the normal samples are from adjacent tissue or have some contamination tumor DNA.
The more normal samples are used to construct this panel, the higher the power will be to detect and remove rare artifacts. Therefore, we typically used all the normal samples readily available. The results presented here were obtained by using a panel of whole-genome sequencing data from blood normal samples of 125 patients with solid tumor cancer. The samples used as part of the virtual-tumor approach were not included in this panel.
Variant classification.
To perform this classification, we used a similar classifier to the one described above. In this case, f in 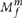 was conservatively set to 0.5 for a germ-line heterozygous variant. Thus we have
was conservatively set to 0.5 for a germ-line heterozygous variant. Thus we have

which can be rewritten as
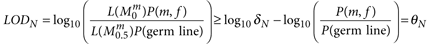
Note that here the terms are inverted because we want to be confident that alteration was not present. For δN, we set a threshold of 10, which is higher than the threshold for δT because we want to be more confident in our variant classification as misclassified germ-line events will quickly appear to be significant in downstream somatic analysis owing to their elevated population frequency at recurrent sites as compared to real somatic events.
To calculate P(germ line) we distinguished two cases: (i) sites which are known to be variant in the population and (ii) all other sites. We used the public dbSNP database41 to make this distinction.
There are ∼30 × 106 sites known to be variant in the human population according to dbSNP release 134, which is ∼1,000 variants/Mb. A given individual typically has ∼3 × 106 variants in their genome, 95% of which are in dbSNP sites41,42. Therefore we expect ∼50 variants/Mb not at dbSNP sites, that is, P(germ line|non-dbSNP site) = 5 × 10−5 and therefore we use θN|non–dbSNP site = 2.2. At dbSNP sites, however, we expect 95% of the ∼3 × 106 variants to occur in the 30 × 106 sites in the dbSNP database, yielding P(germ line|dbSNP site) = 0.095, hence θN|dbSNP site = 5.5.
Sensitivity calculation.
To calculate the sensitivity to detect a mutation with allelic fraction f using n reads having a Phred-like quality score q (and hence a base error, e, of  ), we first calculated k, the minimum number of reads with the alternate allele that will trigger a variant call using
), we first calculated k, the minimum number of reads with the alternate allele that will trigger a variant call using

The sensitivity is then the probability of observing k or more reads given the allelic fraction and depth. The marginal distribution of the number of reads with the alternate allele, either originating from the alternate base or a misread reference base, follows a binomial distribution with a frequency that reflects the true underlying allelic fraction f and the probability of error e (note that here we take the worst case in which all misread bases convert to the same alternate allele). Therefore we can calculate the probability of having observed k or more reads as
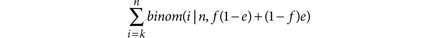
References
Cancer Genome Atlas Research Network. Integrated genomic analyses of ovarian carcinoma. Nature 474, 609–615 (2011).
Cancer Genome Atlas Research Network. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 455, 1061–1068 (2008).
Banerji, S. et al. Sequence analysis of mutations and translocations across breast cancer subtypes. Nature 486, 405–409 (2012).
Stransky, N. et al. The mutational landscape of head and neck squamous cell carcinoma. Science 333, 1157–1160 (2011).
Ding, L. et al. Somatic mutations affect key pathways in lung adenocarcinoma. Nature 455, 1069–1075 (2008).
Berger, M.F. et al. Melanoma genome sequencing reveals frequent PREX2 mutations. Nature 485, 502–506 (2012).
Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature 487, 330–337 (2012).
Carter, S.L. et al. Absolute quantification of somatic DNA alterations in human cancer. Nat. Biotechnol. 30, 413–421 (2012).
Walter, M.J. et al. Clonal architecture of secondary acute myeloid leukemia. N. Engl. J. Med. 366, 1090–1098 (2012).
Park, S.Y., Gönen, M., Kim, H.J., Michor, F. & Polyak, K. Cellular and genetic diversity in the progression of in situ human breast carcinomas to an invasive phenotype. J. Clin. Invest. 120, 636 (2010).
Nik-Zainal, S. et al. The life history of 21 breast cancers. Cell 149, 994–1007 (2012).
Ding, L. et al. Clonal evolution in relapsed acute myeloid leukaemia revealed by whole-genome sequencing. Nature 481, 506–510 (2012).
Landau, D.A. et al. Evolution and impact of subclonal mutations in chronic lymphocytic leukemia. Cell advance online publication, doi: 10.1016/j.cell.2013.01.019 (14 February 2013).
Campbell, P.J. et al. The patterns and dynamics of genomic instability in metastatic pancreatic cancer. Nature 467, 1109–1113 (2010).
Navin, N. et al. Tumour evolution inferred by single-cell sequencing. Nature 472, 90–94 (2011).
Hou, Y. et al. Single-cell exome sequencing and monoclonal evolution of a JAK2-negative myeloproliferative neoplasm. Cell 148, 873–885 (2012).
Xu, X. et al. Single-cell exome sequencing reveals single-nucleotide mutation characteristics of a kidney tumor. Cell 148, 886–895 (2012).
International Cancer Genome Consortium et al. International network of cancer genome projects. Nature 464, 993–998 (2010).
Chapman, M.A. et al. Initial genome sequencing and analysis of multiple myeloma. Nature 471, 467–472 (2011).
Getz, G. et al. Comment on “The consensus coding sequences of human breast and colorectal cancers”. Science 317, 1500 (2007).
Larson, D.E. et al. SomaticSniper: identification of somatic point mutations in whole genome sequencing data. Bioinformatics 28, 311–317 (2012).
Roth, A. et al. JointSNVMix: a probabilistic model for accurate detection of somatic mutations in normal/tumour paired next-generation sequencing data. Bioinformatics 28, 907–913 (2012).
Saunders, C.T. et al. Strelka: accurate somatic small-variant calling from sequenced tumor-normal sample pairs. Bioinformatics 28, 1811–1817 (2012).
Barbieri, C.E. et al. Exome sequencing identifies recurrent SPOP, FOXA1 and MED12 mutations in prostate cancer. Nat. Genet. 44, 685–689 (2012).
Bass, A.J. et al. Genomic sequencing of colorectal adenocarcinomas identifies a recurrent VTI1A–TCF7L2 fusion. Nat. Genet. 43, 964–968 (2011).
Wang, L. et al. SF3B1 and other novel cancer genes in chronic lymphocytic leukemia. N. Engl. J. Med. 365, 2497–2506 (2011).
Pugh, T.J. et al. Medulloblastoma exome sequencing uncovers subtype-specific somatic mutations. Nature 488, 106–110 (2012).
Berger, M.F. et al. The genomic complexity of primary human prostate cancer. Nature 470, 214–220 (2011).
Lohr, J.G. et al. Discovery and prioritization of somatic mutations in diffuse large B-cell lymphoma (DLBCL) by whole-exome sequencing. Proc. Natl. Acad. Sci. USA 109, 3879–3884 (2012).
Imielinski, M. et al. Mapping the hallmarks of lung adenocarcinoma with massively parallel sequencing. Cell 150, 1107–1120 (2012).
Wang, P. et al. Mutations in isocitrate dehydrogenase 1 and 2 occur frequently in intrahepatic cholangiocarcinomas and share hypermethylation targets with glioblastomas. Oncogene advance, online publication, doi:10.1038/onc.2012.315 (23 July 2012).
Durinck, S. et al. Temporal dissection of tumorigenesis in primary cancers. Cancer Discov. 1, 137–143 (2011).
Lee, R.S. et al. A remarkably simple genome underlies highly malignant pediatric rhabdoid cancers. J. Clin. Invest. 122, 2983–2988 (2012).
Cancer Genome Atlas Research Network. et al. Comprehensive genomic characterization of squamous cell lung cancers. Nature 489, 519–525 (2012).
Hodis, E. et al. A landscape of driver mutations in melanoma. Cell 150, 251–263 (2012).
Forbes, S.A. et al. COSMIC: mining complete cancer genomes in the catalogue of somatic mutations in cancer. Nucleic Acids Res. 39, D945–D950 (2011).
Pleasance, E.D. et al. A comprehensive catalogue of somatic mutations from a human cancer genome. Nature 463, 191–196 (2010).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
Li, H. et al. The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
DePristo, M.A. et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 43, 491–498 (2011).
Sherry, S.T. et al. dbSNP: the NCBI database of genetic variation. Nucleic Acids Res. 29, 308–311 (2001).
1000 Genomes Project Consortium. A map of human genome variation from population-scale sequencing. Nature 467, 1061–1073 (2010).
Gnerre, S. et al. High-quality draft assemblies of mammalian genomes from massively parallel sequence data. Proc. Natl. Acad. Sci. USA 108, 1513–1518 (2011).
Shah, S.P. et al. Mutational evolution in a lobular breast tumour profiled at single nucleotide resolution. Nature 461, 809–813 (2009).
Yachida, S. et al. Distant metastasis occurs late during the genetic evolution of pancreatic cancer. Nature 467, 1114–1117 (2010).
Cibulskis, K. et al. ContEst: estimating cross-contamination of human samples in next-generation sequencing data. Bioinformatics 27, 2601–2602 (2011).
Acknowledgements
This work was supported by US National Institutes of Health grants U54HG003067 and U24CA143845. We thank the Genome Analysis ToolKit (GATK) group, and our beta test users for their valuable feedback.
Author information
Authors and Affiliations
Contributions
D.J. posed the concept of using a statistical method and filters to detect somatic mutations. G.G. and K.C. conceived and designed MuTect and the analysis. K.C. implemented the algorithm and performed the analysis. M.S.L. conceived of and initially developed the PON filter. G.G. and S.L.C. developed the power calculations and investigated subclonal events detected with MuTect. C.S. and A.S. assisted in the generation and interpretation of validation data. D.J., C.S. and M.M. critically reviewed the manuscript. K.C., G.G. and E.S.L. wrote the manuscript. G.G., M.M., S.G. and E.S.L. led the project.
Corresponding author
Ethics declarations
Competing interests
K.C., G.G. and M.S.L. are inventors on US provisional patent application no. 61/693,987 covering the method described in the paper.
Supplementary information
Supplementary Text and Figures
Supplementary Methods and Supplementary Figures 1–6 (PDF 265 kb)
Supplementary Tables 1 and 4
MuTect Calculated Sensitivity by Mutant Allele Fraction and Tumor Sequencing Depth and Novel Chromosome 20 COLO-829 Mutations Detected By 4 Methods (MuTect, SomaticSniper, JointSNVMix and Strelka) (XLSX 24 kb)
Supplementary Data
MuTect source code (ZIP 101 kb)
Rights and permissions
About this article
Cite this article
Cibulskis, K., Lawrence, M., Carter, S. et al. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat Biotechnol 31, 213–219 (2013). https://doi.org/10.1038/nbt.2514
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/nbt.2514
This article is cited by
-
Multi-omic profiling reveals associations between the gut microbiome, host genome and transcriptome in patients with colorectal cancer
Journal of Translational Medicine (2024)
-
DNA damage response alterations in clear cell renal cell carcinoma: clinical, molecular, and prognostic implications
European Journal of Medical Research (2024)
-
Race-specific coregulatory and transcriptomic profiles associated with DNA methylation and androgen receptor in prostate cancer
Genome Medicine (2024)
-
Genomic features reveal potential benefit of adding anti-PD-1 immunotherapy to treat non-upper aerodigestive tract natural killer/T-cell lymphoma
Leukemia (2024)
-
Children born after assisted reproduction more commonly carry a mitochondrial genotype associating with low birthweight
Nature Communications (2024)
