
Abstract
Intracellular lipid binding proteins (iLBPs) play a role in the transport and cellular uptake of fatty acids and gene expression regulation. The aim of this work was to characterize the iLBP gene family of the Pacific oyster Crassostrea gigas, one of the most cultivated marine bivalves in the world, using bioinformatics and molecular biology approaches. A total of 26 different iLBPs transcripts were identified in the Pacific oyster genome, including alternative splicing and gene duplication events. The oyster iLBP gene family seems to be more expanded than in other invertebrates. Furthermore, 3D structural modeling and molecular docking analysis mapped the main amino acids involved in ligand interactions, and comparisons to available protein structures from vertebrate families revealed new binding cavities. Ten different CgiLBPs were analyzed by quantitative PCR in various tissues of C. gigas, which suggested differential prevalent gene expression of CgiLBPs among tissue groups. The data indicate a wider repertoire of iLBPs in labial palps, a food-sorting tissue. The different gene transcription profiles and reported docking systems suggest that the iLBPs are a non-generalist ligand binding protein family with specific functions.
Similar content being viewed by others

Introduction
Intracellular lipid-binding proteins (iLBPs) are a group of low molecular mass proteins involved in the intracellular transport of fatty acids and other hydrophobic molecules. The iLBPs are a family of fatty acid (FABP), retinol (CRBP) and retinoic acid (CRABP) binding proteins1,2,3. iLBPs from different organisms usually have 130 amino acids, and have a wide variation in amino acid identity (20 to 70%). However, the tertiary structures of these proteins are highly conserved and particularly consist of a cavity formed by ten anti-parallel strands and two helices that can bind and hold lipophilic compounds such as fatty acids3,4,5.
The iLBPs of vertebrates were classified into four subfamilies according to ligand binding preferences. Subfamily I includes CRBP and CRABP, subfamily II includes FABP1 and FABP6, subfamily III includes FABP2, and subfamily IV includes the most members (FABP3, FABP4, FABP5, FABP7, FABP8, FABP9 and FABP12)5,6. However, the inclusion of invertebrate iLBPs, which differ from vertebrate iLBPs, slightly changed the relationships among iLBP family members7. Regardless of several studies about invertebrate iLBPs5,8,9,10, there is scarce information about gene/protein diversities and their 3D structure-function relationships. Despite the recent availability of genomic and transcriptomic public databases, genome-wide surveys of this multigene family in invertebrate species are limited11. Currently, it is reasonable to perform such studies to characterize iLBPs in invertebrates by their diversity and genomic organization.
Crassostrea gigas is one of the most cultivated bivalves in the world and considered a reference species for molecular studies in mollusks12,13. The Pacific oyster is a typical sentinel organism for biomonitoring studies and is widely used to evaluate environmental pollutant effects since it can accumulate and tolerate these compounds14,15,16. Previous studies have demonstrated upregulation of C. gigas FABP intestinal-like gene (GenBank accession ABU41520) after sewage and pharmaceutical exposures17,18,19,20. Despite the genome of C. gigas is publicly available, thorough studies regarding iLBPs are still lacking21.
To evaluate the gene/protein iLBP diversity of the Pacific oyster genome, the present study investigates iLBP features as: exon/intron boundaries, phylogenetic relationships and gene transcription patterns in different tissues. Furthermore, we modeled 3D proteins and docked fatty acids to map the functional amino acids of these iLBP members. This study provides the first characterized molecular catalog of iLBP putative proteins of a bivalve species, using publicly available data to promote deeper knowledge of an important gene family through the use of bioinformatics and molecular biology techniques.
Results and Discussion
RNA-seq mapping, transcript reconstruction and screening for iLBP family members
Data from the Crassostrea gigas genome and transcriptome were used to analyze the genomic structure of iLBPs. The metrics of the short-read mapping and transcript reconstruction were similar or slightly higher than the original work21 (Tables S1 and S2). These differences can be explained by improvements made to the more recent bioinformatics programs.
After sequence annotation, we identified 25 putative iLBP sequences among the transcripts (Table S3). Their respective open reading frames (ORFs) were compared to the Pacific oyster entries deposited in the NCBI non-redundant protein databank (nr) (Table S4). Among the sequences of putative proteins, 21 had 100% coverage and identity with annotated sequences in NCBI nr, three were assigned as possible new transcripts derived from alternative splicing, and one was a new hypothetical pseudogene. These results show that the transcript reconstruction procedure was robust, recovering almost all described C. gigas iLBPs with the exception of a pseudogene annotated as CRBP1 (GenBank accession EKC22532.1), whose sequence was directly retrieved from the NCBI repository. This exception could be explained due to the lack of transcription of the analyzed tissues, and could possibly be a pseudogene detected by ab initio procedures in the genome sequencing study21. By the adopted criteria of gene boundaries (physical localization and common usage of exons, see Methods), Pacific oyster’s iLBPs were classified as 14 different genes, ten transcripts variants (synonymous and non-synonymous) and two pseudogenes (Table S3). C. gigas presents a wide repertoire of iLBP genes compared to the majority of other invertebrates11.
Nomenclature for Pacific oyster iLBPs
There is no official standard for invertebrate iLBP classification. There are implicit difficulties in establishing orthologous relationships among vertebrate and invertebrate iLBPs due to a distinct evolutionary history; most iLBP genes emerged after the event of vertebrate/invertebrate split (~600–700 mya) and are derived from several duplications of a unique ancestral lipocalin gene1,5.
In vertebrates, the initial nomenclature for FABP genes was based on the tissue in which it was originally detected (e.g., fatty acid binding protein, heart-type). However, the current classification uses numerals after the name (e.g., FABP1, FABP2)22. In invertebrates, several approaches are found in the literature. A common one uses FABP preceded by the abbreviation for the species name, for example, EgFABP1 and EgFABP2 from Echinoccocus23. An alternative is to adopt the -like term after the name of the corresponding putative homolog vertebrate gene, such as the FABP2-like gene found in C. gigas17. However, the automatized functional annotation of recently sequenced genomes and transcriptomes uses homology-based annotation and is responsible for the majority of invertebrate iLBP descriptions in public databases, such as NCBI nr. In general, invertebrate iLBPs have more similarity with vertebrate FABP3 (hearth-like) genes5, creating a bias in automated annotation. Functional characterization of this protein family in invertebrates is a troublesome task, especially in the superphylum Lophotrochozoa, which includes mollusks. Several invertebrate species do not have a well-annotated, publicly available genome to compare and establish reliable orthologous relationships. Thus, a homology-only based annotation of iLBP members of mollusks seems to be inappropriate. In a recent study, new FABPs were identified in a great number of invertebrate species, and authors arbitrarily named them using a sequential order for the newfound FABP genes11. Here, a similar approach was used for several iLBPs identified after searching the Pacific oyster genome. However, instead of CgFABPs, we used CgiLBPs as codification of these proteins to prevent misconceptions when talking about functional characterization, since most of them lack experimental and deeper in silico characterization. The sequential nomenclature respected the scaffold order, as shown in Fig. 1.

Scaffold sizes, gene length and gene position are not drawn to scale. CgiLBPs were named following scaffold order, using letters for gene duplications.
Phylogeny
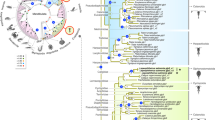
We constructed phylogenetic trees for iLBPs from vertebrate and invertebrate species. Both Bayesian and maximum likelihood approaches resulted in similar topologies (Fig. 2, Figure S3). In general, phylogenies were well supported for more recent divergence events, but not for deeper nodes. Within the iLBP gene family, there is little sequence similarity and few linear motifs5, which is difficulty associated with this type of analysis.

Tree drawn using FigTree setting Lipocalin 1 gene from H. sapiens as outgroup. Hs stands for Homo sapiens (red), Dm for Drosophila melanogaster (green), Sm for Schistosoma mansoni (green) and Lg for Lottia gigantea (purple). CgiLBPs (blue) were named according to this study. Posterior probabilities higher than 50 are shown. Vertebrate iLBP subfamilies I to IV are depicted in red boxes followed by their respective subfamily number.
First, the phylogenetic model used for iLBP genes in vertebrates was maintained in the phylogram, with iLBPs from humans forming its respective subfamilies, in accordance with previous inferences1. Considering iLPB subfamily I in the literature, no CRBP or CRABP was described in bivalves (C. gigas and Lottia gigantea), and only one CRABP was reported in invertebrates24. The results here are similar, with no invertebrate iLBP clustering with vertebrate subfamily I. Similarly, no invertebrate iLBP is clustered in subfamily II, composed of FABPs that bind and transport cholesterol and bile acids. Subfamily III is an interesting example, as CgiLBP4 have higher levels of homology via sequence comparison with FABP2 from vertebrates. In the proposed phylogenetic tree in Fig. 2, there is good support for clustering CgiLBP4 and FABP2 from humans, at least in the Bayesian approach (Fig. 2). In addition to CgiLBP3, CgiLBP4, CgiLBP5 and CgiLBP6, all positioned at the same scaffold (Fig. 1), CgiLBP10, CgiLBP11, CgiLBP12 and some L. gigantea representatives are also clustered in vertebrate subfamily III. Data suggests a putative expansion of subfamily III in bivalves comparing to vertebrate species. Subfamily IV of iLBPs is mainly observed in superior vertebrates1, so only convergent evolution could explain similarities among iLBPs from this subfamily in mollusks. Drosophila melanogaster and Schistosoma mansoni iLBPs, representing invertebrate species other than Lophotrochozoan animals, formed a cluster independent of mollusks and closer to vertebrate subfamily IV. Sm14 was the first FABP described in platyhelminths25 and considered a sister group of subfamily IV of vertebrates7, as depicted in the Bayesian and ML trees (Fig. 2, Figure S3).
Other bivalve iLBPs and one S. mansoni putative FABP clustered, forming several subgroups other than vertebrates. However, due to low support, it should be further investigated using more invertebrate species in future studies. One subgroup was composed of CgiLBP1A and CgiLBP1B, the most derived proteins due to total lack of signature motifs or domains (Table S3), and clustered together with CgiLBP2, CgiLBP7A and one iLBP from L. gigantea. The Pacific oyster iLBP gene repertoire is apparently wider than those of other studied invertebrate species, which suggests different phylogenetic relations with vertebrate iLBPs and even to Arthropoda and Platyhelminth FABPs. Until there are more detailed studies of iLBP evolutionary history in invertebrates, it is not recommended to name bivalve genes using homology with vertebrate FABPs, as several mollusks genes probably derived independently after the vertebrate/invertebrate split at 600–700 mya. More studies focusing on orthologous relationships among invertebrate species could shed light on these questions and yield a more accurate classification.
Genomic organization
The genomic structures of identified iLBPs were similar to the canonical organization of four exons and three introns, with the exception of the two pseudogenes (Table S3). A recent review about invertebrate FABPs shows that FABP genes usually follow a similar genomic configuration to vertebrates11. For example, the mollusk L. gigantea has the majority of FABP genes structured as the canonical organization. The size of identified CgiLBPs ranged from 131 to 143 aa, very close to the average size for this protein family11. Again, the exceptions were the pseudogenes, which lacked one exon and generated translated sequences approximately 100 aa long.
Concerning Crassostrea gigas iLBP splicing variants, eight genes showed no evidence of alternative splicing in analyzed tissues. CgiLBP10 (Figure S1), CgiLBP11 and CgiLBP12 (Figure S2) present identical variants at nucleotide level and are closely located at scaffold 43244 (Fig. 1), with CgiLBP11 and CgiBLP12 overlapping each other (Figure S2). Identities in protein sequences are approximately 45% between CgiLBP10 and the other two genes, and approximately 50% between CgiLBP11 and CgiLBP12. These levels of identity are considered above average in CgiLBPs, suggesting recent local genomic duplications.
Three genes showed splicing variants that resulted in alterations in both nucleotide and amino acid sequences: CgiLBP1A, CgiLBP4 and CgiLBP5. These genes present different paradigms regarding genomic structure and the use of alternative splicing. The CgiLBP5 gene shows a typical mutually exclusive exon alternative splicing mode (Fig. 3A). The third exon either suffered a small local duplication or is a vestige of whole gene duplication. This gene presents two possible transcripts in this region (Fig. 3B). Another gene that shows patterns of alternative splicing is CgiLBP4. This gene appears to be a mix of very recent gene duplication (~90% identity) and common use of exons (Fig. 4A). The variant CgiLBP4.1 unites distant exons, and this genomic region is characterized as one gene. CgiLBP4 gene has been considered a biomarker for exposure to contaminants such as domestic sewage and ibuprofen17,18,19,20. Named as FABP2-like or FABP2 intestinal type in such studies, the primer pairs for PCR quantification predicts amplification of both CgiLBP4.1 and CgiLBP4.4 putative transcripts. It is not clear whether these proteins are involved in another biological role other than fatty acid transportation. The hypothesis that CgiLBPs participate directly in response to xenobiotic exposure needs to be evaluated; the higher levels of CgiLBP4 transcripts observed in these studies could be involved mobilizing and transporting lipids to enhance energy production to cope with the metabolic demands during and after stress caused by contaminant exposure. The probable duplication events that occurred in this region and the preservation of copies in the Pacific oyster genome suggest an important role for the CgiLBP4 gene. Such duplications of stress related genes were significantly retained in the Pacific oyster genome21. Studies should explore how all variants respond to contaminant exposure, or the mechanisms involved in their regulation.

(A) CgiLBP5 genomic structure, depicting the mutually exclusive exon splicing. (B) MUSCLE alignment of amino acid sequence of transcripts CgiLBP5.1 and CgiLBP5.2, showing non-synonymous alterations in the mutually exclusive exons. Graphic representation generated in Jalview v2.1. (C) 3D structural models show the amino acid differences on the surface of the proteins between transcripts CgiLBP5.1 and CgiLBP5.2, and charged surface plots show the positive patch (in blue) and negative patch (in red) of CgiLBP5.1 and CgiLBP5.2.

(A) CgiLBP4 genomic structure, depicting the recent duplications and common usage of exons. (B) MUSCLE alignment of amino acid sequence of transcripts CgiLBP4.1, CgiLBP4.2, CgiLBP4.3 and CgiLBP4.4. Graphic representation generated in Jalview v2.1. (C) 3D structural models showing amino acid substitutions in each isoform. Particularly positions 27 and 132 may tune the lipid binding sites.
Lastly, we describe CgiLBP1A as another example of alternative splicing of iLBPs in Crassostrea gigas (Fig. 5). Similar to CgiLBP4 region, it probably underwent gene duplication. CgiLBP1B sequence shows 71.76%, 85.50%, 87.02% amino acid identity to CgiLBP1A.1, CgiLBP1A.2 and CgiLBP1A.3 transcripts, respectively. CgiLBP1A and CgiLBP1B genes were both detected at scaffold 208, separated by ~4 Kb. An interesting observation is that these genes lack the typical Lipocalin domain (CL0116) from Pfam libraries, and the three FATTYACIDBP motifs (PR0078) from PRINTS database. The total lack of any domain/motif was exclusive of CgiLBP1 duplicated genes, as other CgiLBPs also failed to detect some of these signature domains or motifs (Table S3). This region probably offers a wide range of CgiLBP1 functionalities, which would account for functional gene duplicates and the presence of alternative splicing in CgiLBP1A.

(A) CgiLBP1 genomic structure, depicting the recent duplications and common usage of exons. (B) MUSCLE alignment of amino acid sequence of transcripts CgiLBP1.1, CgiLBP1.2 and CgiLBP1.3. Graphic representation generated in Jalview v2.1. (C) 3D structural models showing amino acid substitutions inside the lipid binding cavities.
Molecular Modeling
Understanding the iLBPs functionalities is a challenging task, considering different binding capacities, multiple ligand binding sites, cavity flexibility and cellular localization26,27. Two different approaches were used here for 3D structural modeling. The first was a hybrid methodology using threading and homology modeling from the I-Tasser suite, which is able to predict structural features of non-conserved regions by fragment assembly simulations28. The second was a homology modeling-only method from the SWISS-MODEL suite29, which preserves the similarities from a single template protein structure. SWISS-MODEL was only used for modeling the most conserved protein cavities, required in molecular docking analysis, as discussed below. Despite the low sequence identity among C. gigas and mammalian iLBPs, 3D models obtained by threading-homology modeling showed high quality models (TM-score higher then 0.7). All members display a conserved FABP structural fold with 2 α-helices and 10 β-strands, except for CgiLBP1B, which exhibits a shorter N-terminal region, and α-helix 2 was not modeled on the helix-loop-helix motif region (Figure S4). Therefore, CgiLBP1B seems to encode a truncated protein compared to CgiLBP1A. CgiLBP1, CgiLBP2, CgiLBP3, CgiLBP4, CgiLBP5, CgiLBP10, CgiLBP11 and CgiLBP12 each showed an additional N-terminal helix. Several FABP structures deposited in Protein Data Bank (PDB), such as FABP3 (3WVM), FABP5 (4LKP), FABP8 (4BVM) and FABP9 (4A60), present the N-terminal 3.10 helices that are relevant for folding and binding since they are located at the “backdoor” of ligand cavity. The FABP ligand portal entrance, reported in vertebrates, is composed of α-helix 2 and the loops that connect β-strands CD and β-strands EF30. Compared to those FABPs, almost all CgiLBPs seem to have the portal entrance.
CgiLBP5 3D models (Fig. 3C) show the amino acid differences at the surface region of its splicing variants. The charged surface was illustrated to show those differences; a negative patch on CgiLBP5.2 represents the substitution of three amino acids with glutamic acid (GLU99, GLU114 and GLU121) when compared to CgiLBP5.1. The iLBP family has been related to many molecular interaction partners, including nuclear receptors for gene expression regulation27; therefore, we can speculate that those surface modifications may reflect different biological roles.
The nuclear localization signal (NLS), which is related to lipid delivery to nuclear receptors, was identified in the CgiLBP family. The typical vertebrate NLS involves residues K21, R29, and K30 in CRABP-II, and K21, R30, and R31 in FABP4, all located within the protein helix-loop-helix26,31. Based on sequence analysis and 3D models, CgiLBP5, CgiLBP6, CgiLBP10, CgiLBP11 and CgiLBP14 have exposed basic residues at these positions and may be involved in nuclear translocation. It is not clear if the basic residue triad is the only feature associated with nuclear lipid delivery. FABP1 and FABP2 were able to modulate PPARα receptor activation32 and do not have the complete basic triad residues. Interestingly, the human FABP2 isoform pattern (E/R/K) at helix-loop-helix is also found in C. gigas iLBPs and it is exclusively found in CgiLBP4 isoforms. BLAST analysis strongly suggests CgiLBP4 is a FABP2 vertebrate homolog and the NLS signature reinforces this correlation, in addition to clustering by phylogenetic inference (Fig. 2).
CgiLBP4 presents four non-synonymic transcript variants. Figure 4C shows structural models that emphasize the differences between those variants. The differences at positions THR132MET inside the cavity, MET27LEU on the portal region and VAL29LYS may change the binding cavity proprieties. CgiLBP1A also has differences in the amino acids inside the cavity: LEU21MET, PHE29TYR, LYS61GLN and ALA34LYS (Fig. 5C). We suggest that iLBPs, particularly the CgiLBP1A sequences, which are the most divergent molecules of the iLBP repertoire, are good targets for experimental structural data collection and biochemical analysis.
Docking Analysis
To evaluate ligand binding properties of C. gigas iLBPs, we used comparative 3D modeling approach. Ligand bound PDB structures were selected as templates for docking analysis and only the CgiLBP structures most similar to PDB templates were selected for analysis (see methods). Palmitic acid, a saturated fatty acid, is found at high concentrations in C. gigas33 tissues and was chosen as model ligand to identify saturated fatty acid protein transporters such as vertebrate FABP226. All CgiLBPs analyzed were able to bind palmitic acid. Figure S5 shows the main positions in each protein involved in palmitic acid binding.
The key residues involved in ligand head group and hydrophobic interactions were highlighted (Fig. 6B). The conserved ARG residue of β-strand 8 preferentially participates as a hydrogen donor; alternatively, the ARG of β-strand 10 can be substituted as a hydrogen donor. The residues acting as hydrogen donors may determine the fatty acid positioning within the cavity. Typically saturated fatty acids present their linearly shaped tail into the protein cavity, similar to vertebrate FABP2 bound to palmitic acid26. In those structures the fatty acid head group is deep inside the cavity and the tail is linear. We found a similar pattern in CgiLBP3, CgiLBP4, CgiLBP5.1, CgiLBP6, CgiLBP9 and CgiLBP13. In these CgiLBP models, the amino acid ARG from β-strand 8 was always the hydrogen-donor to the ligand carboxyl group (Fig. 6A, Figure S5). U-shaped fatty acids positioned in FABP cavities of vertebrates, representative of most of the FABP family, are characterized by a hydrogen bond with the ligand, which involves at least one residue from the pattern ARG/X/TYR of β-strand 10 on C-terminal vertebrate iLBPs22. Some CgiLBPs present patterns similar to vertebrate U-shape poses, involving similar binding residues. Those structures are CgiLBP2, CgiLBP5.2, CgiLBP10, CgiLBP12 and CgiLBP14, where we have found ARG or TYR at the C-terminal positions. All those sequences (except CgiLBP2) have ARG in the C-terminal region, which may reflect some preferences for binding (Fig. 6B, Figure S5).

(A) 3D ligand complexes docked with palmitic acid showing the linear shaped (CgiLBP3) and U-shaped (CgiLBP14) tails. (B) Sequence alignment of CgiLBPs docked with palmitic acid. Black and gray represent polar and hydrophobic contacts to the ligand, respectively. Secondary structure from CgiLBP 4.1, is depicted schematically above the sequence alignment by pink cylinders (β-strands) and blue arrows (α-helices) representing iLBP fold. The graphical representation was generated in Jalview v2.1.
Interestingly, the hydrogen bonding pattern in vertebrate FABP2 involves mainly the ARG from β-strand 8, despite the presence of ARG in the C-terminal region34. At least in Crassostrea gigas, our data show evidence that the C-terminal ARG does not seem to compete for binding. Instead, when the β-strand 10 pattern is observed, the main hydrogen donor is transferred to the C-terminal part of the protein. This binding mode is usually related to unsaturated fatty acids26 and those C. gigas proteins may be interesting candidates for the higher demand of PUFAs (poly unsaturated fatty acids) in marine organisms35. Our approach was able to highlight the main residues and can be used for mining new sequences with the same pattern in different organisms.
All CgiLBP4s bind palmitic acid similarly to vertebrate FABP236. As shown in CgiLBP4.4 (Fig. 4), the residue MET132, which is substituted for THR in CgiLBP4 to interact with ligands, shows evidence of small differences in binding capacities between CgiLBP4 isoforms. Probably, due to recent gene duplication events, the Crassostrea gigas genome had an expansion of members involved in the typical saturated fatty acid binding mode from vertebrates, represented by the first binding mode group reported in this work.
The reported bigger cavities identified in FABP1 and FABP6, that may bind cholesterol and derivatives, even two fatty acids in the same cavity26,36,37,38,37, were not found in CgiLBP structural models due to lower sequence similarities with those vertebrate members. Concerning phylogenetic analysis, none of the invertebrate iLBPs clustered with vertebrate subfamily II, which includes FABP1 and FABP6.
Gene expression profiles
CgiLBPs transcript levels were evaluated in different tissues of C. gigas (Fig. 7). The bivalve feeding process involves several tissues/organs. The filter feeding pathway begins with particle uptake through the gills and transport to the labial palps, which are involved in food selection. The labial palps, in conjunction with the mantle, are also responsible for pseudofeces rejection38.

An example of prevalent variants in each tissue and their corresponding function are presented from panel A to E. The feeding pathway (green dash arrow) is represented by (A) labial palps, (B) mantle and (C) gills. Digestive gland is shown in (D) and adductor muscle in (E).
The prevalent transcripts found in gills were CgiLBP1A, CgiLBP14 and CgiLBP6. Oyster gills are directly in contact with the external environment and it is known that the bivalve Dreissena polymorpha and Crassostrea virginica can uptake lipids directly from water39,40. Therefore, the function of these CgiLBP in gills may be related to lipid uptake from the water column. Other functions for these genes may be related to xenobiotic sensing and transcriptional regulation. The gene products of CgiLBP1A, CgiLBP14 and CgiLBP6 may bind lipophilic xenobiotics absorbed by the gills and trigger intracellular signaling cascades leading to transcription of biotransformation genes. Crassostrea gigas has been used as a sentinel for aquatic pollution41,42. High transcript levels of FABPs, classified as CgiLBP4 by the present study, were found in the gills of oysters exposed to sewage17,18,20 and ibuprofen19. In this study, CgiLBP4 was highly expressed in the labial palps. Considering the use of iLBPs as biomarkers of aquatic pollution, we suggest investigating CgiLBP4 in the labial palps.
Remarkably, the labial palps exhibit many differentially expressed iLBP members (CgiLBP1A, CgiLBP6, CgiLBP4, CgiLBP3 and CgiLBP14). It is important to note that CgiLBP1A and CgiLBP4 have non-synonymous splice variants, presenting a wider repertoire in this tissue, since iLBP members usually have different ligand binding affinities, which may be relevant for food selection. Considering positive correlation between labial palp size and efficiency on particle selection and its capacity to distinguish between different nutrients, nitrogen/carbon or carbon only sources43,44, we also suggest a lipid uptake function for this tissue. The labial palps are complex organs in bivalves38 and iLBP gene expression needs to be evaluated with different tissue segments and closely related species taken into consideration.
In addition to pseudofeces rejection, mantle tissue is associated with energy storage, shell formation and gametogenesis45,46,47. These functions may involve CgiLBP3 and CgiLBP6 proteins since the transcript levels of these genes were significantly higher in this tissue. CgiLBP12, CgiLBP1B and CgiLBP13 transcript rates were 3,064-fold, 629-fold and 21.09-fold, respectively, higher in digestive gland compared to the other tissues, showing 3,064-fold, 629-fold and 21.09-fold respectively. These isoforms may be related to a high energy metabolism and lipid storage48.
CgiLBP9 and CgiLBP2 were highly expressed in adductor muscle. In bivalves, the main function of this tissue in bivalves is to control the closure of the shells, keep the valves tightly closed for a long time, and make constant, slow valve movements49. It is known that bivalve muscle tissue contains limited amounts of stored substrate to generate sufficient energy for these movements, generally sufficient to support contractions for up to three minutes under aerobic conditions and up to 30 seconds under anaerobic conditions. The transcript levels of these iLBPs in adductor muscle of C. gigas may be related to energy metabolism to maintain the valve movements. In insects, FABP from muscle tissues are also involved in energy metabolism to maintain flight4.
No prevalence was found in heart and no difference was found between tissues for CgiLBP11 (Figure S6). In addition to higher sequence identities (~80%) between the CgiLBP1 group, distinct patterns were revealed when CgiLBP1A and CgiLBP1B expression profiles were analyzed. CgiLBP1A was prevalent in the gills, as opposed to CgiLBP1B, which had higher transcript levels in the digestive gland and encodes a truncated protein. One of the many ways that organisms preserve gene duplications is through subfunctionalization, which leads to tissue specialization regarding gene expression profiles50 in many cases. Protein 3D modeling also showed many differences between these isoforms and suggests different functions in respective organs.
Concluding Remarks
Crassostrea gigas presents a wide variety of iLBP proteins, resulting from a process of several duplications and some alternative splicing mechanisms. We reinforce the need for more experimental studies focusing on functional and structural research, as the Pacific oyster’s iLBPs show a distinct evolutionary history when compared to vertebrate’s iLBPs, especially regarding the lack of representatives from classical subfamilies. In addition, CgiLBP1A and CgiLBP1B divergence and the loss of detectable domains suggest a possible new class of iLBPs derived from FABP, and deserves further attention, as qPCR assays demonstrated different gene transcription profiles in some tissues. In light of these observations, we hope that our study will initiate further discussions about iLBPs from Lophotrochozoa species and that a consensus regarding iLBP evolution and functionalities will be reached shortly to benefit both iLBP biology and taxonomy.
Methods
Genome screening for iLBP family members
Pacific oyster’s genome assembly (version 9.0) and transcriptome data from RNA-Seq of five different tissues (the gills, digestive gland, labial palps, mantle and adductor muscle) were retrieved from GigaDB (gigadb.org/dataset/100030). Paired-end reads from each issue were separately mapped into available genomic scaffolds using splice-aware aligner TopHat2 v2.1.051 with Bowtie2 mapper v2.2.452, and the parameter --mate-inner-dist was set to 200. Cufflinks v2.2.153 reconstructed the transcripts from each mapping file and Cuffmerge v2.2.153 joined the resulting GTF files into a single unified transcript catalog. Members of the iLBP family were identified by comparison to NCBI’s non-redundant (nr) proteins, Pfam-A v29.054 and PRINTS v42.055 databanks. To compare with nr, blastx option from BLAST + v2.2.3056 was used with an e-value filter of 1e-05. PRINTS motifs were searched online (bioinf.manchester.ac.uk/cgi-bin/dbbrowser/fingerPRINTScan/FPScan_fam.cgi) using default parameters. Pfam domains were retrieved using HMMSCAN v3.1b257 with 1e-03 as e-value threshold. Nomenclature of iLBPs adopted in this study for transcripts and genes followed scaffold order, without any functional or evolutionary aspects. Duplications were considered at >70% identity between two genes, and named with letters afterwards.
Pacific oyster iLBP family genomic organization
iLBPs were initially described using transcript and gene information from Cufflinks53 as an initial template of Crassostrea gigas iLBPs’ genomic structure. Putative iLBP homologs previously selected were manually curated using the GTF file generated from Cuffmerge53 and genomic scaffolds of C. gigas in the Integrated Genome Viewer v2.3 (IGV)58 to determine their genetic structures and allow correct grouping of transcripts into genes. In this study, the presence of transcription in the same genomic region (physical location in scaffolds and common usage of exons) was considered the main criteria for establishing an iLBP gene. Transcripts were translated and had their most probable open reading frames (ORFs) manually extracted and verified using Expasy translate tool (web.expasy.org/translate/). Amino acid sequences of iLBPs were first aligned against the other members transcribed in the same genomic region (therefore the same gene as our established criteria) using MUSCLE59 to filter alternative transcripts showing synonymous and non-synonymous differences.
Phylogeny
Protein datasets from Homo sapiens, Drosophila melanogaster, Schistosoma mansoni and Lottia gigantea were retrieved from NCBI GenBank. HMMSCAN v3.1b257 and Pfam-A v29.054 were used to scan for Lipocalin domains, in the same manner as C. gigas iLBPs were identified. A reciprocal best BLAST hit procedure60 was used to search for putative orthologues among the species to complement the datasets.
Amino acid sequences were aligned using MUSCLE59. Human Lipocalin 1 (GenBank access NP_002288.1) was selected as an outgroup. The resulting alignment was imported into TOPALi v2.561 and then submitted to Model Selection tool (MrBayes and PhyML), to determine the best substitution model, using BIC (Bayesian information criterion) values to select the models that best fit the data. Phylogenetic trees were generated by PhyML approach through TOPALi v2.561 with 1000 bootstraps, and by MrBayes v3.262 with two runs of 10,000,000 generations, sample rate of 1000, burn-in of 25%. Both procedures used WAG + G as substitution model. Trees were drawn using FigTree v1.4.2 (tree.bio.ed.ac.uk/software/figtree/).
3D Modeling and Molecular Docking
The 3D models were built using I-Tasser28 suite and SWISS-MODEL29. I-Tasser was used with default parameters as a threading assembly approach for protein fold characterization. Models with TM-score higher than 0.5 were selected for analysis. SWISS-MODEL was used on alignment mode to build models for molecular docking. Target iLBP sequences were first submitted to blastp analysis against the SWISS-PROT databank and the best hits (10 different sequences) were used for alignments using Clustal omega v1.2.163. The best hit against PDB with ligand and from a non-NMR structure was selected for molecular modeling. Quality of PDB structural models was checked by using Global Model Quality Estimation score (QMQE). iLBP structural modes with QMQE score higher than 0.6 and with sequence identities against the template PDB structure higher than 30% were used for analysis. SWISS-DOCK software64 was used for molecular docking. The iLBP models were prepared for docking using Chimera v10.165 at default parameters and AMBER force field66. Palmitate ligand was selected from the Zinc database67. An accurate mode and 3 Å sidechain flexibility was used for running dock analysis. The lower full fitness pose inside iLBP cavity was selected for ligand binding analysis with LigPlot v4.5.3 software68.
qPCR analysis
iLBP transcript levels were evaluated using quantitative PCR (qPCR). To characterize the expression of a unique genomic region, some genes with one or more alternative transcripts were analyzed using primers for the most common exon. Oligonucleotide primers for qPCR were designed using Primer Quest software available at www.idtdna.com (IDT). Selected genes and their respective primer pairs are shown in Table S5.
Oyster (C. gigas) samples of heart (HT), adductor muscle (MS), digestive gland (DG), mantle (MT), gills (GL) and labial palps (LP) were collected and frozen in liquid nitrogen. Total RNA from tissues (n = 10) was isolated using Qiazol reagent (Qiagen) following the supplier’s protocol with minor modifications. Briefly, 100 mg of each sample was mechanically disrupted in 1 mL Qiazol using a homogenizer (Tissue-Tearor, BioSpec Products). For heart samples, pools of three animals were made to obtain 100 mg of tissue. To check RNA concentration and purity, samples were measured using a NanoDrop ND-1000 Spectrophotometer (Thermo Scientific, Wilmington, DE, USA). 1 μg of total RNA per sample was reverse transcribed using a QuantiTect Reverse Transcription kit (Qiagen).
The quantitative PCR (qPCR) reactions were performed with Quantifast SYBER Green kit (Qiagen) in a Rotor-Gene TM 6000 thermocycler (Qiagen), according to the manufacturer’s instructions. qPCR efficiency (E) was determined for each primer pair and checked by running a cDNA calibration curve. Samples were normalized by Ribosomal_60 s gene, chosen by the 2−Cq method69. The 2−ΔCq method was applied to the other genes. All data were calibrated by heart group. Statistical analysis was performed using Grubb’s test to detect outliers, and data normality and homoscedasticity were tested by D’Agostino & Pearson and Levene’s test, respectively. When necessary, data were submitted to logarithmic transformation. One-way ANOVA analysis of variance followed by Tukey’s Multiple Comparison’s test was used to compare transcript levels between tissues. Statistics were calculated using Statistica 7 and GraphPad Prism v5.0 software. Differences were considered statistically significant for p < 0.05.
Additional Information
How to cite this article: de Toledo-Silva, G. et al. Intracellular lipid binding protein family diversity from Oyster Crassostrea gigas: genomic and structural features of invertebrate lipid transporters. Sci. Rep. 7, 46486; doi: 10.1038/srep46486 (2017).
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
Schaap, F. G., van der Vusse, G. J. & Glatz, J. F. C. Evolution of the family of intracellular lipid binding proteins in vertebrates. Mol. Cell. Biochem. 239, 69–77 (2002).
Haunerland, N. H. & Spener, F. Fatty acid-binding proteins--insights from genetic manipulations. Prog. Lipid Res. 43, 328–49 (2004).
Chmurzyńska, A. The multigene family of fatty acid-binding proteins (FABPs): function, structure and polymorphism. J. Appl. Genet. 47, 39–48 (2006).
Zhang, J. & Haunerland, N. H. Transcriptional regulation of FABP expression in flight muscle of the desert locust, Schistocerca gregaria. Insect Biochem. Mol. Biol. 28, 683–691 (1998).
Esteves, A. & Ehrlich, R. Invertebrate intracellular fatty acid binding proteins. Comp. Biochem. Physiol. C. Toxicol. Pharmacol. 142, 262–274 (2006).
Liu, R.-Z., Li, X. & Godbout, R. A novel fatty acid-binding protein (FABP) gene resulting from tandem gene duplication in mammals: transcription in rat retina and testis. Genomics 92, 436–445 (2008).
Esteves, A., Joseph, L., Paulino, M. & Ehrlich, R. Remarks on the phylogeny and structure of fatty acid binding proteins from parasitic platyhelminths. Int. J. Parasitol. 27, 1013–23 (1997).
Gong, Y.-N. et al. Molecular cloning and tissue expression of the fatty acid-binding protein (Es-FABP) gene in female Chinese mitten crab (Eriocheir sinensis). BMC Mol. Biol. 11, 71 (2010).
Folli, C., Ramazzina, I., Percudani, R. & Berni, R. Ligand-binding specificity of an invertebrate (Manduca sexta) putative cellular retinoic acid binding protein. Biochim. Biophys. Acta - Proteins Proteomics 1747, 229–237 (2005).
Soderhall, I. et al. Characterization of a hemocyte intracellular fatty acid-binding protein from crayfish (Pacifastacus leniusculus) and shrimp (Penaeus monodon). Febs J 273, 2902–2912 (2006).
Zheng, Y., Blair, D. & Bradley, J. E. Phyletic Distribution of Fatty Acid-Binding Protein Genes. PLoS One 8, 1–9 (2013).
Mao, Y., Zhou, Y., Yang, H. & Wang, R. Seasonal variation in metabolism of cultured Pacific oyster, Crassostrea gigas, in Sanggou Bay, China. Aquaculture 253, 322–333 (2006).
Saavedra, C. & Bachère, E. Bivalve genomics. Aquaculture 256, 1–14 (2006).
Bayen, S., Kee Lee, H. & Philip Obbard, J. Exposure and response of aquacultured oysters, Crassostrea gigas, to marine contaminants. Environ. Res. 103, 375–382 (2007).
Rodrigues-Silva, C., Flores-Nunes, F., Vernal, J. I., Cargnin-Ferreira, E. & Bainy, A. C. D. Expression and immunohistochemical localization of the cytochrome P450 isoform 356A1 (CYP356A1) in oyster Crassostrea gigas . Aquat. Toxicol. 159, 267–275 (2015).
Zhang, G. et al. Molecular basis for adaptation of oysters to stressful marine intertidal environments. Annu. Rev. Anim. Biosci. 4, 2.1–2.25 (2016).
Medeiros, I. D. et al. Induced gene expression in oyster Crassostrea gigas exposed to sewage. Environ. Toxicol. Pharmacol. 26, 362–365 (2008).
Medeiros, I. D. et al. Differential gene expression in oyster exposed to sewage. Mar. Environ. Res. 66, 156–157 (2008).
Serrano, M. A. S. et al. Differential gene transcription, biochemical responses, and cytotoxicity assessment in Pacific oyster Crassostrea gigas exposed to ibuprofen. Environ. Sci. Pollut. Res. 22, 17375–17385 (2015).
Flores-Nunes, F. et al. Effect of linear alkylbenzene mixtures and sanitary sewage in biochemical and molecular responses in pacific oyster Crassostrea gigas . Environ. Sci. Pollut. Res. 22, 17386–17396 (2015).
Zhang, G. et al. The oyster genome reveals stress adaptation and complexity of shell formation. Nature 490, 49–54 (2012).
Vogel Hertzel, A. & Bernlohr, D. A. The mammalian fatty acid-binding protein multigene family: Molecular and genetic insights into function. Trends Endocrinol. Metab. 11, 175–180 (2000).
Esteves, A., Portillo, V. & Ehrlich, R. Genomic structure and expression of a gene coding for a new fatty acid binding protein from Echinococcus granulosus . Biochim. Biophys. Acta - Mol. Cell Biol. Lipids 1631, 26–34 (2003).
Gu, P.-L., Gunawardene, Y. I. N. S., Chow, B. C., He, J. G. & Chan, S.-M. Characterization of a novel cellular retinoic acid/retinol binding protein from shrimp: expression of the recombinant protein for immunohistochemical detection and binding assay. Gene 288, 77–84 (2002).
Moser, D., Tendler, M., Griffiths, G. & Klinkert, M. Q. A 14-kDa Schistosoma mansoni polypeptide is homologous to a gene family of fatty acid binding proteins. J. Biol. Chem. 266, 8447–8454 (1991).
Furuhashi, M. & Hotamisligil, G. S. Fatty acid-binding proteins: role in metabolic diseases and potential as drug targets. Nat. Rev. 7, 489–503 (2008).
Hotamisligil, G. S. & Bernlohr, D. A. Metabolic functions of FABPs—mechanisms and therapeutic implications. Nat. Rev. Endocrinol. 11, 592–605 (2015).
Yang, J. & Zhang, Y. I-TASSER server: new development for protein structure and function predictions. Nucleic Acids Res. 43, W174–181 (2015).
Arnold, K., Bordoli, L., Kopp, J. & Schwede, T. The SWISS-MODEL workspace: A web-based environment for protein structure homology modelling. Bioinformatics 22, 195–201 (2006).
Matsuoka, S. et al. Water-mediated recognition of simple alkyl chains by heart-type fatty-acid-binding protein. Angew. Chemie - Int. Ed. 54, 1508–1511 (2015).
Amber-Vitos, O., Kucherenko, N., Nachliel, E., Gutman, M. & Tsfadia, Y. The interaction of FABP with kapa. PLoS One 10, 1–24 (2015).
Hughes, M. L. R. et al. Fatty acid-binding proteins 1 and 2 differentially modulate the activation of peroxisome proliferator-activated receptor?? in a ligand-selective manner. J. Biol. Chem. 290, 13895–13906 (2015).
Linehan, L., O’Connor, T. & Burnell, G. Seasonal variation in the chemical composition and fatty acid profile of Pacific oysters (Crassostrea gigas). Food Chem. 64, 211–214 (1999).
Sacchettini, J. C., Gordon, J. I. & Banaszak, L. J. Crystal structure of rat intestinal fatty-acid-binding protein. Refinement and analysis of the Escherichia coli-derived protein with bound palmitate. J. Mol. Biol. 208, 327–339 (1989).
Flore Dagorn, Aurélie, Couzinet-Mossion, Melha, Kendel, Peter G. & Beninger Vony Rabesaotra, Gilles Barnathan and G. W.-C. Exploitable lipids and fatty acids in the invasive oyster Crassostrea gigas on the French Atlantic coast. 4662–4697, doi: 10.3390/md11114662 (2013).
Hanhoff, T., Lücke, C. & Spener, F. Insights into binding of fatty acids by fatty acid binding proteins 45–54 (2002).
Wang, L. et al. Molecular characterization and different expression patterns of the FABP gene family during goat skeletal muscle development. Mol. Biol. Rep. 42, 201–207 (2014).
Beninger, P. G. & St-Jean, S. D. The role of mucus in particle processing by suspension-feeding marine bivalves: Unifying principles. Mar. Biol. 129, 389–397 (1997).
Baines, S. B., Fisher, N. S. & Cole, J. J. Uptake of dissolved organic matter (DOM) and its importance to metabolic requirements of the zebra mussel, Dreissena polymorpha . Limnol. Oceanogr. 50, 36–47 (2005).
Bunde, T. A. & Fried, M. The uptake of dissolved free fatty acids from seawater by a marine filter feeder, Crassostrea virginica . Comp. Biochem. Physiol. Part A Physiol. 60, 139–144 (1978).
Jenny, M. J. et al. A cDNA microarray for Crassostrea virginica and C. gigas . Mar. Biotechnol. 9, 577–591 (2007).
Collin, H., Meistertzheim, A.-L., David, E., Moraga, D. & Boutet, I. Response of the Pacific oyster Crassostrea gigas, Thunberg 1793, to pesticide exposure under experimental conditions. J. Exp. Biol. 213, 4010–4017 (2010).
Newel, R. I. E. & Jordan, S. J. Preferential ingestion of organic material by the American oyster Crassostrea virginica . Mar. Ecol. Prog. Ser. 13, 47–53 (1983).
Kiorboe, T. & Mohlenberg, F. Particle Selection in Suspension-Feeding Bivalves. Mar. Ecol. Prog. Ser. 5, 291–296 (1981).
Kennedy, W. J., Taylor, J. D. & Hall, A. Environmental and Biological Controls on Bivalve Shell Mineralogy. Biol. Rev. 44, 499–530 (1969).
Lobo-Da-Cunha, A., Kádár, E. & Serrão Santos, R. Histochemical and ultrastructural characterisation of mantle storage cells in the hydrothermal-vent bivalve Bathymodiolus azoricus . Mar. Biol. 150, 253–260 (2006).
Mathieu, M. & Lubet, P. Storage tissue metabolism and reproduction in marine bivalves—a brief review. Invertebr. Reprod. Dev. 23, 123–129 (1993).
Perrat, E., Couzinet-Mossion, A., Fossi Tankoua, O., Amiard-Triquet, C. & Wielgosz-Collin, G. Variation of content of lipid classes, sterols and fatty acids in gonads and digestive glands of Scrobicularia plana in relation to environment pollution levels. Ecotoxicol. Environ. Saf. 90, 112–120 (2013).
Zhao, C., Ren, L., Liu, Q. & Liu, T. Morphological and confocal laser scanning microscopic investigations of the adductor muscle-shell interface in scallop. Microsc. Res. Tech. 78, 761–770 (2015).
Ding, Y., Zhou, Q. & Wang, W. Origins of New Genes and Evolution of Their Novel Functions. Annu. Rev. Ecol. Evol. Syst. 43, 345–363 (2013).
Kim, D. et al. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 14, R36 (2013).
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012).
Trapnell, C. et al. Differential analysis of gene regulation at transcript resolution with RNA-seq. Nat. Biotechnol. 31, 46–53 (2013).
Finn, R. D. et al. The Pfam protein families database: towards a more sustainable future. Nucleic Acids Res. 44, D279–D285 (2016).
Attwood, T. K. The PRINTS database: a resource for identification of protein families. Brief. Bioinform. 3, 252–63 (2002).
Camacho, C. et al. BLAST+: architecture and applications. BMC Bioinformatics 10, 421 (2009).
Johnson, L. S., Eddy, S. R. & Portugaly, E. Hidden Markov model speed heuristic and iterative HMM search procedure. BMC Bioinformatics 11, 431 (2010).
Robinson, J. T. et al. Integrative genomics viewer. Nat. Biotechnol. 29, 24–26 (2011).
Edgar, R. C. MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinformatics 5, 113 (2004).
Moreno-Hagelsieb, G. & Latimer, K. Choosing BLAST options for better detection of orthologs as reciprocal best hits. Bioinformatics 24, 319–324 (2008).
Milne, I. et al. TOPALi: software for automatic identification of recombinant sequences within DNA multiple alignments. Bioinformatics 20, 1806–1807 (2004).
Ronquist, F. et al. MrBayes 3.2: Efficient Bayesian Phylogenetic Inference and Model Choice Across a Large Model Space. Syst. Biol. 61, 539–542 (2012).
Sievers, F. et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 7, 539–539 (2014).
Grosdidier, A., Zoete, V. & Michielin, O. SwissDock, a protein-small molecule docking web service based on EADock DSS. Nucleic Acids Res. 39, W270–W277 (2011).
Pettersen, E. F. et al. UCSF Chimera: A visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 (2004).
Wang, J., Wolf, R. M., Caldwell, J. W., Kollman, P. A. & Case, D. A. Development and testing of a general amber force field. J. Comput. Chem. 25, 1157–1174 (2004).
Irwin, J. J. & Shoichet, B. K. ZINC − A Free Database of Commercially Available Compounds for Virtual Screening ZINC - A Free Database of Commercially Available Compounds for Virtual Screening. J. Chem. Inf. Model 45, 177–182 (2005).
Wallace, A. C., Laskowski, R. A. & Thornton, J. M. LIGPLOT: a program to generate schematic diagrams of protein-ligand interactions. Protein Eng. 8, 127–34 (1995).
Schmittgen, T. D. & Livak, K. J. Analyzing real-time PCR data by the comparative C(T) method. Nat. Protoc. 3, 1101–1108 (2008).
Acknowledgements
Fellowships of G.T.S., F.L.Z. and N.C.W. from CAPES are sincerely appreciated. A.C.D.B. is a recipient of the CNPq productivity fellowship.
Author information
Authors and Affiliations
Contributions
G.T.S., G.R. and A.C.D.B., conceived the idea and wrote the manuscript. G.T.S., N.C.W. and G.R. performed bioinformatics analysis, F.L.Z. conducted genetic expression experiments and data analysis, J.J.M. supported general analysis of results. All authors reviewed the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
de Toledo-Silva, G., Razzera, G., Zacchi, F. et al. Intracellular lipid binding protein family diversity from Oyster Crassostrea gigas: genomic and structural features of invertebrate lipid transporters. Sci Rep 7, 46486 (2017). https://doi.org/10.1038/srep46486
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep46486
This article is cited by
-
Characterization of a fatty acid-binding protein from the Pacific oyster (Crassostrea gigas): pharmaceutical and toxicological implications
Environmental Science and Pollution Research (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
