
Abstract
Mediators from peripheral tissues can influence the development and progression of heart failure (HF). For example, in obesity, an altered profile of adipokines secreted from adipose tissue increases the incidence of myocardial infarction (MI). Less appreciated is that heart remodeling releases cardiokines, which can strongly impact various peripheral tissues. Inflammation, and, in particular, activation of the nucleotide-binding oligomerization domain-like receptors with pyrin domain (NLRP3) inflammasome are likely to have a central role in cardiac remodeling and mediating crosstalk with other organs. Activation of the NLRP3 inflammasome in response to cardiac injury induces the production and secretion of the inflammatory cytokines interleukin (IL)-1β and IL-18. In addition to having local effects in the myocardium, these pro-inflammatory cytokines are released into circulation and cause remodeling in the spleen, kidney, skeletal muscle and adipose tissue. The collective effects of various cardiokines on peripheral organs depend on the degree and duration of myocardial injury, with systematic inflammation and peripheral tissue damage observed as HF progresses. In this article, we review mechanisms regulating myocardial inflammation in HF and the role of factors secreted by the heart in communication with peripheral tissues.
Similar content being viewed by others
Introduction
Heart failure and the role of inflammation
Cardiovascular diseases are the leading cause of death worldwide, and heart failure (HF) is an important contributor to this statistic.1 When the heart is under stress or injured, it undergoes structural and functional changes termed cardiac remodeling.2 These include cardiac hypertrophy, fibrosis, apoptosis and altered metabolism.3 When an individual suffers from myocardial ischemia, it is intuitively important to re-perfuse the damaged area and re-establish the supply of blood to the damaged area. However, it has also been realized that some cellular events which occur during reperfusion may lead to worse outcomes, a phenomenon termed myocardial ischemia/reperfusion (I/R) injury.4
The various mechanisms underlying the detrimental effects of ischemia and subsequent reperfusion are complex and are not fully understood. Nevertheless, a number of clinical and animal studies suggest that inflammation is a key contributor to adverse myocardial remodeling.4 Broadly speaking, inflammation is a wound-healing process mediated by innate immune cells that recognize microbial and non-microbial sources of danger/stress. Inflammation triggered in the absence of infection is termed ‘sterile inflammation’. Multiple studies have highlighted the importance of targeting sterile inflammation in HF.5, 6, 7 Sterile inflammation involves the secretion of inflammatory cytokines and recruitment of innate immune cells, such as neutrophils and monocytes/macrophages. However, prolonged exposure to inflammatory cytokines will exacerbate adverse remodeling and enhance myocardial damage.8 Importantly, in addition to local adverse effects on cardiac remodeling, ischemia- or I/R-induced inflammation in the heart releases pro-inflammatory cytokines, such as interleukin (IL)-1β and IL-18, into circulation. These, and other so-called cardiokines, can have significant endocrine effects on other tissues, leading to damage in multiple peripheral organs.9 For example, prolonged exposure to IL-1β and IL-18 can lead to caspase-1-dependent cell death via pyroptosis.10, 11 Thus, crosstalk from the heart to other tissues can elicit multi-organ damage as a consequence of ischemia-induced inflammation.9 This review highlights the current knowledge of inflammasome activation in the heart and its consequences on other organs.
Mechanisms regulating cardiac inflammation in HF, focus on the NLRP3 inflammasome
The nucleotide-binding oligomerization domain-like receptors with pyrin domain (NLRP3) inflammasome is a cytoplasmic protein complex composed of NLRP, apoptosis-associated speck-like protein containing CARD (ASC), a caspase recruitment domain and pro-caspase-1.12, 13 NLRP is composed of C-terminal leucine-rich repeats, a central nucleotide domain (NACHT) and N-terminal effector pyrin domain. Upon recognizing patterns, either from a pathogenic source (pathogen-associated molecular patterns) or from a non-pathogenic source (danger/damage-associated molecular patterns, DAMPs), NLRP will recruit ASC, which, in turn, recruits pro-caspase 1, which will then get activated.14 Inflammasomes are classified based on NLRPs, which recognize or sense different stimuli.15 The NLRP3 inflammasome is the most widely studied to date due to its ability to recognize various cellular stressors and its strong relationship with diseases such as HF.16 The key consequence of inflammasome activation is maturation of pro-inflammatory cytokines, in particular IL-1β and IL-18. The generation of active forms of IL-1β and IL-18 is regulated at two steps: expression of pro-IL-1β and pro-IL-18 is mediated by nuclear factor kappa-light chain enhancer of activated B cells (NF-κB), and processing to the mature form of IL-1β and IL-18 is mediated by active caspase-1 in the inflammasome.14
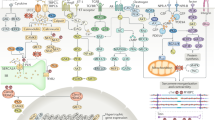
Multiple DAMPs have been found to activate NLRP3 inflammasomes, including monosodium urate, calcium phosphate crystals, cholesterol crystals, amyloid β, hyaluronan, islet amyloid polypeptide, asbestos and silica.14 However, in HF, we suggest that mitochondria have a critical role in initiating inflammasome activation.17, 18 In HF-associated inflammasome activation, the three main triggers are adenosine triphosphate (ATP), mitochondrial DNA (mtDNA), and reactive oxygen species (ROS) (see Figure 1). When cells undergo death they release ATP. Multiple studies have suggested that ATP directly activates the NLRP3 inflammasome.19, 20 High extracellular ATP levels activate P2X7 purinergic receptors to cause potassium efflux. Low intracellular levels of potassium promote the assembly of NLRP3 and ASC. In addition, it has been suggested that low intracellular potassium will also promote pannexin-1 membrane pore formation, further easing the access of inflammasome activating agents.21 mtDNA has been established as a DAMP when liberated into the extracellular space.22, 23 It was shown24 that the translocation of mtDNA to the cytosol was associated with subsequent inflammasome activation. A DNAse treatment reduced secretion of IL-1β in macrophages. It has also been reported25 that mitochondrial dysfunction and oxidized mtDNA directly activate the NLRP3 inflammasome. Macrophages lacking mtDNA or treated with the oxidized nucleoside 8-OH-dG to confer competitive inhibition had severely attenuated IL-1β secretion. Mitochondrial marker and NLRP3 inflammasome colocalization and a significant activation of NLRP3 inflammasome upon mitochondrial membrane disruption have been shown.26 It was reported27 that activation of the NLRP3 inflammasome in macrophages occurred due to an ATP-mediated ROS-dependent activation of phosphoinositide 3 kinase signaling. ROS stimulates the activation of NF-kB and increases the expression of pro-IL-1β and pro-IL-18.21

Mechanisms of NLRP3 inflammasome activation in heart failure. Myocardial infarction (MI), ischemia or ischemia/reperfusion (I/R) injury induces cardiomyocytes to release ROS, ATP and mtDNA. ROS mediates autocrine and paracrine activation and nuclear translocation of NF-κB, which regulates the transcription of pro-IL-1β and pro-IL-18. mtDNA directly primes NLRP3 and ATP via binding to P2X7 receptors and leads to potassium efflux, a trigger for the assembly of NLRP3 inflammasome. These collective effects result in activation of the NLRP3 inflammasome-associated caspase-1, which processes pro-IL-1β and pro-IL-18 into mature IL-1β and IL-18 and can exacerbate local inflammation. It can also be released into circulation to mediate endocrine effects. ATP, adenosine triphosphate; IL, interleukin; mtDNA, mitochondrial DNA; NF-κB, nuclear factor kappa-light-chain-enhancer of activated B cells; NLRP3, NLR family, pyrin domain containing 3; ROS, reactive oxygen species; K+, potassium.
Mitochondrial regulation by autophagy in the heart
Because all the mediators discussed above (ATP, mtDNA and ROS) may come from mitochondria, our hypothesis is that this organelle has a vital role in inflammasome activation. Thus, mitochondrial integrity is a key limiting factor for NLRP3 inflammasome activation. Furthermore, this may be especially relevant in the heart where cardiomyocytes have a higher mitochondrial content relative to other cell types. Overall, the heart is likely to be highly susceptible to mitochondria-derived DAMPs. Thus, effective regulation of damaged mitochondria is critical. Autophagy is a quality control system mediating degradation of protein aggregates and damaged organelles. Multiple studies have documented the importance of mitochondrial regulation by autophagy, specifically referred to as mitophagy, in HF.28 Multiples studies have also now established a strong association between autophagy and inflammasome activation. First, stimulating autophagy in macrophages using rapamycin can directly target precursors of IL-1β for degradation. Mice pretreated with rapamycin showed reduced circulating levels of IL-1β following a challenge with an inflammatory stimulus.29 Using ATG16L1-deficient cells, it was shown that autophagy was involved in endotoxin (lipopolysaccharide)-induced inflammasome activation and increased IL-1β and IL-18 secretion.30 Another study31 reported that inflammasomes can be directly sequestered into autophagosomes and destined for autophagic degradation. Nakahira et al.24 reported on the regulation of mtDNA-driven inflammasome activation by autophagy. They deleted genes encoding key autophagy proteins LC3B and Beclin1, and found a significant enhancement in caspase-1 activation and secretion of IL-1β and IL-18. Thus, defective autophagy-mediated quality control mechanisms resulted in enhanced inflammasome activation via the accumulation of damaged mitochondria and reduced inflammasome clearance in both in vitro and in vivo settings.
Distinct roles of cardiomyocytes, fibroblasts and immune cells in cardiac inflammasome activation
Numerous studies have now established a strong association between inflammasome activation and adverse remodeling in HF. For example, both ASC-KO and caspase-1-KO mice exhibited a significant reduction in infarct zone and fibrosis, as well as improved cardiac function after myocardial I/R injury.32 As highlighted in Figure 1, it has been proposed that activation of the inflammasome occurs via cell-to-cell communication within heterogeneous cell populations of heart tissue, including cardiomyocytes, fibroblasts and innate immune cells.6, 33 Kawaguchi et al.32 identified that both hematopoietic and non-hematopoietic cells are responsible for secreting IL-1β after myocardial I/R injury, because only chimeric mice with ASC-KO bone marrow on an ASC-KO background showed reduction in infarct zone. They followed up with in vitro experiments in which hypoxia/reoxygenation stimulated inflammasome activation in cardiac fibroblasts, but not in cardiomyocytes. This notion was supported by studies in adult cardiomyocytes in which NLRP3 inflammasome activation was inhibited using either siRNA or pharmacological inhibitors. This resulted in fewer cell deaths but not IL-1β secretion.34 Upon permanent myocardial ischemia in both murine and rat models, myocardial fibroblasts were shown to be the primary source of IL-1β secretion in response to ATP released from damaged neighboring cells.35 Further work36 has also supported the notion of non-immune cell-mediated IL-1β and IL-18 secretion. This work concluded that mitochondrial ROS from cardiomyocytes acts as a trigger to prime the NLRP3 inflammasome. Taken together, the data suggest that cardiomyocytes, cardiac fibroblasts and infiltrating immune cells contribute via different roles toward inflammation and cardiac remodeling in myocardial infarction (MI) (Figure 1).
Crosstalk between the heart and adipose tissue
Alterations in adipokine profiles influence the development of HF
There is a well-documented association between obesity and HF.2 Adipose tissue is clearly an important contributor to inflammation in HF. Multiple studies have established both pro- and anti-inflammatory effects of adipokines.37, 38 In obesity, adipose tissue undergoes changes induced by metabolic stress. It releases more pro-inflammatory cytokines, including IL-6, IL-8, monocyte chemoattractant protein-1 (MCP-1), and less of the anti-inflammatory cytokines, including IL-10 and adiponectin.39, 40, 41 Visceral fat is the most important depot, which responds to metabolic stress in this way. There is a well-established positive correlation between visceral fat levels and HF.42 However, it is interesting to note that epicardial and pericardial fat depots exhibit a similar phenotype to visceral fat and have been strongly correlated with the progression of adverse cardiac remodeling.43 McKenney et al.44 observed increased epicardial adipose tissue after MIs, which correlated with a reduced adiponectin level after MI. In their study they compared pigs with or without adipectomy subjected to MI. They observed that the progression of adverse remodeling after the MI was attenuated, and the infarct zone size was diminished in adipectomized animals. This correlates with a previous observation in which a pig with myocardial I/R injury developed improved cardiac function, reduced infarct size and less tumor necrosis factor alpha (TNFα) production with a greater production of IL-10 after intracoronary administration of adiponectin.45 In summary, whereas it is generally accepted that in obesity, the profile of adipokines from various fat depots mediates detrimental effects on the myocardium, the obesity paradox suggests that these adipokines can confer beneficial effects during post-MI stages of remodeling.2
Extensive epidemiological and clinical data suggest that type 2 diabetes increases the risk for HF independently of other risk factors, such as hypertension.46, 47 One potential mechanism is that type 2 diabetes, often associated with obesity, leads to myocardial lipotoxicity that contributes to cell death, and thus, to cardiac dysfunction. Diabetic cardiomyopathy is also characterized by interstitial and perivascular fibrosis. A significant increase in collagen deposition was found around intramural vessels and between myofibers in heart biopsies in patients with diabetes.48, 49, 50 Given that fibrosis is one consequence of inflammation, IL-1β and other inflammatory markers, such as fibrosis, signal the onset and progression of HF in this way.51, 52, 53, 54
Cardiokine and endocrine effects on peripheral tissues in HF
In recent years, there has been an increased realization of endocrine effects mediated by factors produced and secreted by the heart.55 Collectively, these are referred to as cardiokines. Ischemic stress results in a substantial change in the profile of cardiokines secreted from the myocardium.9, 55 In particular, upon activation of the inflammasome and infiltration of splenocytes in the infarct zone, the heart will release more pro-inflammatory cytokines.9 Cardiac fibroblasts have been proposed as the principal source of inflammatory signals in pathological conditions, although cardiomyocytes also contribute to the pro-inflammatory environment in the myocardium by producing different cytokines and chemokines.56, 57 Injured cardiac cells release damage-associated molecular pattern molecules, such as high-mobility group box 1, DNA fragments, heat-shock proteins and matricellular proteins, which instruct surrounding healthy cardiomyocytes to produce inflammatory mediators. These mediators, mainly IL-1β, IL-18, IL-6, MCP-1 and TNFα, in turn activate versatile signaling networks within surviving cardiomyocytes and trigger leukocyte activation and recruitment.
Evidence for myocardial production of TNFα has been controversial.58, 59 However, it is now clear that TNFα can be produced by isolated cardiomyocytes under certain conditions, such as treatment with lipopolysaccharide.60, 61, 62, 63 Similarly, increased expression of TNFα in cardiac myocytes and fibroblasts isolated from failing hearts suggests that if exposed to pathophysiological stimuli, the heart has the capacity to produce TNFα.64, 65 IL-6 can be produced in most cells in the heart, including cardiomyocytes66, 67 and fibroblasts.68, 69 A lipopolysaccharide treatment or hypoxia-reoxygenation stimulated the production of IL-1β in isolated cardiac fibroblasts, while isolated cardiomyocytes did not respond to either treatment.32 A co-culture of cardiomyocytes with fibroblasts induced by an angiotensin-II treatment secreted much greater levels of IL-6 and TNFα than cultures of fibroblasts alone, indicating that a paracrine action has a vital role in the production of pro-inflammatory cytokines.70
Another good example of a cardiokine is atrial natriuretic peptide (ANP), which is produced mainly in the myocardium. Its expression is enhanced during myocardial stretching.71 ANP has a beneficial role in cardiac remodeling by acting in an autocrine or paracrine manner. For example, treatment with cultured cardiac myocytes with an antagonist of ANP receptor HS-142-1 increased expression of contractile protein genes, such as skeletal-actin and beta-myosin heavy chain, as well as the size of cardiomyocytes.72 ANP also contributes to oxytocin-induced protection in myocardial ischemia-reperfusion injury by reducing lipid peroxidation in a nitric oxide-dependent mechanism.73
ANP receptors are found in adipose tissue and mediate effects, including enhanced lipolysis and energy expenditure, as well as altering adipokine production and release.74, 75, 76 Thus, natriuretic peptides can definitely influence peripheral metabolism by acting on adipose tissue. Therapeutically targeting ANP action may confer metabolic and cardiovascular benefits in the future.74
Crosstalk between the heart and spleen: the cardio-splenic axis
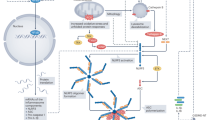
Neutrophil activation and leukocyte infiltration in the heart are prominent features of MIs that exacerbate inflammatory cytokine release and tissue damage.77 Indeed, the mononuclear phagocyte network undergoes extensive remodeling after MI. There are different subpopulations of monocytes residing in mice. These are converted from one to another upon inflammatory responses after an MI. Monocytes are generally classified into two categories: migratory monocytes with inflammatory characteristics, which express high levels of Ly6C and CC chemokine receptor CCR2 and low levels of fractalkine receptor CX3CR1 (Ly-6Chi,CCR2hiCX3CR1low), and reparative monocytes with anti-inflammatory profiles (Ly-6Clow,CCR2lowCX3CR1high).78 The exact mechanism of how each phenotype of monocytes regulates the inflammatory response during an MI is complicated and is not resolved.79 However, under acute MI conditions, monocyte recruitment to the heart is very dynamic and largely dependent on the spleen. The spleen is one of most important lymphoid tissues. It has a role in filtering blood and regulating immune responses to circulating agents.80 The spleen contains large pools of undifferentiated monocyte reservoirs80, 81 that can undergo splenic hematopoiesis, increasing motility and pro-inflammatory characteristics (Ly-6Chi).81 Recruitment of reparative monocytes (Ly-6low) ultimately helps resolve inflammation and promote tissue healing.81, 82
Dendritic cells (DCs), specialized for presenting antigens to T cells, also have an important role in the immune response to an MI.83 In an acute MI, both DCs and monocytes/macrophages have been shown to positively contribute to tissue healing. Upon an initial cardiac inflammatory response, DCs infiltrate the infarcted area to confer a protective role. This is demonstrated with DC-ablated mice exhibiting greater adverse cardiac remodeling after an MI.84 In these mice, there was sustained expression of pro-inflammatory cytokines, IL-1β, IL-18 and TNFα, yet reduced IL-10 expression.84
Interestingly, under chronic MI conditions, the protective roles of splenocytes become detrimental. Mice with a long-term MI (8 weeks) showed profound splenic remodeling with a prolonged existence of pro-inflammatory monocytes (Ly-6Chi) and increased expression of alarmins.85 A splenectomy was performed to investigate the role of splenocytes (splenic monocytes/macrophages and DCs) in the progression of HF-associated inflammation and post-MI remodeling. Intriguingly, mice without spleens showed less cardiac dysfunction. This was associated with attenuated monocytes/DC infiltration in the heart.85 When splenocytes from the mice with an MI were injected into normal mice, the recipient mice developed left ventricule (LV) dilation, cardiac hypertrophy, systolic dysfunction, myocardial apoptosis and fibrosis.85 However, the recipient mice did not reveal changes in pro-inflammatory cytokines in circulation, indicating that the adverse cardiac remodeling in chronic MI model was specifically due to splenocytes. Therefore, there is a clear sequential activation of inflammatory responses depending on the duration of MI via splenocyte-mediated crosstalk to the heart (Figure 2).

Crosstalk mechanisms in the cardio-splenic axis in heart failure, and their functional consequences on peripheral tissues. In acute myocardial infarction (MI), splenocytes (splenic monocytes/macrophages and dendritic cells) migrate to the heart and mediate protective effects during the inflammatory response. In chronic MI, cardiokines induce dramatic changes in the spleen, such that splenocytes develop inflammatory profiles. This exacerbates existing inflammation in the heart and promotes adverse cardiac remodeling leading to cardiac dysfunction. Inflammatory splenocytes also lead to peripheral organ damage. For example, kidney inflammation results in enhanced activation of the renin-angiotensin system (RAS) and release of lipocalin-2. In vasculature, inflammation results in adverse alterations in vascular tone and vascular cell proliferation. Skeletal muscle is also strongly affected by heart failure, at least in part via inflammation resulting in cellular changes, such as sphingosine accumulation and muscle wasting. In adipose tissue, inflammation results in reduced adiponectin levels and further increased levels of pro-inflammatory adipokines (IL-6, MCP-1, IL-10). IL, interleukin; MCP-1, monocyte chemotactic protein 1.
Crosstalk between the heart and kidney: cardio-renal axis
Multiple clinical studies have suggested that patients with chronic kidney disease experience extremely high mortality rates following acute MI.86, 87, 88, 89 This suggests that there is a strong crosstalk between the kidneys and heart. The association between end-stage renal disease and cardiovascular disease is often termed cardio-renal syndrome.90 The renin-angiotensin system (RAS), a signaling cascade responsible for regulating blood pressure, has a well-established critical role in cardio-renal syndrome.91 Ogawa et al.92 reported that nephrectomy in mice with an MI influenced cardiac remodeling after the MI. The combination of nephrectomy and MI resulted in deteriorated left ventricular remodeling and RAS activation, oxidative stress and MCP-1. This observation was similar to transgenic mice overexpressing renin and angiotensinogen after a coronary artery ligation (CAL) surgery.92 This correlates with previous findings that cardiomyocytes increase the expression of TNFα and IL-1 family through activation of NF-kB and activator protein 1 transcription factor in response to angiotensin II.93, 94, 95
Other than RAS, a new biomarker has been identified that strongly correlated with cardio-renal syndrome. HF patients with declined renal function exhibit elevated levels of neutrophil gelatinase-associated lipocalin (also known as lipocalin-2)96. Neutrophil gelatinase-associated lipocalin levels are strongly correlated with inflammation and cardiac remodeling in HF patients with renal dysfunction.96 Pro-inflammatory effects of lipocalin-2 are also known to induce endothelial dysfunction97, 98 and promote apoptosis in cardiomyocytes.99, 100
In addition, cardiokines, such as ANP, can mediate endocrine effects on the kidney.101 They have effects on electrolyte balance and water excretion in the kidney by increasing glomerular permeability and filtration rate. ANP also antagonizes the deleterious effects of the renin-angiotensin-aldosterone system activation.101, 102, 103 Furthermore, crosstalk between the heart and kidney are evident from the observation that worsening renal function manifests only in end-stage HF and is strongly related to mortality.104 Although cardio-renal interactions in HF are well established, many questions, especially mechanistic, remain unanswered.
Crosstalk between the heart and skeletal muscle
We now appreciate that HF is strongly associated with skeletal muscle wasting, which is typically not associated with general weight loss.105, 106 Skeletal muscle in congestive HF patients shows increased fatigability as well as decreased endurance and exercise capacity.105, 106 Changes evident in muscle include metabolic imbalance, increased degradation of myofibrils and myocyte apoptosis. The signals mediating crosstalk from the heart need to be comprehensively identified.107, 108 One possibility is that generation of TNFα from the failing heart has a detrimental effect on several processes in skeletal muscle. NF-κB is rapidly activated by TNFα in differentiated skeletal muscle cells, which directly induces skeletal muscle protein loss.109 Another proposed mechanism is that TNFα induces sphingosine production, which then leads to induction of apoptosis in these cells.110 In addition, exercise attenuates the local expression of TNFα, IL-1β and inducible Nitric Oxide Synthase (iNOS) in skeletal muscle and decreases the catabolic wasting process in HF patients.34, 111, 112 Angiotensin-II is also produced by the heart under conditions of stress and contributes to cardiac hypertrophy and fibrosis.113 Studies have shown that there is a catabolic effect of angiotensin-II on skeletal muscle, suggesting its role in muscle wasting in HF.114, 115
Therapeutic approaches targeting inflammation in HF
As outlined above, myocardial inflammation in HF is often detrimental to peripheral tissues. There have been several studies addressing consequences of manipulating HF-associated inflammation.116 Targeting TNFα has been extensively studied in numerous clinical trials. Patients who already have severe inflammatory conditions, such as rheumatoid arthritis (RA), were treated with TNFα inhibitors (etanercept, infliximab and adalimumab), which effectively reduced the inflammatory activity and reduced the prevalence of HF complications.117 The data from two-large-scale trials with more than 2000 HF patients showed that etanercept treatment reduced the risk of mortality or morbidity in HF.64 Indeed, the US Food and Drug Administration has issued a directive concerning the use of etanercept in the population with HF.118 However, targeting TNFα using a neutralizing antibody (infliximab) showed no improvement and perhaps even worsened the clinical condition of patients with chronic HF.119 Other studies indicated that patients treated with high-dose infliximab continued to show a worse outcome compared with other groups.57, 120 Several other agents have also been suggested to have potential as therapeutic tools for chronic HF because of their inhibitory effect on TNFα, including the glutamic acid derivative thalidomide.121 Thalidomide prevents the accumulation of TNFα by inducing the degradation of TNFα messenger ribonucleic acid transcripts, and thus, protein production.121 The xanthine derivative pentoxifylline has also been reported to have a role in therapeutic TNFα modulation. Reduced TNFα in the serum of patients treated with pentoxifylline was observed. This correlated with improved peripheral vasodilation and blood hemodynamics.122 Other studies demonstrated a significant improvement in NYHA functional class in patients treated with pentoxifylline.123, 124 However, the results were not reproduced by another group,125 suggesting that the significance of targeting TNFα is controversial and requires further exploration.126 This may be because pleiotropic TNFα effects may be involved in many beneficial physiologic, as well as pathologic, processes. For example, TNFα provides endogenous cyto-protective signals that prevent cardiomyocyte apoptosis following ischemic injury.127 Additionally, TNF type 1 receptor deficiency was associated with accelerated myocardial death.128 Overall, the cardiomodulatory effects of TNFα and other cytokines likely depend on factors such as cell type and timing and extent of inhibition.
We propose that IL-1β is a major mediator between HF and peripheral tissues. In fact, multiple studies have targeted IL-1β. Canakinumab, a neutralizing antibody against IL-1β, and anakinra, a recombinant IL-1 receptor antagonist, were shown to exert beneficial effects on acute MI in animal models.129, 130 Several clinical trials identified therapeutic benefits by blocking IL-1β.131, 132 Anakinra successfully reduced adverse remodeling in patients with an MI and reduced the level of C-reactive protein, a common biomarker used to determine the severity of inflammation.133, 134 Patients diagnosed with HF were treated with Anakinra and subjected to exercise performance testing. Two weeks of the Anakinra treatment significantly increased oxygen consumption, decreased carbon dioxide retention and exercise performance with significant reduction in IL-1β, C-reactive protein and IL-6 serum profiles.135 These correlated with a previous study in which patients with RA treated with Anakinra had improved cardiac function. A single injection of Anakinra resulted in increased blood flow in 3 h.136 The commercial usage of Anakinra was approved by Food and Drug Administration in 2001. However, it was for treating patients with RA not chronic HF, although multiple studies demonstrated cardiac benefits of Anakinra in treating RA.137, 138, 139
There also have been therapeutic efforts to target IL-18 and inflammasome activation. A recombinant human IL-18 binding protein and neutralizing antibody for IL-18 have been developed, and initial clinical trials to treat patients with RAs are ongoing.140, 141 In subjects with moderate to severe RA, IL-18 binding protein shows a favorable safety profile and is well tolerated in healthy volunteers. Because IL-18 binding protein stays in circulation much longer than any other inhibitors previously described, it has attracted a lot of attention.141, 142 Antagonists targeting P2X7 receptors have also been tested to potentially block inflammasome activation. Many successful cases have been shown; they limit neuronal damage and lung, liver and kidney injury in several animal models.143, 144, 145, 146 Currently, the safety and efficacy of P2X7 receptor antagonists are being investigated and have progressed to phase 2 clinical trials. However, their main use is to target inflammatory bowel disease, RA and chronic obstructive airway disease.147
Concluding Remarks
Myocardial ischemia- and I/R-induced inflammation involve NLRP3 inflammasome activation. One principal trigger for inflammasome activation is the recognition of mitochondrial DAMPs. This results in the production and secretion of pro-inflammatory cytokines, including IL-1β and IL-18 (Table 1). These and other factors produced by the heart during inflammation can have local effects. They can also crosstalk with other peripheral tissues via endocrine effects. For example, HF is associated with dramatic changes in the spleen, skeletal muscle wasting, alterations in adipose metabolism and kidney function. The extent and significance of bidirectional crosstalk between the heart and other organs may have been underappreciated, but is now becoming more established and may represent a logical focus of therapeutic interventions in the future.
References
Nabel EG, Braunwald E . A tale of coronary artery disease and myocardial infarction. N Engl J Med 2012; 366: 54–63.
Abel ED, Litwin SE, Sweeney G . Cardiac remodeling in obesity. Physiol Rev 2008; 88: 389–419.
Heusch G, Libby P, Gersh B, Yellon D, Bohm M, Lopaschuk G et al. Cardiovascular remodelling in coronary artery disease and heart failure. Lancet 2014; 383: 1933–1943.
Mozaffari MS, Liu JY, Abebe W, Baban B . Mechanisms of load dependency of myocardial ischemia reperfusion injury. Am J Cardiovasc Dis 2013; 3: 180–196.
Chen GY, Nunez G . Sterile inflammation: sensing and reacting to damage. Nat Rev Immunol 2010; 10: 826–837.
Nakayama H, Otsu K . Translation of hemodynamic stress to sterile inflammation in the heart. Trends Endocrinol Metab 2013; 24: 546–553.
Takahashi M . NLRP3 inflammasome as a novel player in myocardial infarction. Int Heart J 2014; 55: 101–105.
Marchant DJ, Boyd JH, Lin DC, Granville DJ, Garmaroudi FS, McManus BM . Inflammation in myocardial diseases. Circ Res 2012; 110: 126–144.
Doroudgar S, Glembotski CC . The cardiokine story unfolds: ischemic stress-induced protein secretion in the heart. Trends Mol Med 2011; 17: 207–214.
Zheng Y, Gardner SE, Clarke MC . Cell death, damage-associated molecular patterns, and sterile inflammation in cardiovascular disease. Arterioscler Thromb Vasc Biol 2011; 31: 2781–2786.
Bergsbaken T, Fink SL, Cookson BT . Pyroptosis: host cell death and inflammation. Nat Rev Microbiol 2009; 7: 99–109.
Lu A, Wu H . Structural mechanisms of inflammasome assembly. FEBS J 2015; 282: 435–444.
Lechtenberg BC, Mace PD, Riedl SJ . Structural mechanisms in NLR inflammasome signaling. Curr Opin Struct Biol 2014; 29: 17–25.
Latz E, Xiao TS, Stutz A . Activation and regulation of the inflammasomes. Nat Rev Immunol 2013; 13: 397–411.
de Zoete MR, Palm NW, Zhu S, Flavell RA . Inflammasomes. Cold Spring Harb Perspect Biol 2014; 6: a016287.
Abderrazak A, Syrovets T, Couchie D, El Hadri K, Friguet B, Simmet T et al. NLRP3 inflammasome: from a danger signal sensor to a regulatory node of oxidative stress and inflammatory diseases. Redox Biol 2015; 4: 296–307.
Rosca MG, Tandler B, Hoppel CL . Mitochondria in cardiac hypertrophy and heart failure. J Mol Cell Cardiol 2013; 55: 31–41.
Hollander JM, Thapa D, Shepherd DL . Physiological and structural differences in spatially distinct subpopulations of cardiac mitochondria: influence of cardiac pathologies. Am J Physiol Heart Circ Physiol 2014; 307: H1–H14.
Franchi L, Eigenbrod T, Nunez G . Cutting edge: TNF-alpha mediates sensitization to ATP and silica via the NLRP3 inflammasome in the absence of microbial stimulation. J Immunol 2009; 183: 792–796.
Bauernfeind FG, Horvath G, Stutz A, Alnemri ES, MacDonald K, Speert D et al. Cutting edge: NF-kappaB activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression. J Immunol 2009; 183: 787–791.
Kanneganti TD, Lamkanfi M, Kim YG, Chen G, Park JH, Franchi L et al. Pannexin-1-mediated recognition of bacterial molecules activates the cryopyrin inflammasome independent of Toll-like receptor signaling. Immunity 2007; 26: 433–443.
Zhou R, Tardivel A, Thorens B, Choi I, Tschopp J . Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nat Immunol 2010; 11: 136–140.
Zhang Q, Raoof M, Chen Y, Sumi Y, Sursal T, Junger W et al. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature 2010; 464: 104–107.
Nakahira K, Haspel JA, Rathinam VA, Lee SJ, Dolinay T, Lam HC et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat Immunol 2011; 12: 222–230.
Shimada K, Crother TR, Karlin J, Dagvadorj J, Chiba N, Chen S et al. Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity 2012; 36: 401–414.
Zhou R, Yazdi AS, Menu P, Tschopp J . A role for mitochondria in NLRP3 inflammasome activation. Nature 2011; 469: 221–225.
Cruz CM, Rinna A, Forman HJ, Ventura AL, Persechini PM, Ojcius DM . ATP activates a reactive oxygen species-dependent oxidative stress response and secretion of proinflammatory cytokines in macrophages. J Biol Chem 2007; 282: 2871–2879.
Shires SE, Gustafsson AB . Mitophagy and heart failure. J Mol Med (Berl) 2015; 93: 253–262.
Harris J, Hartman M, Roche C, Zeng SG, O'Shea A, Sharp FA et al. Autophagy controls IL-1beta secretion by targeting pro-IL-1beta for degradation. J Biol Chem 2011; 286: 9587–9597.
Saitoh T, Fujita N, Jang MH, Uematsu S, Yang BG, Satoh T et al. Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1beta production. Nature 2008; 456: 264–268.
Shi CS, Shenderov K, Huang NN, Kabat J, Abu-Asab M, Fitzgerald KA et al. Activation of autophagy by inflammatory signals limits IL-1beta production by targeting ubiquitinated inflammasomes for destruction. Nat Immunol 2012; 13: 255–263.
Kawaguchi M, Takahashi M, Hata T, Kashima Y, Usui F, Morimoto H et al. Inflammasome activation of cardiac fibroblasts is essential for myocardial ischemia/reperfusion injury. Circulation 2011; 123: 594–604.
Shinde AV, Frangogiannis NG . Fibroblasts in myocardial infarction: a role in inflammation and repair. J Mol Cell Cardiol 2014; 70: 74–82.
Mezzaroma E, Toldo S, Farkas D, Seropian IM, Van Tassell BW, Salloum FN et al. The inflammasome promotes adverse cardiac remodeling following acute myocardial infarction in the mouse. Proc Natl Acad Sci USA 2011; 108: 19725–19730.
Sandanger O, Ranheim T, Vinge LE, Bliksoen M, Alfsnes K, Finsen AV et al. The NLRP3 inflammasome is up-regulated in cardiac fibroblasts and mediates myocardial ischaemia-reperfusion injury. Cardiovasc Res 2013; 99: 164–174.
Bracey NA, Gershkovich B, Chun J, Vilaysane A, Meijndert HC, Wright JR Jr. et al. Mitochondrial NLRP3 protein induces reactive oxygen species to promote Smad protein signaling and fibrosis independent from the inflammasome. J Biol Chem 2014; 289: 19571–19584.
Park M, Sweeney G . Direct effects of adipokines on the heart: focus on adiponectin. Heart Fail Rev 2013; 18: 631–644.
Turer AT, Hill JA, Elmquist JK, Scherer PE . Adipose tissue biology and cardiomyopathy: translational implications. Circ Res 2012; 111: 1565–1577.
Romacho T, Elsen M, Rohrborn D, Eckel J . Adipose tissue and its role in organ crosstalk. Acta Physiol (Oxf) 2014; 210: 733–753.
Rutkowski JM, Stern JH, Scherer PE . The cell biology of fat expansion. J Cell Biol 2015; 208: 501–512.
Sun K, Kusminski CM, Scherer PE . Adipose tissue remodeling and obesity. J Clin Invest 2011; 121: 2094–2101.
Amato MC, Giordano C . Visceral adiposity index: an indicator of adipose tissue dysfunction. Int J Endocrinol 2014; 2014: 730827.
Britton KA, Fox CS . Ectopic fat depots and cardiovascular disease. Circulation 2011; 124: e837–e841.
McKenney ML, Schultz KA, Boyd JH, Byrd JP, Alloosh M, Teague SD et al. Epicardial adipose excision slows the progression of porcine coronary atherosclerosis. J Cardiothorac Surg 2014; 9: 2.
Kondo K, Shibata R, Unno K, Shimano M, Ishii M, Kito T et al. Impact of a single intracoronary administration of adiponectin on myocardial ischemia/reperfusion injury in a pig model. Circ Cardiovasc Interv 2010; 3: 166–173.
Boudina S, Abel ED . Diabetic cardiomyopathy, causes and effects. Rev Endocr Metab Disord 2010; 11: 31–39.
Wilson PW . Diabetes mellitus and coronary heart disease. Am J Kidney Dis 1998; 32 (5 Suppl 3): S89–S100.
Boudina S, Sena S, Theobald H, Sheng X, Wright JJ, Hu XX et al. Mitochondrial energetics in the heart in obesity-related diabetes: direct evidence for increased uncoupled respiration and activation of uncoupling proteins. Diabetes 2007; 56: 2457–2466.
Regan TJ, Lyons MM, Ahmed SS, Levinson GE, Oldewurtel HA, Ahmad MR et al. Evidence for cardiomyopathy in familial diabetes mellitus. J Clin Invest 1977; 60: 884–899.
Shimizu M, Umeda K, Sugihara N, Yoshio H, Ino H, Takeda R et al. Collagen remodelling in myocardia of patients with diabetes. J Clin Path 1993; 46: 32–36.
Maedler K, Dharmadhikari G, Schumann DM, Storling J . Interleukin-1 beta targeted therapy for type 2 diabetes. Expert Opin Biol 2009; 9: 1177–1188.
Bujak M, Frangogiannis NG . The role of IL-1 in the pathogenesis of heart disease. Arch Immunol Ther Exp (Warsz.) 2009; 57: 165–176.
Mori MA, Bezy O, Kahn CR . Metabolic syndrome: is Nlrp3 inflammasome a trigger or a target of insulin resistance? Circ Res 2011; 108: 1160–1162.
Fuentes-Antras J, Ioan AM, Tunon J, Egido J, Lorenzo O . Activation of toll-like receptors and inflammasome complexes in the diabetic cardiomyopathy-associated inflammation. Int J Endocrinol Metab 2014; 2014: 847827.
Shimano M, Ouchi N, Walsh K . Cardiokines: recent progress in elucidating the cardiac secretome. Circulation 2012; 126: e327–e332.
Aoyagi T, Matsui T . The cardiomyocyte as a source of cytokines in cardiac injury. J Cell Sci Ther 2011; 2012: 003.
Pugh PJ, Jones RD, Jones TH, Channer KS . Heart failure as an inflammatory condition: potential role for androgens as immune modulators. Eur J Heart Fail 2002; 4: 673–680.
Espel E, Garcia-Sanz JA, Aubert V, Menoud V, Sperisen P, Fernandez N et al. Transcriptional and translational control of TNF-alpha gene expression in human monocytes by major histocompatibility complex class II ligands. Eur J Immunol 1996; 26: 2417–2424.
Trede NS, Geha RS, Chatila T . Transcriptional activation of IL-1 beta and tumor necrosis factor-alpha genes by MHC class II ligands. J Immunol 1991; 146: 2310–2315.
Atefi G, Zetoune FS, Herron TJ, Jalife J, Bosmann M, Al-Aref R et al. Complement dependency of cardiomyocyte release of mediators during sepsis. FASEB J 2011; 25: 2500–2508.
Song X, Kusakari Y, Xiao C-Y, Kinsella SD, Rosenberg MA, Scherrer-Crosbie M et al. mTOR attenuates the inflammatory response in cardiomyocytes and prevents cardiac dysfunction in pathological hypertrophy. Am J Physiol Cell Physiol 2010; 299: C1256–C1266.
Comstock KL, Krown KA, Page MT, Martin D, Ho P, Pedraza M et al. LPS-induced TNF-alpha release from and apoptosis in rat cardiomyocytes: obligatory role for CD14 in mediating the LPS response. J Mol Cell Cardiol 1998; 30: 2761–2775.
Kapadia S, Torre-Amione G, Yokoyama T, Mann DL . Soluble TNF binding proteins modulate the negative inotropic properties of TNF-alpha in vitro. Am J Physiol 1995; 268 (2 Pt 2): H517–H525.
Coletta AP, Clark AL, Banarjee P, Cleland JG . Clinical trials update: RENEWAL (RENAISSANCE and RECOVER) and ATTACH. Eur J Heart Fail 2002; 4: 559–561.
Yokoyama T, Sekiguchi K, Tanaka T, Tomaru K, Arai M, Suzuki T et al. Angiotensin II and mechanical stretch induce production of tumor necrosis factor in cardiac fibroblasts. Am J Physiol 1999; 276 (6 Pt 2): H1968–H1976.
Atefi G, Zetoune FS, Herron TJ, Jalife J, Bosmann M, Al-Aref R et al. Complement dependency of cardiomyocyte release of mediators during sepsis. Faseb j 2011; 25: 2500–2508.
Yamauchi-Takihara K, Ihara Y, Ogata A, Yoshizaki K, Azuma J, Kishimoto T . Hypoxic stress induces cardiac myocyte-derived interleukin-6. Circulation 1995; 91: 1520–1524.
Fredj S, Bescond J, Louault C, Potreau D . Interactions between cardiac cells enhance cardiomyocyte hypertrophy and increase fibroblast proliferation. J Cell Physiol 2005; 202: 891–899.
Shioi T, Matsumori A, Kihara Y, Inoko M, Ono K, Iwanaga Y et al. Increased expression of interleukin-1 beta and monocyte chemotactic and activating factor/monocyte chemoattractant protein-1 in the hypertrophied and failing heart with pressure overload. Circ Res 1997; 81: 664–671.
Sarkar S, Vellaichamy E, Young D, Sen S . Influence of cytokines and growth factors in ANG II-mediated collagen upregulation by fibroblasts in rats: role of myocytes. Am J Physiol Heart Circ Physiol 2004; 287: H107–H117.
Koller KJ, Goeddel DV . Molecular biology of the natriuretic peptides and their receptors. Circulation 1992; 86: 1081–1088.
Horio T, Nishikimi T, Yoshihara F, Matsuo H, Takishita S, Kangawa K . Inhibitory regulation of hypertrophy by endogenous atrial natriuretic peptide in cultured cardiac myocytes. Hypertension 2000; 35: 19–24.
Houshmand F, Faghihi M, Zahediasl S . Role of atrial natriuretic peptide in oxytocin induced cardioprotection. Heart Lung Circ 2015; 24: 86–93.
Gruden G, Landi A, Bruno G . Natriuretic peptides, heart, and adipose tissue: new findings and future developments for diabetes research. Diabetes Care 2014; 37: 2899–2908.
Szabo T, Postrach E, Mahler A, Kung T, Turhan G, von Haehling S et al. Increased catabolic activity in adipose tissue of patients with chronic heart failure. Eur J Heart Fail 2013; 15: 1131–1137.
Fenzl M, Schnizer W, Aebli N, Schlegel C, Villiger B, Disch A et al. Release of ANP and fat oxidation in overweight persons during aerobic exercise in water. Int J Sports Med 2013; 34: 795–799.
Bujak M, Dobaczewski M, Chatila K, Mendoza LH, Li N, Reddy A et al. Interleukin-1 receptor type I signaling critically regulates infarct healing and cardiac remodeling. Am J Pathol 2008; 173: 57–67.
Si Y, Tsou CL, Croft K, Charo IF . CCR2 mediates hematopoietic stem and progenitor cell trafficking to sites of inflammation in mice. J Clin Invest 2010; 120: 1192–1203.
Frangogiannis NG . Regulation of the inflammatory response in cardiac repair. Circ Res 2012; 110: 159–173.
Cesta MF . Normal structure, function, and histology of the spleen. Toxicol Pathol 2006; 34: 455–465.
Swirski FK, Nahrendorf M, Etzrodt M, Wildgruber M, Cortez-Retamozo V, Panizzi P et al. Identification of splenic reservoir monocytes and their deployment to inflammatory sites. Science 2009; 325: 612–616.
Nahrendorf M, Pittet MJ, Swirski FK . Monocytes: protagonists of infarct inflammation and repair after myocardial infarction. Circulation 2010; 121: 2437–2445.
Mahnke K, Schmitt E, Bonifaz L, Enk AH, Jonuleit H . Immature, but not inactive: the tolerogenic function of immature dendritic cells. Immunol Cell Biol 2002; 80: 477–483.
Anzai A, Anzai T, Nagai S, Maekawa Y, Naito K, Kaneko H et al. Regulatory role of dendritic cells in postinfarction healing and left ventricular remodeling. Circulation 2012; 125: 1234–1245.
Ismahil MA, Hamid T, Bansal SS, Patel B, Kingery JR, Prabhu SD . Remodeling of the mononuclear phagocyte network underlies chronic inflammation and disease progression in heart failure: critical importance of the cardiosplenic axis. Circ Res 2014; 114: 266–282.
Smolina K, Wright FL, Rayner M, Goldacre MJ . Determinants of the decline in mortality from acute myocardial infarction in England between 2002 and 2010: linked national database study. BMJ 2012; 344: d8059.
Tonelli M, Muntner P, Lloyd A, Manns BJ, Klarenbach S, Pannu N et al. Risk of coronary events in people with chronic kidney disease compared with those with diabetes: a population-level cohort study. Lancet 2012; 380: 807–814.
Anavekar NS, McMurray JJ, Velazquez EJ, Solomon SD, Kober L, Rouleau JL et al. Relation between renal dysfunction and cardiovascular outcomes after myocardial infarction. N Engl J Med 2004; 351: 1285–1295.
Szummer K, Lundman P, Jacobson SH, Schon S, Lindback J, Stenestrand U et al. Influence of renal function on the effects of early revascularization in non-ST-elevation myocardial infarction: data from the Swedish Web-System for Enhancement and Development of Evidence-Based Care in Heart Disease Evaluated According to Recommended Therapies (SWEDEHEART). Circulation 2009; 120: 851–858.
Go AS, Chertow GM, Fan D, McCulloch CE, Hsu CY . Chronic kidney disease and the risks of death, cardiovascular events, and hospitalization. N Engl J Med 2004; 351: 1296–1305.
Bock JS, Gottlieb SS . Cardiorenal syndrome: new perspectives. Circulation 2010; 121: 2592–2600.
Ogawa M, Suzuki J, Takayama K, Senbonmatsu T, Hirata Y, Nagai R et al. Impaired post-infarction cardiac remodeling in chronic kidney disease is due to excessive renin release. Lab Invest 2012; 92: 1766–1776.
Ruiz-Ortega M, Ruperez M, Lorenzo O, Esteban V, Blanco J, Mezzano S et al. Angiotensin II regulates the synthesis of proinflammatory cytokines and chemokines in the kidney. Kidney Int Suppl 2002; 82: S12–S22.
Kalra D, Sivasubramanian N, Mann DL . Angiotensin II induces tumor necrosis factor biosynthesis in the adult mammalian heart through a protein kinase C-dependent pathway. Circulation 2002; 105: 2198–2205.
Moriyama T, Fujibayashi M, Fujiwara Y, Kaneko T, Xia C, Imai E et al. Angiotensin II stimulates interleukin-6 release from cultured mouse mesangial cells. J Am Soc Nephrol 1995; 6: 95–101.
Siasos G, Tousoulis D, Michalea S, Oikonomou E, Vavuranakis M, Athanasiou D et al. Novel biomarkers assessing renal function in heart failure: relation to inflammatory status and cardiac remodelling. Curr Med Chem 2014; 21: 3976–3983.
Song E, Fan P, Huang B, Deng HB, Cheung BM, Feletou M et al. Deamidated lipocalin-2 induces endothelial dysfunction and hypertension in dietary obese mice. J Am Heart Assoc 2014; 3: e000837.
Liu JT, Song E, Xu A, Berger T, Mak TW, Tse HF et al. Lipocalin-2 deficiency prevents endothelial dysfunction associated with dietary obesity: role of cytochrome P450 2C inhibition. Br J Pharmacol 2012; 165: 520–531.
Xu G, Ahn J, Chang S, Eguchi M, Ogier A, Han S et al. Lipocalin-2 induces cardiomyocyte apoptosis by increasing intracellular iron accumulation. J Biol Chem 2012; 287: 4808–4817.
Sung H, Choi JY, Lee SA, Lee KM, Han S, Jeon S et al. The association between the preoperative serum levels of lipocalin-2 and matrix metalloproteinase-9 (MMP-9) and prognosis of breast cancer. BMC Cancer 2012; 12: 193.
Theilig F, Wu Q . ANP-induced signaling cascade and its implications in renal pathophysiology. Am J Physiol Renal Physiol 2015; 308: F1047–F1055.
O'Tierney PF, Komolova M, Tse MY, Adams MA, Pang SC . Altered regulation of renal interstitial hydrostatic pressure and the renal renin-angiotensin system in the absence of atrial natriuretic peptide. J Hypertens 2008; 26: 303–311.
Abu-Amarah I, Balment RJ . Vascular, renal, and endocrine responses to low-dose atrial natriuretic peptide in the fluid-balanced New Zealand genetically hypertensive rats with and without endogenous arginine vasopressin. Can J Physiol Pharmacol 1999; 77: 102–110.
Damman K, Testani JM . The kidney in heart failure: an update. Eur Heart J 2015; 36: 1437–1444.
Ebner N, Elsner S, Springer J, von Haehling S . Molecular mechanisms and treatment targets of muscle wasting and cachexia in heart failure: an overview. Curr Opin Support Palliat Care 2014; 8: 15–24.
von Haehling S, Steinbeck L, Doehner W, Springer J, Anker SD . Muscle wasting in heart failure: an overview. Int J Biochem Cell Biol 2013; 45: 2257–2265.
Marzetti E, Calvani R, Cesari M, Buford TW, Lorenzi M, Behnke BJ et al. Mitochondrial dysfunction and sarcopenia of aging: from signaling pathways to clinical trials. Int J Biochem Cell Biol 2013; 45: 2288–2301.
Sakuma K, Aoi W, Yamaguchi A . Current understanding of sarcopenia: possible candidates modulating muscle mass. Pflugers Arch 2015; 467: 213–229.
Li YP, Schwartz RJ, Waddell ID, Holloway BR, Reid MB . Skeletal muscle myocytes undergo protein loss and reactive oxygen-mediated NF-kappaB activation in response to tumor necrosis factor alpha. Faseb j 1998; 12: 871–880.
Dalla Libera L, Sabbadini R, Renken C, Ravara B, Sandri M, Betto R et al. Apoptosis in the skeletal muscle of rats with heart failure is associated with increased serum levels of TNF-alpha and sphingosine. J Mol Cell Cardiol 2001; 33: 1871–1878.
Gielen S, Adams V, Mobius-Winkler S, Linke A, Erbs S, Yu J et al. Anti-inflammatory effects of exercise training in the skeletal muscle of patients with chronic heart failure. J Am Coll Cardiol 2003; 42: 861–868.
Adams V, Spate U, Krankel N, Schulze PC, Linke A, Schuler G et al. Nuclear factor-kappa B activation in skeletal muscle of patients with chronic heart failure: correlation with the expression of inducible nitric oxide synthase. Eur J Cardiovasc Prev Rehabil 2003; 10: 273–277.
Crowley SD, Gurley SB, Herrera MJ, Ruiz P, Griffiths R, Kumar AP et al. Angiotensin II causes hypertension and cardiac hypertrophy through its receptors in the kidney. Proc Natl Acad Sci USA 2006; 103: 17985–17990.
Sukhanov S, Semprun-Prieto L, Yoshida T, Michael Tabony A, Higashi Y, Galvez S et al. Angiotensin II, oxidative stress and skeletal muscle wasting. Am J Med Sci 2011; 342: 143–147.
Delafontaine P, Akao M . Angiotensin II as candidate of cardiac cachexia. Curr Opin Clin Nutr Metab Care 2006; 9: 220–224.
Saxena A, Russo I, Frangogiannis NG . Inflammation as a therapeutic target in myocardial infarction: learning from past failures to meet future challenges. Transl Res 2016; 167: 152–166.
Listing J, Strangfeld A, Kekow J, Schneider M, Kapelle A, Wassenberg S et al. Does tumor necrosis factor alpha inhibition promote or prevent heart failure in patients with rheumatoid arthritis? Arthritis Rheum 2008; 58: 667–677.
Behnam SM, Behnam SE, Koo JY . TNF-alpha inhibitors and congestive heart failure. Skinmed 2005; 4: 363–368.
Chung ES, Packer M, Lo KH, Fasanmade AA, Willerson JT . Randomized, double-blind, placebo-controlled, pilot trial of infliximab, a chimeric monoclonal antibody to tumor necrosis factor-alpha, in patients with moderate-to-severe heart failure: results of the anti-TNF Therapy Against Congestive Heart Failure (ATTACH) trial. Circulation 2003; 107: 3133–3140.
Pugh PJ, Jones TH, Channer KS . Acute haemodynamic effects of testosterone in men with chronic heart failure. Eur Heart J 2003; 24: 909–915.
Moreira AL, Sampaio EP, Zmuidzinas A, Frindt P, Smith KA, Kaplan G . Thalidomide exerts its inhibitory action on tumor necrosis factor alpha by enhancing mRNA degradation. J Exp Med 1993; 177: 1675–1680.
Schandené L, Vandenbussche P, Crusiaux A, Alègre ML, Abramowicz D, Dupont E et al. Differential effects of pentoxifylline on the production of tumour necrosis factor-alpha (TNF-alpha) and interleukin-6 (IL-6) by monocytes and T cells. Immunology 1992; 76: 30–34.
Sliwa K, Skudicky D, Candy G, Wisenbaugh T, Sareli P . Randomised investigation of effects of pentoxifylline on left-ventricular performance in idiopathic dilated cardiomyopathy. Lancet 1998; 351: 1091–1093.
Skudicky D, Sliwa K, Bergemann A, Candy G, Sareli P . Reduction in Fas/APO-1 plasma concentrations correlates with improvement in left ventricular function in patients with idiopathic dilated cardiomyopathy treated with pentoxifylline. Heart 2000; 84: 438–441.
Bahrmann P, Hengst UM, Richartz BM, Figulla HR . Pentoxifylline in ischemic, hypertensive and idiopathic-dilated cardiomyopathy: effects on left-ventricular function, inflammatory cytokines and symptoms. Eur J Heart Fail 2004; 6: 195–201.
Fildes JE, Shaw SM, Yonan N, Williams SG . The immune system and chronic heart failure: is the heart in control? J Am Coll Cardiol 2009; 53: 1013–1020.
Misra A, Haudek SB, Knuefermann P, Vallejo JG, Chen ZJ, Michael LH et al. Nuclear factor-κB protects the adult cardiac myocyte against ischemia-induced apoptosis in a murine model of acute myocardial infarction. Circulation 2003; 108: 3075–3078.
Monden Y, Kubota T, Inoue T, Tsutsumi T, Kawano S, Ide T et al. Tumor necrosis factor-α is toxic via receptor 1 and protective via receptor 2 in a murine model of myocardial infarction. Am J Physiol Heart Circ Physiol 2007; 293: H743–H753.
Hwang MW, Matsumori A, Furukawa Y, Ono K, Okada M, Iwasaki A et al. Neutralization of interleukin-1beta in the acute phase of myocardial infarction promotes the progression of left ventricular remodeling. J Am Coll Cardiol 2001; 38: 1546–1553.
Abbate A, Salloum FN, Vecile E, Das A, Hoke NN, Straino S et al. Anakinra, a recombinant human interleukin-1 receptor antagonist, inhibits apoptosis in experimental acute myocardial infarction. Circulation 2008; 117: 2670–2683.
Dinarello CA, Simon A, van der Meer JW . Treating inflammation by blocking interleukin-1 in a broad spectrum of diseases. Nat Rev Drug Discov 2012; 11: 633–652.
Dinarello CA, van der Meer JW . Treating inflammation by blocking interleukin-1 in humans. Semin Immunol 2013; 25: 469–484.
Abbate A, Van Tassell BW, Biondi-Zoccai G, Kontos MC, Grizzard JD, Spillman DW et al. Effects of interleukin-1 blockade with anakinra on adverse cardiac remodeling and heart failure after acute myocardial infarction [from the Virginia Commonwealth University-Anakinra Remodeling Trial (2) (VCU-ART2) pilot study]. Am J Cardiol 2013; 111: 1394–1400.
Abbate A, Van Tassell BW, Seropian IM, Toldo S, Robati R, Varma A et al. Interleukin-1beta modulation using a genetically engineered antibody prevents adverse cardiac remodelling following acute myocardial infarction in the mouse. Eur J Heart Fail 2010; 12: 319–322.
Van Tassell BW, Arena RA, Toldo S, Mezzaroma E, Azam T, Seropian IM et al. Enhanced interleukin-1 activity contributes to exercise intolerance in patients with systolic heart failure. PLoS ONE 2012; 7: e33438.
Ikonomidis I, Lekakis JP, Nikolaou M, Paraskevaidis I, Andreadou I, Kaplanoglou T et al. Inhibition of interleukin-1 by anakinra improves vascular and left ventricular function in patients with rheumatoid arthritis. Circulation 2008; 117: 2662–2669.
Clark W, Jobanputra P, Barton P, Burls A . The clinical and cost-effectiveness of anakinra for the treatment of rheumatoid arthritis in adults: a systematic review and economic analysis. Health Technol Assess 2004; 8: iii-iv, ix-x, 1–105.
Bresnihan B, Cobby M . Clinical and radiological effects of anakinra in patients with rheumatoid arthritis. Rheumatology (Oxford) 2003; 42 (Suppl 2): ii22–ii28.
Furst DE . Anakinra: review of recombinant human interleukin-I receptor antagonist in the treatment of rheumatoid arthritis. Clin Ther 2004; 26: 1960–1975.
Plater-Zyberk C, Joosten LA, Helsen MM, Sattonnet-Roche P, Siegfried C, Alouani S et al. Therapeutic effect of neutralizing endogenous IL-18 activity in the collagen-induced model of arthritis. J Clin Invest 2001; 108: 1825–1832.
Tak PP, Bacchi M, Bertolino M . Pharmacokinetics of IL-18 binding protein in healthy volunteers and subjects with rheumatoid arthritis or plaque psoriasis. Eur J Drug Metab Pharmacokinet 2006; 31: 109–116.
Marchetti C, Chojnacki J, Toldo S, Mezzaroma E, Tranchida N, Rose SW et al. A novel pharmacologic inhibitor of the NLRP3 inflammasome limits myocardial injury after ischemia-reperfusion in the mouse. J Cardiovasc Pharmacol 2014; 63: 316–322.
Gourine AV, Poputnikov DM, Zhernosek N, Melenchuk EV, Gerstberger R, Spyer KM et al. P2 receptor blockade attenuates fever and cytokine responses induced by lipopolysaccharide in rats. Br J Pharmacol 2005; 146: 139–145.
Csolle C, Sperlagh B . Peripheral origin of IL-1beta production in the rodent hippocampus under in vivo systemic bacterial lipopolysaccharide (LPS) challenge and its regulation by P2X(7) receptors. J Neuroimmunol 2010; 219: 38–46.
Riteau N, Gasse P, Fauconnier L, Gombault A, Couegnat M, Fick L et al. Extracellular ATP is a danger signal activating P2X7 receptor in lung inflammation and fibrosis. Am J Respir Crit Care Med 2010; 182: 774–783.
Sugiyama T, Oku H, Shibata M, Fukuhara M, Yoshida H, Ikeda T . Involvement of P2X7 receptors in the hypoxia-induced death of rat retinal neurons. Invest Ophthalmol Vis Sci 2010; 51: 3236–3243.
Arulkumaran N, Unwin RJ, Tam FW . A potential therapeutic role for P2X7 receptor (P2X7R) antagonists in the treatment of inflammatory diseases. Expert Opin Investig Drugs 2011; 20: 897–915.
He J, Lu Y, Xia H, Liang Y, Wang X, Bao W et al. Circulating Mitochondrial DAMPs Are Not Effective Inducers of Proteinuria and Kidney Injury in Rodents. PLoS ONE 2015; 10: e0124469.
Bergsbaken T, Cookson BT . Macrophage activation redirects yersinia-infected host cell death from apoptosis to caspase-1-dependent pyroptosis. PLoS Pathog 2007; 3: e161.
Gruden G, Thomas S, Burt D, Zhou W, Chusney G, Gnudi L et al. Interaction of angiotensin II and mechanical stretch on vascular endothelial growth factor production by human mesangial cells. J Am Soc Nephrol 1999; 10: 730–737.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
About this article

Cite this article
Jahng, J., Song, E. & Sweeney, G. Crosstalk between the heart and peripheral organs in heart failure. Exp Mol Med 48, e217 (2016). https://doi.org/10.1038/emm.2016.20
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/emm.2016.20
This article is cited by
-
Cardiac-to-adipose axis in metabolic homeostasis and diseases: special instructions from the heart
Cell & Bioscience (2023)
-
Ketones and the cardiovascular system
Nature Cardiovascular Research (2023)
-
Relationship between spleen size and exercise tolerance in advanced heart failure patients with a left ventricular assist device
BMC Research Notes (2022)
-
Proteomics Reveals Long-Term Alterations in Signaling and Metabolic Pathways Following Both Myocardial Infarction and Chemically Induced Denervation
Neurochemical Research (2022)
-
Splenic size as an indicator of hemodynamics and prognosis in patients with heart failure
Heart and Vessels (2022)
